Article Text

Abstract
Background and aims CD8 T cells are central to the control of hepatitis C virus (HCV) although the key features of a successful CD8 T cell response remain to be defined. In a cohort of Irish women infected by a single source, a strong association between viral clearance and the human lecucocyte (HLA)-A*03 allele has been described, and the aim of this study was to define the protective nature of the associated CD8 T cell response.
Methods A sequence-led approach was used to identify HLA-A*03-restricted epitopes. We examine the CD8 T cell response associated with this gene and address the likely mechanism underpinning this protective effect in this special cohort, using viral sequencing, T cell assays and analysis of fitness of viral mutants.
Results A strong ‘HLA footprint’ in a novel NS3 epitope (TVYHGAGTK) was observed. A lysine (K) to arginine (R) substitution at position 9 (K1088R) was seen in a significant number of A*03-positive patients (9/12) compared with the control group (1/33, p=0.0003). Threonine (T) was also substituted with alanine (A) at position 8 (T1087A) more frequently in A*03-positive patients (6/12) compared with controls (2/33, p=0.01), and the double substitution of TK to AR was also observed predominantly in HLA-A*03-positive patients (p=0.004). Epitope-specific CD8 T cell responses were observed in 60% of patients three decades after exposure and the mutants selected in vivo impacted on recognition in vitro. Using HCV replicons matched to the viral sequences, viral fitness was found to be markedly reduced by the K1088R substitution but restored by the second substitution T1087A.
Conclusions It is proposed that at least part of the protective effect of HLA-A*03 results from targeting of this key epitope in a functional site: the requirement for two mutations to balance fitness and escape provides an initial host advantage. This study highlights the potential protective impact of common HLA-A alleles against persistent viruses, with important implications for HCV vaccine studies.
- (MeSH)
- T cells
- viral fitness
- footprints
- cellular immunity
- chronic viral hepatitis
- hepatitis c
- HLA
- immune response
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- (MeSH)
- T cells
- viral fitness
- footprints
- cellular immunity
- chronic viral hepatitis
- hepatitis c
- HLA
- immune response
Significance of this study
What is already known about this subject?
Hepatitis C virus (HCV) infection is a leading cause of cirrhosis and liver disease globally.
Functional CD4 and CD8 T cells are thought to be critical to the process of viral clearance, although many aspects of the nature of a successful response remain to be defined.
The occurrence of a large single-source outbreak of HCV in Ireland has provided clear insights into the natural history of HCV, and genetic studies of this unque cohort have previously identifed the class I allele human leucocyte antigen (HLA)-A*03 as an independent and significant determinant of viral clearance (OR 2.8).
We examine the CD8 T cell response associated with this gene and address the likely mechanism underpinning this protective effect in this special cohort.
What are the new findings in this study?
We identify a novel T cell epitope restricted by HLA-A*03 (NS3 1080–1088) which is commonly targeted by women in this cohort (60%).
Immune pressure imposed by CD8 T cells appears to drive the selection of an escape mutation at a key anchor site within the epitope (K1088R, p=0.0003). A second common substitition is also observed (T1087A, p=0.01) in this group.
This study shows that the K1088R substitution is associated with a significant loss of fitness; however, the second substitution (T1087A) appears to compensate this and restores viral fitness.
The protective effect of the HLA-A*03 allele may thus potentially be explained by the requirement for two mutations which balance fitness and escape.
How might this study impact on clinical practice in the future?
Unlike other protective alleles, HLA-A*03 is a prevalent allele (∼20% of the population). Identifying important epitopes linked to protection in HCV has important implications with respect to HCV vaccine studies.
Introduction
Hepatitis C virus (HCV) is a single-stranded RNA virus and replication occurs through the NS5B-encoded RNA polymerase which lacks proof reading ability.1 As a consequence, mutations are readily generated and are subject to immune selection pressure. Mutations that facilitate immune escape are used by the virus to establish persistence.2 3 Approximately 20% of those exposed will clear the infection spontaneously.1 The factors that determine outcome after HCV infection have been extensively investigated and there is substantial immunological evidence that the cellular immune response is a key component in viral clearance. Studies have shown that the appearance of the T cell response within the liver coincides temporally with control of viral replication.4 Furthermore, antibody-mediated depletion of CD4 and CD8 T cells in chimpanzees re-challenged with autologous virus was associated with persistent viraemia.5 6 A study which examined the relationship between major histocompatibility complex (MHC) class I alleles and viral outcome also strongly supports a key role for T cells in determining viral clearance. This study was performed on a group of Irish women who were iatrogenically infected with HCV in 1977 through the receipt of contaminated anti-D immunoglobulin.7 Unlike other class I association studies in HCV, this was unique in that all patients were of a single gender, of similar age and ethnic background and had all been infected by a single genotype (1b) from a common donor. HLA-B*27, HLA-A*03 and HLA-Cw*01 as well as a number of class II alleles were associated with viral clearance.
