Article Text

Abstract
Hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, also called statins, are commonly prescribed medications that lower serum cholesterol and decrease cardiac morbidity and mortality. They also possess beneficial effects beyond their cholesterol-lowering properties. Preclinical data suggest statins exhibit pleiotropic antineoplastic effects in a variety of tumours, but clinical studies have provided conflicting data as to whether statins influence the risk of cancer. The biological underpinning of potential effects of statins in colorectal cancer and their role in its prevention or as adjuvant therapy are reviewed. Following a meta-analysis of both randomised clinical trials and epidemiological studies, it is concluded that available clinical data only support a modest, although statistically significant, protective effect of statins in colorectal cancer. Statins may impact on outcomes by decreasing the invasiveness or metastatic properties of colorectal cancer. The data supporting these hypotheses, however, are few and further studies are required to better assess these hypotheses. Statins may also exert a beneficial effect on colorectal cancer by sensitising the tumour to chemotherapeutic agents. Further research is needed to better define the role of statins in overcoming chemoresistance. The combination of statins with other drugs, such as low-dose aspirin or safer non-steroidal anti-inflammatory medications, may be useful in both the prevention and treatment of colorectal cancer.
- Colorectal cancer
- statins
- HMG CoA reductase inhibitors
- chemoprevention
- colorectal adenomas
- meta-analysis
- apoptosis
- cancer prevention
- carcinogenesis
Statistics from Altmetric.com
- Colorectal cancer
- statins
- HMG CoA reductase inhibitors
- chemoprevention
- colorectal adenomas
- meta-analysis
- apoptosis
- cancer prevention
- carcinogenesis
Introduction
In Western countries, the cumulative lifetime risk of developing colorectal cancer (CRC) approximates 5% in the general population.1 Moreover, CRC remains the third leading cause of cancer-related death after lung and prostate cancers in men and lung and breast cancers in women, in the United States.2 The 10–15 years required for an adenomatous polyp to evolve into clinically invasive cancer in most patients,3 and the favourable prognosis of cancers detected at early stages, have both justified the important role of screening, with different means, in this condition.4 Faecal occult blood testing (FOBT) reduces the risk of CRC mortality by 16% (RR 0.84, 95% CI 0.78 to 0.90).5 The protective effect is even greater (RR 0.75, 95% CI 0.66 to 0.84) for those attending at least one round of screening using FOBT. Alternate preventative strategies have included chemoprevention, which involves the long-term use of a variety of oral agents that can delay, prevent, or even reverse the development of adenomas in the large bowel, and interferes with the multistep progression from adenoma to carcinoma. Chemoprevention is of particular importance to individuals with a hereditary predisposition to colorectal neoplasia6 7 and to those who are especially susceptible to the environmental triggers of CRC.
Statins exert their effects through the inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase). HMG-CoA reductase catalyses the conversion of HMG-CoA into mevalonate in the mevalonate biosynthetic pathway, the rate-limiting step in the cholesterol biosynthetic pathway.8 Statins, by inhibiting cholesterol biosynthesis, emerged as one of the most important drugs responsible for lowering the incidence of cardiovascular disease, even in apparently healthy persons without hyperlipidaemia but with increased high-sensitivity C-reactive protein levels.9 10 In addition to reducing cholesterol levels, statins inhibit the generation of other products of the mevalonate pathway, including mevalonate and the downstream isoprenoids (farnesyl pyrophosphate and geranylgeranyl pyrophosphate). Post-translational isoprenylation is important in determining the membrane localisation and function of many cellular proteins, including small GTPases such as Ras and Rho.11 This pathway, with its potential roles in carcinogenesis, is summarised in figure 1. Because Ras mutations are frequent in tumours,12 and Rho proteins participate in growth-factor signalling,13 the study of the action of statins in tumour cells has largely focused on their ability to inhibit small GTPases,14 even if there is evidence that this may not be the only mechanism by which statins may inhibit proliferation and induce apoptosis. On the other hand, since cholesterol is the main structural component of cell membranes, any compound decreasing cholesterol may in turn affect various cellular events and impair homeostasis. This hypothesis has been supported by the observed increases in both non-cardiovascular-related mortality following statin therapy, and cancer risk in patients with low cholesterol levels.15 Even if these findings have since been challenged,16 17 the possible effect of statins on the incidence of various cancers has been assessed as part of safety analyses of cholesterol lowering trials and will also be addressed.

Mevalonate pathway. Statins inhibit the conversion of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) to mevalonate. Mevalonate is then phosphorylated to form pyrophosphomevalonate, which is then converted to isopentenyl pyrophosphate (isopentenyl-PP). Isopentenyl-PP and dimethylallyl pyrophosphate (dimethylallyl-PP) can then be combined to form the 10-carbon isoprenoid geranyl pyrophosphate (geranyl-PP). Additional isopentenyl-PPs can be added to produce farnesyl pyrophosphate (farnesyl-PP), the 15-carbon isoprenoid, and geranylgeranyl pyrophosphate (geranylgeranyl-PP), the 20-carbon isoprenoid. Inhibition of this pathway by statins prevents the formation of both mevalonate and its downstream product, isopentenyl-PP. Several other branches of this pathway can convert farnesyl-PP into various other products, including cholesterol. In general, farnesyl-PP helps prenylate proteins in the Ras family, whereas geranylgeranyl-PP helps prenylate proteins in the Rho and Rac families. Ras, Rho and Rac proteins have several physiological functions potentially involved in carcinogenesis.