The reasons why T cells fail to control viraemia remains a critical question. One of the key immune evasion strategies employed by the virus is the development of escape mutations in T cell epitopes. The occurrence of escape mutations in targeted CD8 T cell epitopes is documented in HCV and has been shown to occur in epitopes restricted by a number of alleles.8 9 Escape mutations may also provide critical information about the nature of the T cell response within a given population. Significant T cell responses within a population will result in selection pressure and results in mutations emerging in an HLA-dependent fashion (‘footprints’). This identifies crucial responses linked to the presenting allele and can be an important tool in recognising T cell epitopes. In the case of a single-source outbreak where the inoculating sequence is known and genetic divergence is limited, ‘footprints’ are particularly useful for this purpose. Escape mutations may abrogate the T cell response and promote viral persistence; however, since mutations may also lead to impairment of viral fitness, their overall impact on disease outcome is still unclear. Viral mutations which lead to impairment of replicative fitness may still provide overall selective advantage for the pathogen in the face of strong T cell pressure. Furthermore, compensatory mutations may arise, which lead to recovery of fitness, although this requires further viral adaptation. In the case of HIV this may take some time to develop, providing the host with a prolonged period of protection.10 Since in HCV the virus has a relatively short window in which to establish persistence, the issue of fitness cost and the requirement for compensatory mutations could be of high importance in defining the protective nature of a T cell response and thus the HLA allele.
While much attention, especially in the HIV field, has been paid to rare alleles, particularly from the HLA-B locus11, HLA-A*03 is relatively common (∼20% in Caucasian populations) (http://www.allelefrequencies.net), and thus understanding how this allele exerts its protective effect has important implications for the population with respect to vaccine design. In this study, we aimed to understand the mechanism of protection afforded by HLA-A*03 by identifying key epitopes associated with it. We have identified a key epitope close to the active site of the NS3 protease which is routinely targeted in HLA-A*03-positive patients. Furthermore, we show that a single escape mutation is accompanied by a significant impairment of viral fitness, which may be rescued by mutation at a second covariant site. Our results provide important new data supporting the role of HLA-A*03 as an important driver of protective immune responses in HCV and offers insight into the formidable interplay between host and virus in determining outcome of infection.
Materials and methods
Study population
The study population is derived from a group of Irish women who were exposed to HCV (genotype 1b) through the receipt of contaminated anti-D immunoglobulin in 1977–1979. This group has been described in detail.7 12 Ethics approval for this study was obtained from the Research and Ethics Committees at St James' Hospital and St Vincent's Hospital. Informed consent was obtained from all patients. Peripheral blood mononuclear cells (PBMCs) were isolated by density centrifugation and, following centrifugation of whole blood, plasma was obtained and frozen at −80°C.
HCV and control peptides
Fifteen HLA-A*02-restricted HCV epitopes13 (supplementary table 2 online) were chosen using the predominant amino acid sequences which were either present in the donor (GenBank: AF313916) or circulating in the recipients.14 Control epitopes from influenza, cytomegalovirus (CMV) and Epstein–Barr virus (EBV) were also chosen (Supplementary table 2). A set of 300 20mer overlapping peptides (average purity 86%) were used (Pro-immune, Oxford, UK) and were based on the genotype 1b donor sequence (GenBank: AF313916).
Identification of candidate HLA-A*03-restricted HCV-specific CD8 T cell epitopes
Predicted epitopes were identified using a combination of different prediction programs (Bimas, SYFPEITHI, Jenner, ProPred 1, Rankpep, MAPPP and Net MHC). Epitopes with the highest ranking were chosen (supplementary table 1 online). In order to generate CD8 T cell lines, we stimulated PBMCs with three of the predicted HLA-A*03-restricted HCV epitopes and two EBV-derived epitopes. These were the two ‘footprint’ epitopes NS3 1080–1088 TVYHGAGTK and NS5B 2510–2518 KLTPPHSAK and also a third non-‘footprint’ epitope NS5B 2562–2570 EVFCVQPEK. The control peptides were RLRAEAQVK and RVRAYTYSK (see supplementary table 1 for more details).
Sequencing and in silico protein translation and alignment
Sera samples from cohort subjects were collected between 1996 and 2002 and stored at −80°C (St James' Hospital, Dublin, Ireland). Stored patient sera were available on 15 previously identified HLA-A*03-positive patients.7 Viral RNA was extracted from plasma using the Roche Cobas Amplicor HCV Kit and then used in three one-step reverse transcription–PCR system reactions (SuperScript III One-Step RT-PCR, Invitrogen, Paisley, UK) to amplify the viral genome (from the core to the NS5B region). The first-round template was subsequently used in nested second-round PCRs (Taq DNA polymerase, Roche, Burgess Hill, UK). Resultant PCR products were bulk-sequenced using the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Warrington, UK). Due to the age of the samples and the likely event of variability in parts of the HCV genome targeted by primers, some samples failed to produce a PCR product and, as a result, some individuals did not contribute sequence for NS3 (n=3). Sequence chromatograms were edited using the program Assign (Conexio Genomics, Freemantle, Australia).
In silico protein translation was carried out using CLC Bio Free Workbench (Aarhus, Denmark) software. Using Clustal W multiple alignment algorithms all sequences were aligned with the original donor sequence. Epitopes (supplementary tables 1 and 2) were first identified in the donor sequence and all patient samples. The amino acid sequence of the epitope, throughout all sequences, was investigated for potentially significant substitutions.
Antigen-specific CD8 T cell lines
Fresh PBMCs (4×106) were resuspended in 1 ml of medium and stimulated with 10 μg/ml peptide and 25 ng/ml interleukin 7 (IL-7; R&D systems, Abingdon, UK). IL-2 (20 ng/ml, Sigma Aldrich, Gillingham, UK) and fresh media were added on days 3, 7, 10 and 12. Cells were restimulated with 10 μg/ml of the corresponding peptide on day 7 and harvested for assays on day 14.
The protocol for intracellular cytokine staining is available in the supplementary methods section.
Statistical analysis
To determine the statistical significance of the amino acid substitutions within the defined epitopes, the Fisher exact test was used, and a p value of <0.05 was considered significant. The paired Student t test was used to compare frequencies of tetramer-positive populations, and a p<0.05 was considered significant.
Construction of mutations
The previously described HCV replicon pFK-i341PiLucNS3-3′-ET15 was used for the transient replication assay. PCR-based site-directed mutagenesis was used to obtain the escape mutants. Primer pairs were used as described in the supplementary methods. The PCR fragments were cloned in pGEMT easy vector. Selected clones were sequenced to confirm the presence of the mutation. Fragments were then digested with restriction enzymes NotI and MluI, and ligated into linearised vector.
RNA transcription and electroporation
Mutant plasmids along with the wild type and GND negative control were linearised with PvuI enzyme and transcribed in vitro using the T7 RNAmax (Promega, Southampton, UK) and DNase treated. RNA was extracted with acid phenol chloroform and stored at −20°C. A 20 μg aliquot of purified RNA was mixed with 400 μl of Huh-7 cells (107 cells/ml) using the gene pulser (Bio-Rad, Hemel Hempstead, UK) adjusted to deliver one pulse of 0.25 kV and 960 μFd. Cells were then seeded into 6-well cluster plates and cultured in the presence of complete media. Cell culture conditions are described in the supplementary methods section.
Luciferase assay
The Bright-Glo Luciferase assay system (Promega) was used to analyse viral fitness using luciferase as the reporter. Luciferase readouts were taken at 4, 24, 48 and 72 h post-transfection. Each assay was compared with the wild type (positive control) and the replication-deficient mutant (negative control GND). All values were normalised to the 4 h RNA value (set at 100%) and also further normalised to the wild type. The results are presented as the mean of three independent experiments.
Results
Ex vivo T cell responses are not detectable in patients infected with HCV three decades previously
Ex vivo Elispot analysis of PBMCs from six HLA-A*03-positive patients (table 1) with spontaneous resolved infection showed no significant CD8 T cell responses to 26 HCV epitopes (figure 1A and supplementary table 1). Similar results were obtained when we examined responses to previously identified immunodominant HLA-A*02-restricted epitopes (supplementary table 2) in 17 HLA-A*02 patients (figure 1B and supplementary table 5).
Details of HLA and HCV RNA status of HLA-A*03-positive patients included in the study

Magnitude of CD8 T cell responses as determined by ex vivo interferon γ ELISpot. HCV-specific CD8 T cell responses are absent ex vivo in all patients. (A) HLA-A*03-positive patients, n=6. All patients had spontaneously resolved infection. (B) HLA-A*02-positive patients, n=17 (6 patients had spontaneous resolved infection and 11 had persistent infection. (C) Whole genome assay in HLA diverse individuals, n=8 (4 spontaneous resolvers and 4 persistently infected). Responses are presented as the mean of duplicate wells after substraction of background. Summation of spot-forming units (SFU) is expressed per 1×106 peripheral blood mononuclear cells (PBMCs). CMV, cytomegalovis, EBV, Epstein–Barr virus; HCV, hepatitis C virus; HLA, human leucocyte antigen; PHA, phyto-haemagglutinin.