Methods for the meta-analysis
Search strategy
We performed a comprehensive search on a possible relationship between statins and colorectal or digestive tumours by searching EMBASE, MEDLINE, CENTRAL and the ISI Web of knowledge. The identification of articles was developed using a highly sensitive search strategy to identify reports with a combination of controlled vocabulary and text words related to: (1) statin (hydroxymethylglutaryl-CoA reductase inhibitor, pravastatin, simvastatin, atorvasatin, rosuvastatin, fluvastatin, mevastatin); and (2) cancer (carcinoma, adenoma, tumour, polyp, lesion). In addition recursive searches and cross-references were carried out using a ‘similar articles’ function and hand searches of articles identified after an initial search. We included all adult human studies in French or English and included abstracts, spanning the last 10 years up to September 2009. In the case of studies with incomplete information, we attempted to contact authors to obtain additional data.
Inclusion criteria
We included the following types of articles: (1) randomised clinical trials, case–control and cohort studies that either assessed or reported colorectal, digestive, gastrointestinal, colon and rectal cancer prevalence in subjects taking, or not taking, statins; and (2) articles that contained sufficient detail to reconstruct 2×2 tables expressing colorectal cancer prevalence by statin intake status. Since randomised clinical trials assessed primary cardiovascular outcomes and were not designed to assess CRC, full papers were manually reviewed (MB and MM), to search for CRC data.
Exclusion criteria
We excluded all studies whose designs did not fulfil inclusion criteria, studies not published in English or French, studies without specific cancer site assessment, and studies not assessing digestive tract cancers. If more than one study was included from the same author, we carefully assessed the absence of overlap by using the recruitment periods noted in the manuscript or by contacting the author if the report did not provide these data.
The data were extracted by two independent reviewers (MB and MM) with discrepancies settled by a third investigator (AB). Analyses were first performed separately for randomised controlled trials, and cohort or case–control studies, and then altogether as previously reported by Shrier et al18 who suggested that the advantages of including both observational and randomised studies in a meta-analysis could outweigh the disadvantages in many situations, and that observational studies should not be excluded a priori. To quantify the strength of any observed association, we used the Mantel–Haenszel method from the 2×2 tables defined by the statin group and the incidence of colorectal cancer for the randomised controlled trials. For observational studies, we preferred the multiple risk factor adjustment to the crude risk ratio. The estimates of risk ratio were transformed to their natural logarithm before pooling and the variance was calculated by ((ln(upper CI RR)−ln (lower CI RR))/3.92)2.19 Fixed effect models were applied to all comparisons to determine corresponding overall effect sizes and their confidence intervals, unless heterogeneity was noted, in which case a random effect model was employed. The Higgins I-squared statistic was calculated to quantify the proportion of variation in treatment effects attributable to between-study heterogeneity. Values of I-squared equal to 25%, 50% and 75% represent low, moderate and high heterogeneity, respectively. The presence of heterogeneity across studies was defined using a χ2 test of homogeneity with a 0.10 significance level. To better characterise possible sources of statistical heterogeneity, sensitivity analyses were carried out excluding studies one-by-one. We performed all meta-analyses using the RevMan 5 software.
Statins and the prevention of colorectal cancer
What is the rationale supporting a potential protective effect of statins against CRC?
Several mechanisms may be responsible for an antitumour effect of statins, including induction of apoptosis, inhibition of cell growth or angiogenesis or enhancement of immune response.20
Apoptosis
Colorectal cancer is thought to originate in the expansion of colonic crypt cells as a result of aberrant gene expression caused by transcription factors of the T-cell factor (TCF)/β-catenin family.21 Many of the genetic errors that accumulate during colorectal carcinogenesis affect the control of apoptosis, and many studies have shown that colorectal carcinogenesis is related to the inhibition of apoptosis and the augmentation of proliferative activity.22 Effective chemoprevention strategies for colorectal cancer must thus target these genetic defects and promote or restore apoptosis. Mutation of the APC gene is often the initiating genetic lesion observed in colorectal cancers.23 Depending on the cellular context, loss of APC activates the Wnt signalling pathway, causing immediate widespread apoptosis of colorectal epithelial cells and defects in differentiation and cell migration. Only cells that are inherently resistant to apoptosis survive this initial wave of apoptosis. These surviving cells constitute the epithelial population that develops into adenomas. Two gene targets of the Wnt signalling pathway are of particular relevance to apoptosis: survivin24–26 and the proto-oncogene c-MYC.27–29 Although controversial, survivin may enhance cell proliferation and inhibit apoptosis,26 30 and is overexpressed in colorectal carcinomas.31 c-MYC in normal cells is involved with both the induction of apoptosis and cell proliferation. As these are opposing functions, c-MYC can only induce cell proliferation if apoptosis has first been disabled.
Statins have been shown to induce apoptosis through both extrinsic and intrinsic pathways, leading to cell death.32–35 Although the mechanism of statin-induced apoptosis has not been fully elucidated, statin-induced apoptosis is likely due to the depletion of geranylgeranylated or farnesylated proteins, which may be dependent on cell type or the state of differentiation (see figure 2).36
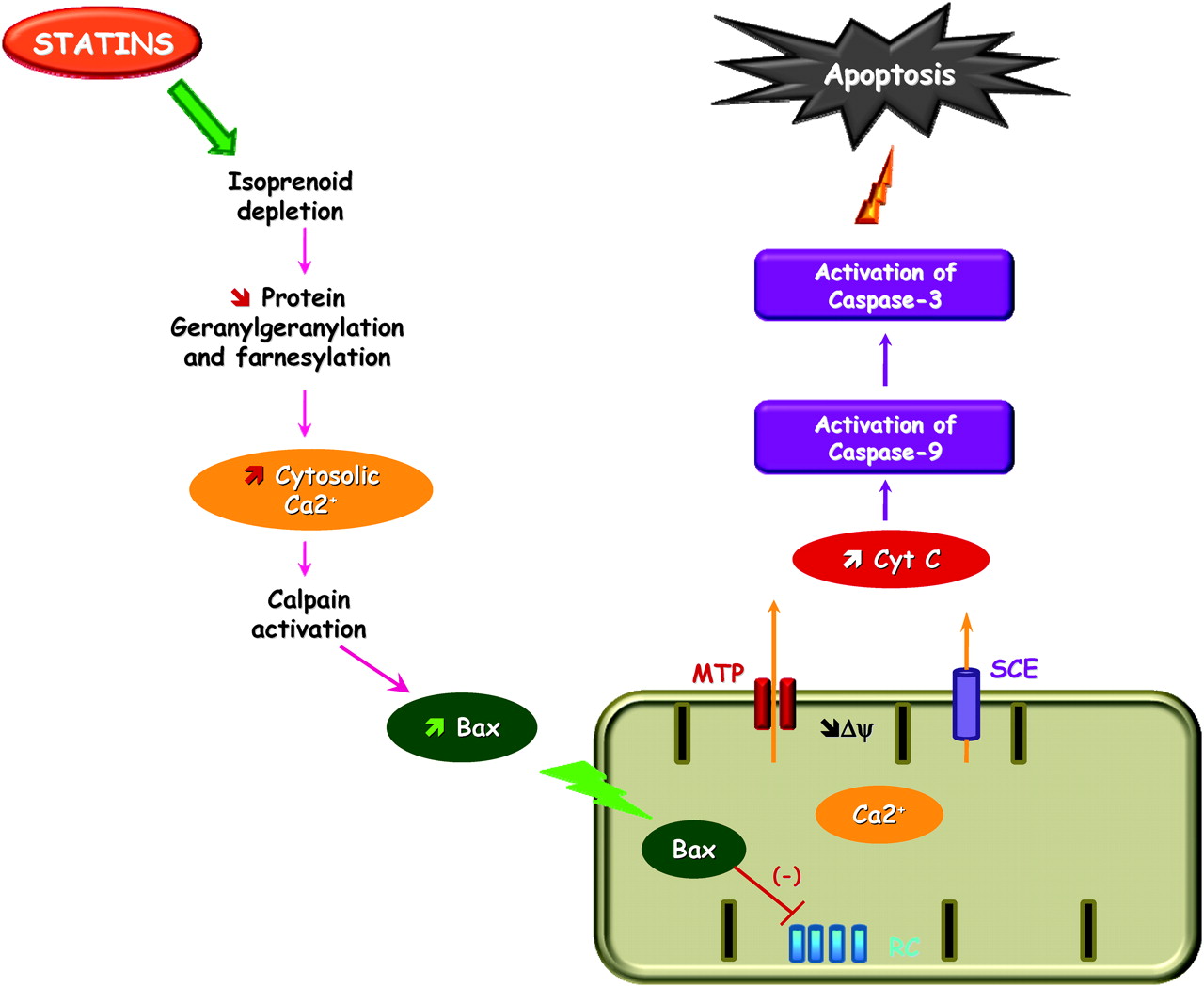
Suggested effect of statin on mitochondria-mediated apoptotic signalling. HMG-CoA reductase inhibition by statins increases cytosolic calcium concentration and expression of the pro-apoptotic protein Bax. Bax in turn inhibits complex I of the mitochondrial respiratory chain (RC). This depolarises the inner membrane (Δψ) triggering a calcium release through the mitochondrial transition pore (MTP) and sodium calcium exchanger (SCE). Cytochrome c (Cyt C) release from the mitochondria leads to activation of procaspase-9. Active caspase-9 cleaves and activates procaspase-3, which leads to apoptosis.
Experiments conducted in spontaneously immortalised rat intestinal epithelial cells, IEC-18 and their K-ras transformed clones, have shown that lovastatin induces morphological changes and apoptosis (not influenced by K-ras mutations), by inhibiting geranylgeranylation of small GTPases of the rho family, thereby inactivating these.32 Shibata et al showed, in a mouse model of metastatic mammary cancer carrying a p53 mutation, that lovastatin induced both a decrease in DNA synthesis and an increase in apoptosis.37 Although this study was not performed on colorectal cancer cells, it suggests a p53-independent pathway, which is of particular interest since APC, K-ras, Smad4 (or DPC4-deleted in pancreatic cancer 4) and p53 genes have all been incriminated in CRC. Nevertheless there are, to our knowledge, no data suggesting that the effect of statins on CRC may depend on K-ras activation status.