In parallel we also screened the entire genome for novel T cell responses. Using a set of overlapping peptides (20mers overlapping by 10 amino acids) spanning the entire HCV polyprotein (AF313916—genotype 1b), Elispot assays confirmed the absence of detectable ex vivo T cell responses in women derived from this cohort (figure 1C and supplementary table 6).
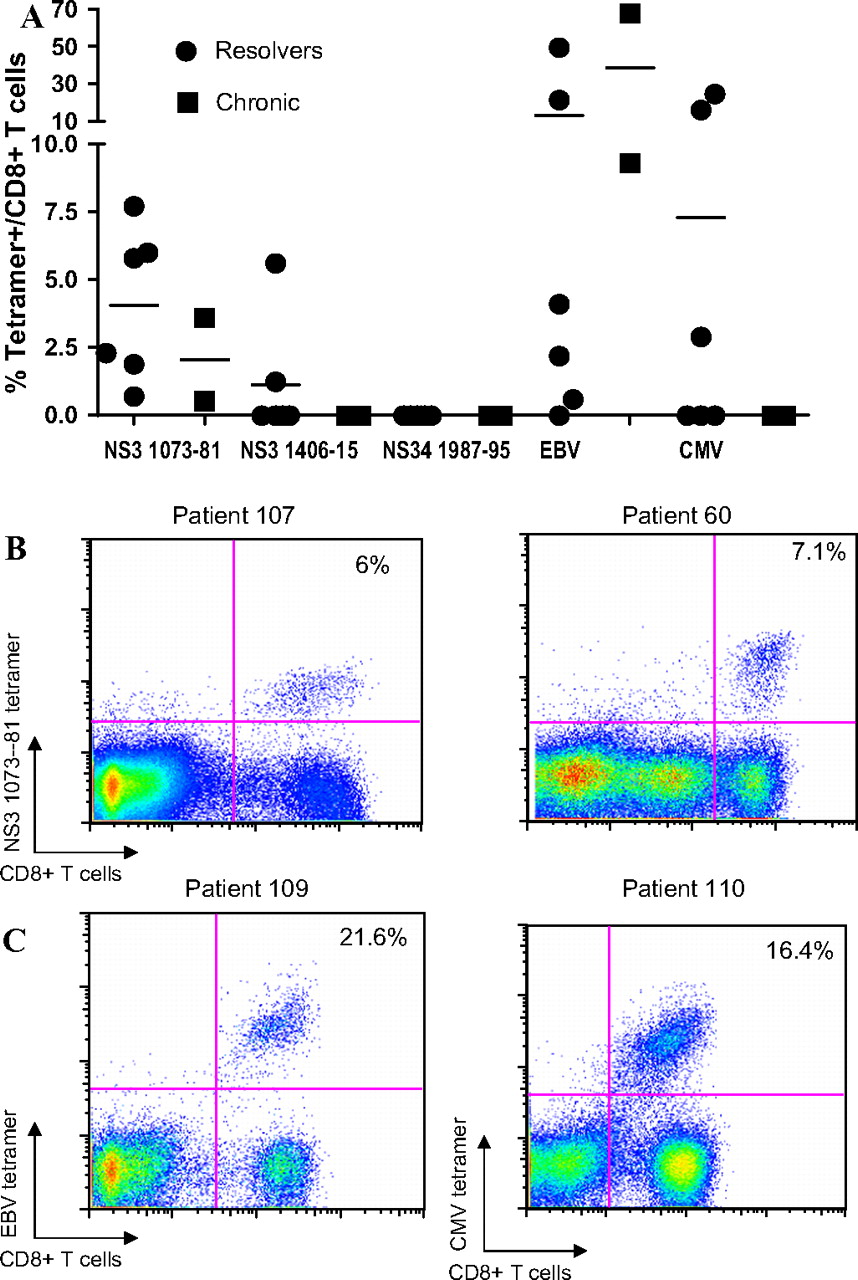
To determine if low-frequency responses were present, peptide-specific cell lines were generated by culturing fresh PBMCs in the presence of peptide for 14 days. Three immunodominant HLA-A*02-restricted epitopes (NS3 1073–1081 CINGVCWTV and CVNGVCWTV, NS3 1406–1415 KLSGLGLNAV, NS4 1987–1995 VLSDFKTWL) as well as control peptide epitopes from CMV and EBV were used to stimulate the cells, and cultured lines were obtained in eight HLA-A*02-positive patients (supplementary table 7). Two epitopes generated detectable CD8 T cell responses (figure 2A,B). Responses targeting the NS3 1073–81 epitope were observed in all 8 (100%) patients by tetramer staining. In addition, 2 of the 8 patients (25%) also had detectable responses to the NS3 1406–15 epitope. When we tested patients for CD8 T cell responses to the same set of tetramers ex vivo, we were unable to observe antigen-specific cells.

CD8 T cell responses to HLA-A*02-restricted epitopes in cultured lines. (A) Group data. (B) Representative dot plots of the CD8 T cell responses to the NS3 1073–81 epitope (CVNGVCWTV or CINGVCWTV) in two patients with resolved infection (patient 107 and 60). (C) Representative dot plots of tetramer responses to HLA-A*02-restricted control EBV and CMV epitopes. CMV, cytomegalovis, EBV, Epstein–Barr virus; HLA, human leucocyte antigen.
HLA ‘footprinting’ reveals novel CD8 T cell responses restricted by HLA-A*03
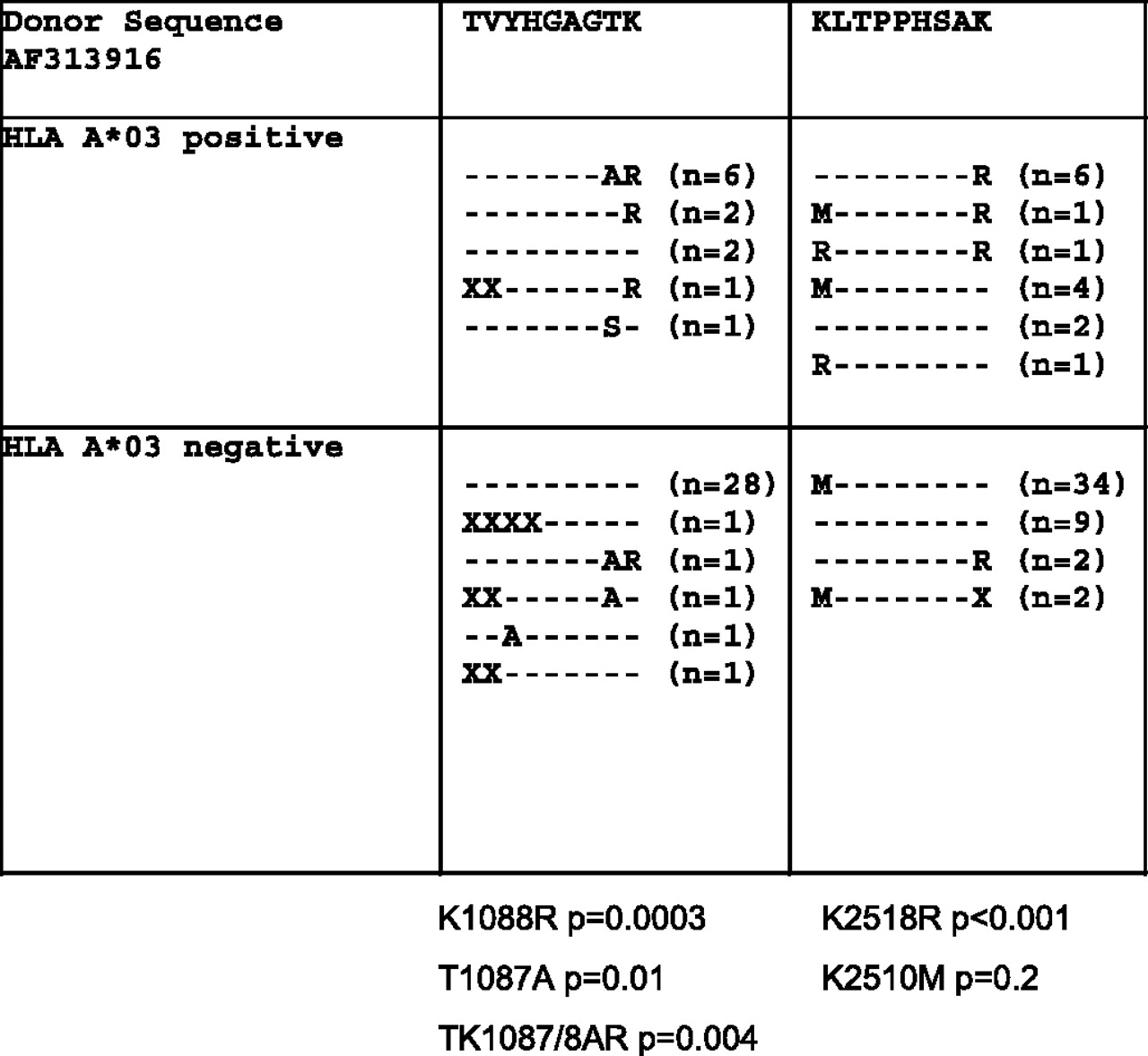
In order to define the HLA-A*03-restricted CD8 T cell response further, we adopted an alternative sequence-directed approach. To assess the impact of HLA-A*03-mediated T cell selection pressure, we determined the bulk sequence of the non-structural region of the viral genome in the serum of HLA-A*03-positive and -negative patients derived from the cohort (table 1 and supplementary table 8). The sequences were then aligned with that of the donor and the 26 HLA-A*03-restricted epitopes (Supplementary table 1) were identified. A comparison of the epitope sequences in HLA-A*03-positive and -negative individuals revealed two clear HLA-A*03 ‘footprints’ (figure 3). The first was located within the NS3 1080–88 TVYHGAGTK epitope. Lysine (K) was substituted with arginine (R) at position 9 (K1088R) in 9/12 (75%) A*03-positive patients compared with 1/33 (3%) controls (p=0.0003). Threonine (T) was also substituted with alanine (A) at position 8 (T1087A) in 6/12 (50%) A*03-positive patients compared with 2/33 (6%) controls (p=0.01). The double substitution of TK to AR is also observed predominantly in HLA-A*03-positive patients (6/12, 50%) compared with 1/33 (3%) in controls and was also found to be significantly linked to A*03 (p=0.004). The second ‘footprint’ was located in the NS5B epitope (NS5B 2510–18 KLTPPHSAK). Lysine (K) was substituted with arginine (R) at position 9 in 8/15 (53%) A*03-positive patients compared with 2/45 (4%) A*03-negative patients (p=0.001). A second common substitution was also observed at position 1 in both HLA-A*03-positive and -negative patients, but was not significant. The 15 HLA-A*02-restricted epitopes (supplementary table 2) were also examined, but no HLA association was observed (supplementary figure 2).