More recently, Cho et al showed that simvastatin induces apoptosis in human colorectal cancer COLO 205 and HCT 116 cell lines in a dose and time-dependent manner, and down-regulates the expression of antiapoptotic proteins Bcl-2, Bcl-xL, cIAP1 and cFLIP.33 These authors also showed that simvastatin is able to reduce tumour development in a colitis-associated colon cancer (CAC) model in C57/BL6 mice.33 In addition they inoculated subcutaneously 5×106 COLO205 cells into BALB/c nu/nu mice, to develop a xenograft model, and observed that tumours from animals treated with simvastatin had smaller volumes, larger necrotic areas, lower expression of VEGF and higher apoptotic scores compared to controls.33 Simvastatin may thus have the potential to inhibit CRC development by inducing apoptosis and suppressing angiogenesis. Using an experimental (APCMin mice) model for familial adenomatous polyposis, Swamy et al38 showed that atorvastatin and celecoxib were both able to induce apoptosis in CRC, with a trend towards a more pronounced effect with atorvastatin. Conversely, Xiao et al34 reported that neither atorvastatin nor celecoxib were able to induce apoptosis in two human colon cancer cell lines (HCT116 and HT29), whereas the combination of both drugs induced apoptosis, but only after a treatment of 48 h or more.34 This apparent discrepancy might be explained by the differing sensitivities of CRC cell lines to statin treatment.39
The BMP (bone morphogenetic protein) pathway has recently been implicated in CRC with the identification of germline mutations in BMPR1a and SMAD4 in families with familial juvenile polyposis syndrome.40 Affected individuals exhibit a greatly increased risk of developing cancer.41 42 BMP2 is a promoter of apoptosis in mature epithelial cells in the colon,43 and Kodach et al39 have shown that statin-sensitive CRC cell lines show inductions of both the BMP pathway and the BMP target gene ID-2. These authors suggested that the beneficial or deleterious effects of statins might depend on the CRC cells line, since they observed in a xenograft mouse model that simvastatin induced inhibition of tumour growth when using sensitive cells, while it promoted tumour growth when insensitive cells were used.39
Kaneko et al44 reported that lovastatin induces apoptosis in the human colon cancer cell line SW480 by blocking the cholesterol synthesis pathway. They observed that, among all antiapoptotic proteins that were assessed, survivin was the only one to be down-regulated by lovastatin. Furthermore, farnesyl pyrophosphate and geranylgeranyl pyrophosphate (see figure 1) simultaneously reversed surviving down-regulation, indicating that the effects of statins are indeed mediated through HMG-CoA reductase inhibition.44
To summarise, statins can, at least theoretically, oppose colorectal cancer growth by inducing apoptosis, through a down-regulation of antiapoptotic proteins such as BCl2 or cIAP1 and an up-regulation of proapoptotic proteins such as BMP, and inhibiting tumour angiogenesis.
Inhibition of cell proliferation
Cell proliferation in the upper part of colonic crypts is a premalignant marker,45 and inhibition of cancer cell proliferation through HMG-CoA reductase inhibition and isoprenoids depletion (figure 1) has been one of the mechanisms implicated in the anticancer effect of statins. For example, Hong et al46 showed that lovastatin decreases cellular proliferation in HCT-116 and HT-29 human colon cancer cells. This effect, mediated through a G0/G1 cell cycle arrest, was also described for mevastatin and atorvastatin47 in Caco-2 colorectal cancer cell lines.48 It has been reported, although not in colorectal cancer cells but in breast cancer cells, that cerivastatin treatment modified the expression of 13 genes that may contribute to the inhibition of both cell proliferation and invasion, either directly or indirectly by stimulating an anti-angiogenic gene, an effect mainly explained by the inhibition of RhoA-dependent cell signalling.49 It is important to note, however, that a statin-induced effect such as the inhibition of cell proliferation, may not be generalisable to other cancer cell lines.50
Inhibition of angiogenesis
Angiogenesis is indispensable for the growth of solid tumours, and without the supply of new blood vessels, the size of a tumour can only reach a volume of about 2 mm3.51 It has been suggested that angiogenic switch occurs at the onset of dysplasia in the adenoma carcinoma sequence.52 Targeting the formation of blood vessels is therefore regarded as a promising strategy in cancer therapy. Statins exhibit both pro-angiogenic effects, which are regarded as beneficial for the treatment of cardiovascular diseases, and anti-angiogenic activities that can be of particular importance in anticancer therapy. Although most of the studies were not performed in cancer models, some mechanisms have been suggested underlying the pro- or anti-angiogenic effects of statins, including an increase in bone marrow-derived endothelial progenitor cells or activation of Akt and nitric oxide synthase.53 It has been suggested that low statin doses induce a pro-angiogenic effect, whereas high statin doses may decrease protein prenylation and inhibit cell growth.54 Additionally, the same dose of a given statin may either promote or inhibit angiogenesis based on the presence of other angiogenesis promoters, such as hypoxia or TNF.55 Moreover, the statin-mediated inhibition of vascular endothelial growth factor (VEGF) synthesis, the major angiogenic mediator, may contribute to the attenuation of angiogenesis. Pravastatin has also been shown, using human umbilical vein endothelial cells, to induce a dose-dependent decrease in the proliferative activity of endothelial cells, which is dependent on the cell cycle arrest to the G1 phase and not on cell apoptosis.56 There exist few data supporting the anti-angiogenic role of statin therapy in CRC. Cho et al33 assessed the anti-angiogenic effect of simvastatin using anti-VEGF antibody in a xenograft model where human colorectal cancer cells (COLO205) were inoculated subcutaneously into mice. They found that simvastatin suppressed VEGF expression both in vitro and in vivo, and suggested that simvastatin inhibits VEGF-mediated angiogenesis via the NF-kB pathway.33
Decrease in metastatic capacity
It has been suggested that the Rho family of small GTPases plays a role in growth and metastatic capacity of various tumours, such as colorectal cancer.57 58 A few years ago, Nubel et al59 showed that lovastatin significantly blocked cytokine-induced adhesion of colon cancer cells to primary human endothelial cells (HUVEC) and that this effect was mediated through an inhibition of Rho-regulated expression of E-selectin by TNF. This is of particular interest since it has been suggested that E-selectin plays an important role in the attachment of tumour cells to the endothelium during the process of metastasis as it has been shown to be crucial not only for initial adhesion and rolling of circulating HT-29 colon cancer cells on the endothelium but also for their subsequent diapedesis.60 Statins inhibit Rho activation (see figure 1) and have been shown, for example in an in vitro model of pancreatic cancer invasiveness, to inhibit cancer cell invasion induced by epidermal growth factor (EGF), in a manner sensitive to C3 transferase, a specific inhibitor of Rho.61 Additionally, using an in vivo model of hepatocellular carcinoma in rats, Parag et al62 have shown that fluvastatin had a dose-dependent inhibitory effect on primary and metastatic tumours, and that the inhibitory effect on growth was more pronounced in metastases than in primary tumours. Nevertheless, convincing data showing a statin-induced decrease in metastatic capacity of colorectal cancer are scarce.
All the aforementioned effects, that may explain in part the beneficial effects of statins in CRC, are largely overlapping, and additional effects, such as the modulation of inflammation or immune responses, may also play important roles. The latter are controversial since statins affect multiple cell populations relevant to the immune response, including B cells, T cells, regulatory T cells, macrophages and dendritic cells; some favour immune tolerance while others an immune response role.
Is there clinical evidence supporting a protective effect of statins in CRC?
The association between statin use and CRC occurrence remains controversial. Following the observations that simvastatin or pravastatin exhibited chemopreventive effects in colon carcinogenesis,63 many studies have since explored the effect of statins, reaching disparate conclusions. However, most of the results from randomised clinical trials were obtained from studies that were not primarily designed and powered to assess the role of statin in CRC prevention.
Several meta-analyses have been published in the past few years; most have not concluded whether or not there is a protective effect of statins in CRC.64–69
Our literature search using the following MeSH terms indentified 4002 citations for a final inclusion of 32 studies. MeSH terms used: Hydroxymethylglutaryl-CoA Reductase Inhibitors; Statins; HMG-CoA reductase inhibitors; Pravastatin; Simvastatin; Atorvasatin; Rosuvastatin; Fluvastatin; Mevastatin; Colorectal Neoplasms; Carcinoma; Adenoma; Tumor; Polyp; Cancer; Lesion. Figure 3 presents the QUORUM diagram.

QUORUM diagram.
Randomised clinical trials
We retrieved 930 references and identified 11 studies, with a total of 95 984 patients (47 884 in the treatment and 48 004 in the control group) that were included in the analysis (table 1). Pooled analysis showed a modest trend towards a protective effect of statins against the occurrence of CRC (RR 0.94; 95% CI 0.86 to 1.04, p=0.23) that failed to reach statistical significance (figure 4)—a result consistent with that of the meta-analyses of Dale et al65 and Bonnovas et al,67 who also found non-significant odds ratios of 1.02 and 0.95, respectively.
Randomised controlled trials included in the meta-analysis
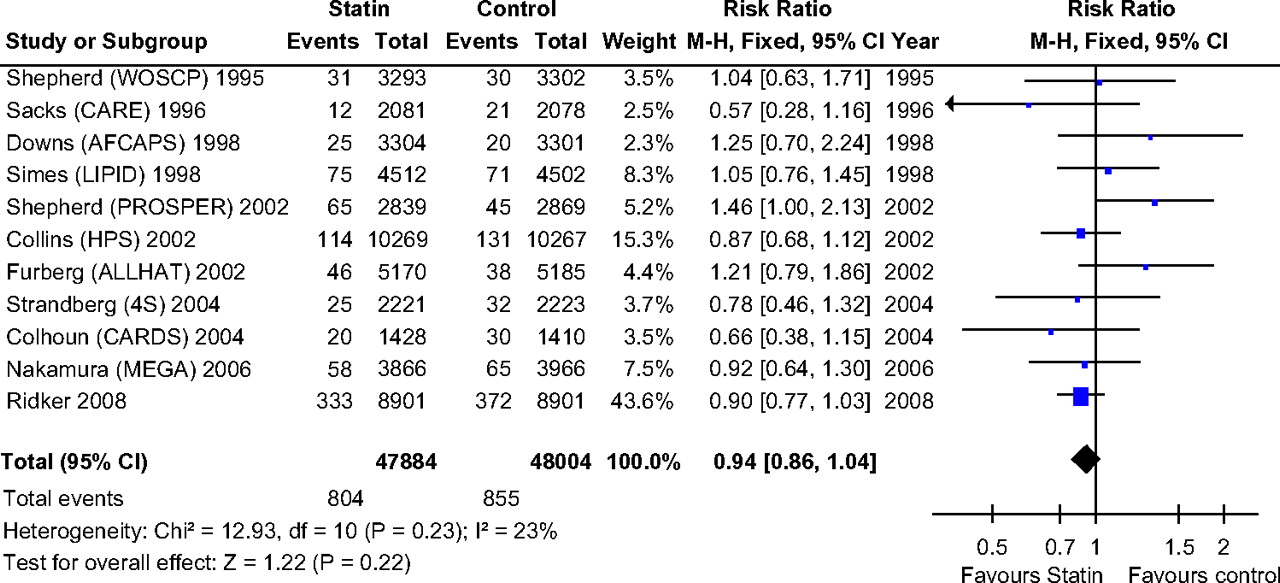
Forrest plot: results from individual studies and meta-analyses of randomised clinical trials. The risk ratio and 95% CI for each study are displayed on a logarithmic scale. Pooled estimates are from a fixed-effect model.