Two HLA-A*03 ‘footprints’ in predicted hepatitis C virus epitopes. Significant sequence variation is seen in the NS3 1080–88 TVYHGAGTK epitope. Lysine (K) is substituted with arginine (R) at position 9 (K1088R) in 9/12 patients (75%) in the A*03-positive group compared with 1/33 (3%) in the control group, p<0.0003. Furthermore, the threonine (T) at position 8 is frequently substituted with alanine (A) (T1087A) in 6/12 (50%) HLA-A*03-positive patients compared with A*03-negative patients (2/33, 6%), p=0.01. The double substitution TK1087AR is also observed in a significant number of HLA A*03-positive patients (50%) compared with controls (3%), p=0.004. A second HLA-A*03 ‘footprint’ is also present in the hepatitis C virus NS5B protein. Significant sequence variation in seen in the NS5B 2510–18 KLTPPHSAK epitope. Lysine (K) is substituted with arginine (R) at position 9 (K2518R) in 8/15 (53%) in the A*03-positive group compared with 2/45 (4%) in the control group (p=0.001). Lysine (K) is also substituted with methionine (M) in both groups. Its association with HLA-A*03 is not significant (p=0.2). X indicates no sequence available. Statistical comparison by the Fisher exact test. HLA, human leucocyte antigen.
HLA-A*03-restricted NS3 1080–88 epitope is targeted by CD8 T cell responses in resolved HCV infection
These HLA-A*03-specific ‘footprints’ provide strong evidence that they are likely to be in vivo targets of the CD8 T cell repertoire. To assess this, we generated peptide-specific T cell lines corresponding to the ‘footprint’ epitopes (NS3 1080–88 TVYHGAGTK and NS5B 2510–18 KLTPPHSAK) and to another predicted epitope NS5B 2562–70 EVFCVQPEK. Two control peptides derived from EBV were also included (supplementary table 1). A total of 6/10 patients (table 1) with resolved infection demonstrated CD8 T cell responses to the NS3 1080–88 TVYHGAGTK epitope by tetramer analysis (figure 4A,B). All six patients had resolved infection either spontaneously or with antiviral treatment. CD8 T cell responses were not detectable in lines stimulated with the other two HCV peptide epitopes (figure 4A).

CD8 T cell responses to the HLA-A*03-restricted ‘footprint’ epitope NS3 1080–88 in cultured cell lines. (A) Overall data set; n=10 (seven patients had spontaneously resolved infection, one patient resolved with antiviral therapy and two patients had chronic infection) (B) Representative dot plots showing tetramer responses to the NS3 1080–88 epitope in two patients (both resolvers) (C) Dot plots demonstrating tetramer responses to the HLA-A*03-restricted control EBV epitopes. EBV, Epstein–Barr virus; HLA, human leucocyte antigen.