The test for heterogeneity was 0%, indicating no variability between studies that cannot be explained by chance, and funnel plot analysis did not reveal significant publication bias. In sensitivity analysis, only the removal of the ALLHAT-LLT70 trial had a significant impact on the RR which then reached statistical significance (0.90 (0.81 to 0.99)) without clear explanation, except that it was the only non-blinded study. Although it has been suggested that different statins might not interfere with cell cycle and apoptosis to the same extent,71 72 none of the assessed statins (pravastatin, lovastatin, simvastatin, atorvastatin and rosuvastatin) appeared to differ in their impact on CRC risk. Overall, 667 patients need to receive long term statin therapy to prevent one CRC. Importantly, even if all but one study had a treatment/follow-up of more than 4 years, most of these clinical trials were not designed to assess CRC incidence, and the duration of follow-up may have been too short. Our meta-analysis did not suggest any trend associating treatment duration and risk reduction for CRC, but most of the studies exhibited a follow-up of about 5 years, with only two extending beyond 8 years. The observed incidence ratio of CRC brings into question the feasibility of a clinical trial dedicated to this question.
Observational studies
From the 930 references retrieved, we included 13 case–control (821 416 patients) and 8 cohort studies (784 818 patients). Details of the studies are presented in tables 2 and 3, respectively.
Case–control studies included in the meta-analysis
Cohort studies included in the meta-analysis
We assessed case–control studies separately and found that statin use was associated with a significant, although modest risk reduction in CRC risk (adjusted RR (aRR) 0.92, 95% CI 0.90 to 0.94, p<0.001) (figure 5). No heterogeneity was found among studies, and sensitivity analysis did not suggest that the omission of any of the included studies altered the magnitude of the observed effect.
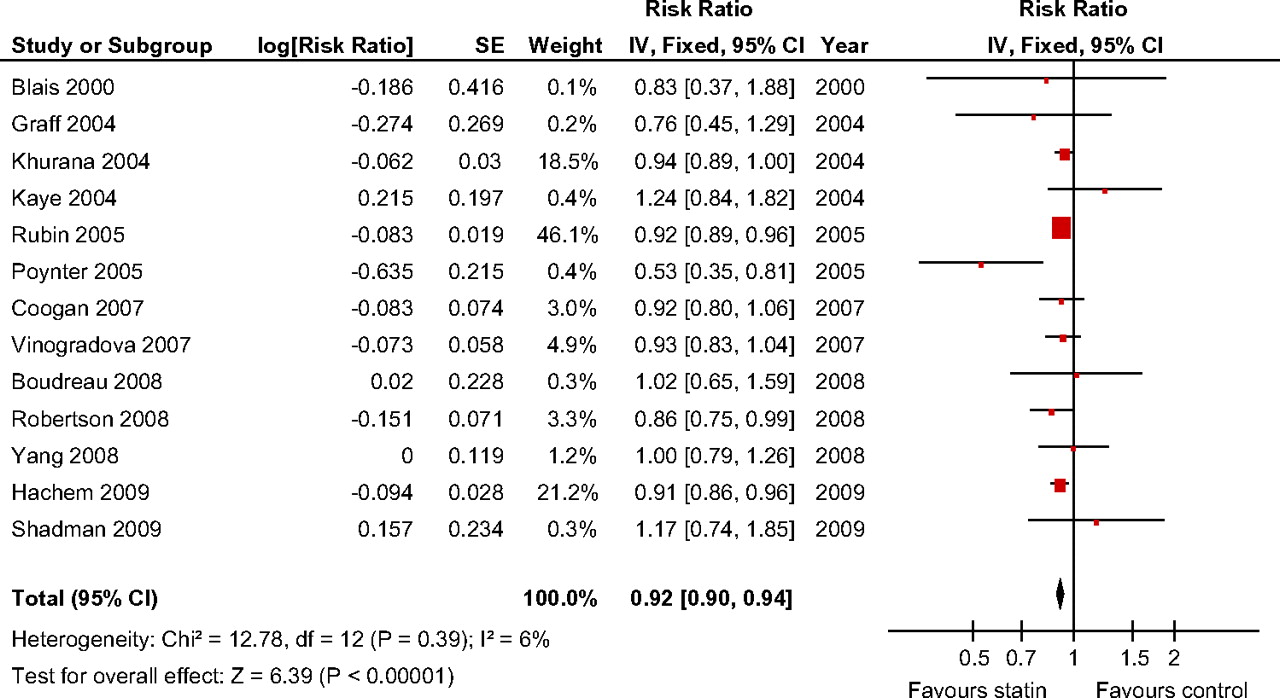
Forrest plot: results from individual studies and meta-analyses of case–control studies. The risk ratio and 95% CI for each study are displayed on a logarithmic scale. Pooled estimates are from a fixed-effect model.
When assessing cohort studies, significant heterogeneity was observed, with a non-significant trend towards a small risk reduction of CRC among statin users (aRR 0.89, 95% CI 0.75 to 1.05, p=0.16) (figure 6). Only the removal of the study by Singh et al,73 the only study with a significant increased risk for CRC in statin exposed compared to non-exposed patients (RR 1.13, 95% CI 1.02 to 1.25), had a significant impact on the observed RR of CRC among statin users, that just reached statistical significance (aRR 0.84, 95% CI 0.71 to 1.00).When pooling case–control and cohort studies together, despite significant heterogeneity, a small but significant risk reduction of CRC in statin users was noted (RR 0.82, 95% CI 0.87 to 0.98).

Forrest plot: results from individual studies and meta-analyses of cohort studies. The risk ratio and 95% CI for each study are displayed on a logarithmic scale. Pooled estimates are from a random-effect model.
Finally we combined randomised clinical trials and observational studies (31 studies, 2 552 943 patients) and found a 9% risk reduction in CRC risk (aRR 0.91, 95% CI 0.87 to 0.96, p<0.001) (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forrest plot: results from individual studies and meta-analyses of all 34 randomised clinical trials and observational studies. The risk ratio and 95% CI for each study are displayed on a logarithmic scale. Pooled estimates are from a random-effect model.
Summary
It appears that combination of both randomised clinical trials and observational studies suggests a small impact on CRC occurrence attributable to chronic statin use. The numbers needed to treat (NNT) are correspondingly modest (varying from ∼ 670 to 4800). These estimates have to be compared to that of statin therapy in cardiovascular prevention where the five-year NNT to prevent heart attack or stroke for statins in people with an annual risk of 3% is about 20. Hundreds of thousands of people would have to be enrolled in randomised trials to demonstrate a protective effect of statin therapy against CRC. Thus available data do not support the recommendation of chronic statin use to prevent CRC occurrence in patients at low or intermediate risk for CRC and there are no data available in high-risk patients with familial adenomatous polyposis (FAP) or Lynch syndrome.
Limitations of non-randomised studies relate to the possible influence of known and unknown factors that may affect outcome and be unequally distributed among cases and controls. One of the most obvious confounding factors, in the case of CRC, relates to lifestyle habits, like smoking and drinking, and dietary risk factors like meat, fat and fruit/vegetables consumption which are likely to be related both to the exposure, statin use, and the outcome, of CRC occurrence. Most of these factors are often unlikely to be captured in population databases, making adjustment unfeasible. One of the limitations of the randomised controlled trials that were included in the meta-analysis is that they were not designed to assess cancer incidence and survival, leading to possible inaccuracies in CRC incidence reporting, that may not necessarily have been equally distributed in both groups.
Is there clinical evidence supporting a protective effect of statins against colorectal adenomas?
Although the data supporting a protective effect of chronic statin use in CRC are not very conclusive, colorectal carcinogenesis is a multi-step process. The relationship between statin use and colorectal adenomas, an earlier step, has thus also been questioned. Surprisingly only few data are available to address this issue.
Statins and adenoma occurrence
In a secondary analysis of data from three large colorectal adenoma chemoprevention trials, statin use was not associated with a reduced adenoma risk (1.03, 95% CI 0.87 to 1.23), nor was it associated with decreased risks of advanced adenoma (1.13, 95% CI 0.70 to 1.81) or multiple adenomas (1.25, 95% CI 0.95 to 1.65).74
A case–control study published in abstract form in 2007 evaluated whether self-reported use of statin was associated with a reduced incidence of colorectal adenomas.75 Data from 1570 subjects (554 cases and 1016 controls), of whom 35.1% reported regular use of statin medications over the previous 5 years, were analysed. No significant association was found linking statin use to a decreased occurrence of colorectal adenomas (adjusted OR 0.96, 95% CI 0.77 to 1.21). On the other hand it was also reported, in a small cohort study published only in abstract form in 2006, that patients using statin were more likely to be diagnosed with adenomatous polyps (118/227, 52%) compared to patients not taking a statin (186/452, 41%) (OR 1.55, 95% CI 1.12 to 2.13).76 These results must be considered with great caution as they remain unpublished as a full paper, and adenoma prevalence was very high.
Statin and prevention of adenoma recurrence
Siddiqui et al77 recently identified 2626 patients (84% men, mean age 62.2 years) through a review of endoscopy and pathology databases with adenomatous polyps removed at an index colonoscopy who had undergone a follow-up colonoscopy 3–5 years later. Statin use was assessed through a review of medical and pharmacy records. Overall, 583 of 1688 patients (35%) who used statins continuously had an adenoma found on follow-up colonoscopy, compared to 477 of 938 patients (51%) who did not (OR 0.51, 95% CI 0.43 to 0.60, p <0.01). From this study, the number needed to treat with a statin to avoid an adenoma was calculated as 6–7. Statins were also associated with decreased polyp numbers (2.6 vs 3.1, p=0.002), smaller polyp size (7.1 vs 7.9 mm, p=0.03), and a significant reduction in the incidence of advanced adenomas (OR 0.74, 95% CI 0.52 to 0.96, p=0.03).