Impact of escape mutations on CD8 T cell recognition
To address further the impact of the amino acid substitutions on T cell recognition, cell lines specific for the epitope were tested for recognition of TVYHGAGTK (wild type) or the escape mutants TVYHGAGAR or TVYHGAGTR in three patients who had detectable CD8 T cell responses. In all patients, the secretion of interferon γ was reduced in response to the peptide with both the escape mutations (TVYHGAGAR or TVYHGAGTR) (figure 5A,B).

Immune escape by selected peptides in NS3 1080–88. (A) Peripheral blood mononuclear cells from three patients with resolved infection were cultured in the presence of the NS3 1080–88 peptide TVYHGAGTK. They were then re-stimulated with the wild-type NS3 TVYHGAGTK or the escape mutant (TVYHGAGTR and TVYHGAGAR) peptides (10 μg/ml final concentration). The ability to secrete interferon γ (IFNγ) was analysed by intra-cellular cytokine staining (ICS). A significant reduction in IFNγ secretion was observed in lines stimulated with the mutant peptides TVYHGAGTR and TVYHGAGAR, *p=0.04 (Student t test). (B) A peptide titration for the wild type and the escape mutants was performed on subject 49. The peptide concentration is presented as μg/ml. (C) The predicted binding ability of the wild-type and variant peptides identified in two bio-informatic programs (NetMHC and BIMAS). The lysine to arginine substitution at position 9 causes a significant reduction in the predicted binding ability. The NetMHC program relies on artificial neural networks which are capable of performing sensitive, quantitative predictions of peptide binding to the MHC (major histocompatibility complex) class I molecules. These systems have been designed to predict binding versus non-binding peptides, and their binding abilities are presented as an estimated affinity in nM.
To assess the binding ability of the epitopes to the HLA-A*03 allele, we used two different bio-informatic programs (Net MHC and BIMAS) to identify the predicted binding of both the wild-type (TVYHGAGTK) and the mutant epitopes (TVYHGAGAK, TVYHGAGTR and TVYHGAGTR. This search also confirmed a significant reduction in the predicted binding ability of the variant epitopes containing the K1088R substitution (figure 5C).
We also cultured PBMCs in the presence of peptides representing either the wild-type epitope (TVYHGAGTK) or mutant peptides which possessed either of the single mutations observed (TVYHGAGTR and TVYHGAGAK) or the double mutant (TVYHGAGAR), and looked for expansion of antigen-specific T cells. After 14 days, these lines were analysed by tetramer staining. Notably in four out of five patients, a reduction in the number of tetramer-positive CD8 T cells was observed in cells stimulated with the single mutant TVYHGAGAK, although this was not significant. Cultured cell lines stimulated with peptides containing the K1088R mutation (TVYHGAGTR or TVYHGAGAR) showed almost no stimulation of antigen-specific CD8 T cells (figure 6A,B and C).

Variant peptides fail to stimulate NS3 1080–88-specific CD8 T cells. Antigen-specific lines were generated in response to the wild-type (WT) peptide (TVYHGAGTK) or variants that contained either a single mutation (TVYHGAGAK or TVYHGAGTR) or the double mutation (TVYHGAGAR). (A) The lysine (K) to arginine (R) mutation at position 9 (TVYHGAGTR) and (TVYHGAGAR) caused a significant reduction in the binding of the WT tetramer (p=0.03), while the threonine (T) to alanine (A) change alone (TVYHGAGAK) led to a reduction in binding in four patients (p=ns). (B) Results after normalisation of the WT (TK-specific) cells to 100%. (C) Representative dot plots (patient 29) are shown.
The escape mutant TVYHGAGTR is associated with fitness cost but the double mutant TVYHGAGAR compensates to restore optimal viral replication
To determine the effect of the mutations observed in the HLA-A*03-positive patients, we assessed their impact on viral replication efficiency. Mutations were introduced into a subgenomic replicon (genotype 1b strain) carrying a firefly luciferase reporter gene. In this strain the wild-type sequence of the epitope carries serine at position 8 (TVYHGAGSK). We therefore introduced the threonine substitution here (TVYHGAGTK) and this mutant construct served as our wild type (as observed in the Irish cohort). Two further epitope mutants were created—one bearing the single K1088R substitution at position 9 and the other bearing the double substitution T1087A and K1088R. The replication efficiency of these mutants was then compared with the wild-type sequence and a replication-defective replicon (GND) at a number of time points (0, 4, 12, 24 and 48 h). We observed a significant reduction in the replication efficiency in mutants bearing the single substitution K1088R at all time points. In contrast, the mutant carrying the double substitution (T1087A and K1088R) was associated with a viral replication efficiency which was ∼98% of that of the wild type at 72 h (figure 7). The previous functional T cell experiments suggest that only the K1088R mutation is necessary to achieve CD8 T cell escape; however, this single substitution is observed only rarely in vivo in the Irish cohort. These data indicate that this substitution is associated with a marked loss of viral fitness. The development of the double substitution appears to restore viral replication efficency and thus supports its importance as a key compensatory mechanism.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The percentage replication efficiency of mutants compared with the wild type (TK). Representative time course of mutants TR and AR compared with the wild type (TK) and the negative mutant (GND) at 48 and 72 h. Replicative capacity is expressed as a percentage after normalisation to the 4 h input RNA value (reflecting transfection efficiency). Wild-type (TK) was normalised to 100%. The mean±SD of three independent experiments is shown. Replication efficiency of the mutant TR is significantly reduced at both time points while the AR mutant replicates almost to wild-type levels, indicating a compensatory role. Student t test was used for statistical analysis.
Discussion
HLA-A*03 was found to be a protective allele in determining viral clearance in HCV infection in a study of patients infected from a single source.7 We hypothesised that this effect may be due to a single epitope or a set of immunodominant epitopes restricted by this allele. In order to identify HLA-A*03-restricted responses in this cohort, we adopted two approaches. In the first, we chose a set of 26 HLA-A*03-restricted epitopes based on previous publications and prediction programs using the known sequence of the inoculating virus. We were unable to detect CD8 T cell responses ex vivo in patients with either resolved or persistent infection irrespective of their HLA type. This result differs from that of Takaki et al who examined ex vivo responses to HLA-A*02-restricted epitopes in women infected from a similar single-source outbreak, where vigorous responses were observed in the resolvers but not in the chronically infected group.16 The differences in the CD8 T cell responses observed may reflect the natural waning of the cellular responses to HCV three decades after a single exposure (as occurs with humoral responses). Other factors such as the genetic background, technical differences and the dose of inocula may also have played a role. To determine if persistent responses were present after re-stimulation, we cultured PBMCs in the presence of specific peptide. We were able to confirm that antigen-specific responses persist in this cohort but that the precursor frequency is low and requires re-stimulation prior to detection.
As ex vivo T cell responses were not detectable in this cohort, we used a sequence-led approach to establish T cell targets. HLA-associated ‘footprint’ epitopes provide a sensitive measure of where the important CD8 T cell-mediated activity is being exerted. We were thus able to identify two clear HLA-A*03 ‘footprints’ in the NS3 1080–88 and the NS5B 2510–18 epitopes.
In the NS3 1080–88 TVYHGAGTK epitope, there are two significant substitutions, K1088R at position 9 (P9) and T1087A at position 8 (P8), in HLA-A*03-positive patients. Furthermore, in a significant number of HLA-A*03 patients, these two mutations (T1087A and K1088R) occur together. Anchor positions represent the preferred amino acid side chain that fits into pockets within the MHC groove. The anchor motifs for HLA-A*03 molecules usually occur at P2 and P917 and thus the K1088R mutation at P9 is likely to facilitate escape from this immunodominant response in vivo. To support this, we have shown that interferon γ production from cell lines stimulated with peptides containing the K1088R mutation is reduced and that mutant peptides failed to stimulate antigen-specific proliferation in vitro.
The above findings support the role of CD8 T cell escape as a major mechanism for viral persistence but do not explain how HLA-A*03 mediates its protective effect. In HIV infection, the protective effects associated with class I alleles, for example HLA-B*27, have been attributed to the targeting of epitopes which lie in structurally or functionally constrained regions. Escape mutations occurring within these epitopes incur a fitness cost which reduces viral replication and attenuates the virus.18–20 The fitness cost of escape mutations has also been studied in HCV although to a much lesser extent. In a recent study, the impact of amino acid substitutions in a HLA-B*08-restricted epitope was analysed and a significant fitness cost was observed.21 The NS3 1080–88 TVYHGAGTK epitope lies within the NS3 region which codes for a bifunctional protein with both protease activity and NTPase helicase activity. Mutations at key catalyic sites including His1083, which lies within the TVYHGAGTK epitope, have been shown to abolish cleavage of the non-structural proteins.22 Thus, substitutions occurring within this region are likely to have an effect on viral processing and replication. Supporting this, the adjacent immunodominant HLA-A*02-restricted epitope NS3 1073–81 CVNGVCWTV is routinely targeted in A*02-positive individuals but is relatively conserved, and a previous study indicates that substitutions within this epitope significantly affect its function.23
We therefore speculated that a dominant CD8 T cell response associated with HLA-A*03 might drive escape mutations that reduce viral fitness and contribute to the favourable outcomes by either delaying the fixation of substitutions or promoting a weakened virus. In the case of several HIV epitopes linked to protective alleles, compensatory mutations arise in adjacent regions of the genome and their function is primarily to restore fitness capacity.10 20 24 In HCV infection, the protective allele HLA-B*27 has been linked to a dominant CD8 T cell response associated with the NS5B 2841–49 ARMILMTHF epitope. Escape from immune recognition in this case appears to require multiple mutations and these mutations do not appear to have a compensatory function.25 We postulated that the development of the T1087A mutation within the NS3 1080–88 epitope may reflect accessory changes that are compensatory in the setting of restoring viral fitness or are occurring to complete the evolution to successful escape from T cell-mediated control—a‘two-hit’ hypothesis. This hypothesis is strongly supported by the functional analysis of the replicons in vitro, which revealed a major compensatory effect of the second T1087A mutation. Furthermore, the structural impact of such mutations is not fully understood. We modelled the wild-type epitope onto the existing structure of HCV NS3 (supplementary figure 3).26 This revealed strong structural constraints on the lysine residue at 1088, which stabilises a helical turn. In contrast, threonine at position 1087 would appear to have much less constraint, and the mechanism for the association—which is very clear at a genetic level in vivo and supported by functional studies in vitro—requires further specific analysis to define its molecular basis.