More recently Bertagnoli et al78 reported the results of a planned secondary analysis of a colorectal adenoma recurrence prevention trial where 679 of 2035 adenoma patients were randomised to receive placebo and the others varying dose of celecoxib. Analyses performed in the placebo arm of this study, i.e. those not receiving celecoxib, suggest that for patients at high risk of colorectal cancer, statins do not protect against colorectal adenoma (RR 1.24, 95% CI 0.99 to 1.56, p=0.065 for those who used statins at any dose during the 5 years follow-up) and may even increase the risk of developing colorectal adenomas (RR 1.39, 95% CI 1.04 to 1.86, p=0.024 for those having used statin for more than 3 years).
Thus, the effect of statins on adenomatous polyps remains controversial, with disparate results stemming from secondary analyses of interventional trials or three separate observational or case–control trials.
Additional mechanisms of statins that may underlie a benefit in CRC other than through a chemopreventive effect
Additionally to a potential chemopreventive effect, statins might also interfere with CRC by inhibiting the ability of cancer cells to metastasise, as it has been reported that, among patients diagnosed with colorectal cancer, statin users had a 30% decreased prevalence of metastasis compared to statin non-users (OR 0.7, 95% CI 0.4 to 0.9, p<0.01)79 or enhancing the response to chemotherapeutic agents. It has been shown that pretreatment of different colon cancer cell lines with lovastatin significantly increases apoptosis induced by 5-fluorouracil (5-FU) or cisplatin,80 an effect described in both drug-sensitive and drug-resistant cell lines.81 Statins have also been shown, although it was not in colorectal cancer cells, to favour reversion of chemoresistance by reducting in P-glycoprotein expression a protein implicated in drug efflux.82 Statin use may thus overcome chemoresistance.
A randomised controlled trial comparing rosuvastatin to placebo in 5011 patients aged 60 or more with New York Heart Association class II, III or IV systolic heart failure followed for up to 36 months83 showed identical cancer mortality rates in both groups (0.8%). Unfortunately the study did not provide results specifically addressing CRC mortality; moreover, included patients had a severely impaired cardiac function with about one third of patients dying within 36 months in both groups, not allowing for long term outcome, such as CRC, assessment. A recent case–control study of 1309 men with a new diagnosis of CRC (mean age 69±1.1 (SE) years; 326 statin users, 983 statin non-users) suggested that in patients who presented to hospital with CRC, long-term use of statins was associated with less advanced tumour stage, a lower frequency of distant metastases, and an improved 5-year survival rate (37% vs 33%; OR 0.7, 95% CI 0.6 to 0.9, p=0.03).79 This study is principally limited by possible recall bias, relatively small sample size, and the heterogeneous evaluation of CRC extension and the absence of information on cause of death. A retrospective study of 349 patients undergoing neoadjuvant chemoradiation for either locally advanced tumours or low-lying tumours requiring abdomino-perineal resection, compared results between patients on or off statins.84 Only 9% used a statin, and overall, 23 non-statin users (7%) were found to have metastatic disease at the time of surgery, compared to none among statin users. In multivariable analysis, the odds ratio for statin use as predictor of complete histological response was 4.2 (95% CI 1.7 to 12.1, p=0.003).
A recent open-label phase II trial evaluating the efficacy and toxicity profile of conventional FOLFIRI chemotherapy in association with simvastatin in metastatic colorectal cancer patients85 suggested this combination exhibited promising antitumour activity.
Other perspectives: the combination of statins with low-dose aspirin or non-steroidal anti-inflammatory drugs
If statin therapy by itself does not appreciably reduce the risk of CRC, the combination of statin plus another drug such as a non-steroidal anti-inflammatory agent (NSAID) or low dose aspirin might be considered. Randomised clinical trials have shown that cyclooxygenase-2 inhibitors can reduce colorectal adenoma recurrence,86 87 and epidemiological studies have suggested that their use is associated with a decreased risk of colorectal carcinoma.88 Some studies have shown that statins and NSAIDs can act synergistically to inhibit cell cycle and promote apoptosis. Xiao et al showed a dramatic increase in growth inhibition of cancer cell lines, from approximately 10% to 60%, when atorvastatin was combined with celecoxib, compared to both drugs used separately—an effect explained by a potentiation of G0/G1 arrest.34 These findings were confirmed and expanded very recently by Yang et al47 and support the need for further studies.
Conclusions
Although secondary analysis of randomised trials or cohort studies has failed to convincingly show a significant benefit attributable to statins in colorectal cancer, these agents have appeared beneficial, although modestly so, in case–control studies or when all studies were pooled together. They may thus have a role to play in colorectal cancer, perhaps more as adjuvant agents to chemotherapy, or in combination with drugs exhibiting different mechanism of action, such as NSAIDs or low-dose aspirin, or with antioxidants, for example.89 Whereas experimental and clinical data suggest some differences in statin sensitivity across several types of cancers, such as of the breast, prostate or liver, no data support the hypothesis that any observable effect may be limited to certain cancers of particular molecular subtypes. Further research is needed to validate these interpretations and postulates, and assess more precisely the effect of statins in the prophylaxis of colorectal adenomas, or as combination agents with adjuvant or neo-adjuvant chemotherapy.
AB is the holder of the DG Kinnear Chair in Gastroenterology at McGill University, and is a Research Scholar (Chercheur National) of the Fonds de la Santé en Recherche du Québec.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.