There were two clear ‘footprints’ associated with HLA-A*03 epitopes. Although we did not find evidence of CD8 T cell responses to the NS5 2510–18 KLTPPHSAK ‘footprint’ epitope in this study, we cannot exclude the possibility that persisting responses are present in the liver or that technical issues may have precluded their identification. The strong ‘footprint’ associated with this epitope in this study is important and may indicate ongoing selection pressure at some level. This epitope has previously been identified in genotype 1a infections27 and a ‘footprint’ has also been identified in other populations (personal communication, Todd Allen, Boston). It is possible that the simultaneous targeting of more than one dominant epitope restricted by HLA-A*03 may be contributing to the overall protection. Longitudinal studies of the T cell responses in HLA-A*03 individuals with acute infection will be necessary to define the kinetics and hierarchy of these responses. The degree to which this epitope is targeted in other populations infected with differing genotypes and subtypes has not yet been assessed and further studies are required to address this. Although we were unable to detect ex vivo CD8 T cell responses to this epitope in the Irish cohort, the highly significant HLA-A*03 ‘footprint’ provides robust evidence that this epitope was likely to have been under considerable T cell-mediated selection pressure relatively early on in the course of infection. Other studies have indicated that the initial 6 months are pivotal to outcome and that escape mutations typically occur during this time.28 Furthermore, these mutations tend to become fixed within the circulating quasispecies, perhaps reflecting ongoing selection pressure in the liver.2 Although we have not looked for T cell responses in longitudinal studies across other populations, several studies have looked for the presence of HLA-associated footprints in diverse populations.29–31 These studies serve as a surrogate marker which can be used to identify areas of the genome under CD8 T cell-mediated selection pressure (putative epitopes). To date, none of these studies has identified significant polymorphisms linked to the NS3 1080–88 epitope. We therefore speculate that the immunodominance of this epitope may be subtype specific and so difficult to identify in the absence of larger studies or subtype-specific studies. Our examination of the sequences of this epitope available in the database also demonstrates that the protective epitope TVYHGAGTK was found in the majority of genotype 1b viral strains but not in genotype 1a (supplementary table 4). It is interesting to note that the protection associated with the HLA-B*27 allele has been linked to an epitope identified in genotype 1 viral strains. Indeed, a protective effect of HLA-B*27 was not identified in genotype 3 infections in German patients and the same epitope was not targeted in genotype 3 infections.32 These data highlight the need to identify a range of protective T cell epitopes across all genotypes.
The highly polymorphic nature of the HLA genes poses significant challenges to vaccine design, and extensive diversity is observed in HLA class I and II associations with susceptibility and protection in HCV infection in different cohorts.33 Other studies have also identified HLA-A*03 as a critical determinant of outcome in HCV and hepatitis B virus (HBV). Wang et al describe an association between viral clearance and HLA-A*03; however, this association was only identified in the non-Caucasian subgroup of the cohort.34 In a Korean study, HLA-A*03 was found significantly more frequently in chronic HCV carriers compared with healthy controls.35 In HBV infection, HLA-A*0301 has also been identified as a protective allele.36 These genetic studies clearly indicate that both the ethnic and geographic distribution of HLA genes associated with HCV outcome are different but point to an important interaction between HLA-A*03 and virus in other cohorts.
While it is well recognised that escape from certain CD8 T cell epitopes is likely to be an important mechanism in determining outcome in HCV infection, it certainly is not a universal finding in persistent infection. Komatsu et al previously examined the viral sequences from a set of chronically HCV-infected patients in whom CD8 T cell responses in the liver had previously been accurately mapped. They found that even in the presence of persistent intrahepatic T cell responses, only about half of the patients studied showed evidence of escape.37 This finding may potentially be explained by differences in binding ability, antigen presentation or functional capabilities. However, the highly significant association between HLA-A*03 and the presence of the K1088R in chronically infected individuals in this study provides strong support for CD8 T cell-mediated selection pressure and indicates that targeting of the wild-type epitope may have contributed to the protective effect found to be associated with HLA-A*03. However, even in the 12 HLA-A*03 individuals with viral sequences available, 25% (3/12) had no evidence of mutation at the 1088 anchor position, indicating that other mechanisms were responsible for viral persistence in these subjects. Recent sequencing of other viral polymorphisms from this cohort in relation to diverse HLA genes has revealed a similar picture in other sites across the HCV NS genes, and this is consistent with previous data.38 39
In conclusion, we have identified a novel CD8 T cell response associated with the protective HLA-A*03 allele through the use of a unique genetic set of information available as a consequence of a single-source outbreak. Furthermore, this setting provides valuable insights into the complex interplay between host and pathogen in a tightly defined population. A*03 is a relatively common allele and thus the protective impact within a population is likely to be substantially greater. These findings have important implications with respect to vaccine design and reveal the power of both host and viral genetic approaches to define the crucial components of the host response.
Acknowledgments
We thank the study participants and the staff working in the Hepatology Department, St James’ Hospital. Special thanks to Barbara Hynes, Jenny Smith and Helena Irish for help recruiting the patients.
References
Supplementary materials
Online only appendix
Files in this Data Supplement:
Footnotes
DK and PK contributed equally to this work.
Funding KF is a recipient of a Wellcome Trust Research Training Fellowship award. This work was also supported by the Health Research Board (Ireland), Higher Education Authority Programme for Research in Third Level Institutions (PRTLI) Cycle 3 (DK), the James Martin School for 21st Century, NIHR Biomedical Research Centre Programme (Oxford) and the Wellcome Trust (PK-WT091663MA).
Competing interests None.
Ethics approval This study was conducted with the approval of the St Vincent's Hospital Ethics committee, Dublin, Ireland and St James' Hospital, Dublin, Ireland.
Provenance and peer review Not commissioned; externally peer reviewed.