Article Text

Abstract
Background and aims Teduglutide, a GLP-2 analogue, may restore intestinal structural and functional integrity by promoting repair and growth of the mucosa and reducing gastric emptying and secretion, thereby increasing fluid and nutrient absorption in patients with short bowel syndrome (SBS). This 24-week placebo-controlled study evaluated the ability of teduglutide to reduce parenteral support in patients with SBS with intestinal failure.
Methods In 83 patients randomised to receive subcutaneous teduglutide 0.10 mg/kg/day (n=32), 0.05 mg/kg/day (n=35) or placebo (n=16) once daily, parenteral fluids were reduced at 4-week intervals if intestinal fluid absorption (48 h urine volumes) increased ≥10%. Responders were subjects who demonstrated reductions of ≥20% in parenteral volumes from baseline at weeks 20 and 24. The primary efficacy end point, a graded response score (GRS), took into account higher levels and earlier onset of response, leading to longer duration of response. The intensity of the response was defined as a reduction from baseline in parenteral volume (from 20% to 100%), and the duration of the response was considered the response at weeks 16, 20 and 24. The results were tested according to a step-down procedure starting with the 0.10 mg/kg/day dose.
Results Using the GRS criteria, teduglutide in a dose of 0.10 mg/kg/day did not have a statistically significant effect compared with placebo (8/32 vs 1/16, p=0.16), while teduglutide in a dose of 0.05 mg/kg/day had a significant effect (16/35, p=0.007). Since parenteral volume reductions were equal (353±475 and 354±334 ml/day), the trend towards higher baseline parenteral volume (1816±1008 vs 1374±639 ml/day, p=0.11) in the 0.10 mg/kg/day group compared with the 0.05 mg/kg/day group may have accounted for this discrepancy. Three teduglutide-treated patients were completely weaned off parenteral support. Serious adverse events were distributed similarly between active treatment groups and placebo. Villus height, plasma citrulline concentration and lean body mass were significantly increased with teduglutide compared with placebo.
Conclusions Teduglutide was safe, well tolerated, intestinotrophic and suggested pro-absorptive effects facilitating reductions in parenteral support in patients with SBS with intestinal failure.
ClinicalTrials.gov number NCT00172185.
- Short bowel syndrome
- glucagon-like peptide 2
- teduglutide
- intestinal failure
- parenteral nutrition
- glucagen-like peptides
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Short bowel syndrome
- glucagon-like peptide 2
- teduglutide
- intestinal failure
- parenteral nutrition
- glucagen-like peptides
Significance of this study
What is already known on this subject?
In an open-label non-placebo controlled 21-day phase 2 study, teduglutide has been shown to increase intestinal wet weight absorption in patients with short bowel syndrome using metabolic balance studies.
What are the new findings?
This is the first long-term (24 weeks) randomised placebo-controlled study of teduglutide in patients with short bowel syndrome dependent on parenteral support.
Teduglutide was safe, well tolerated and led to restoration of intestinal functional and structural integrity through significant intestinotrophic and pro-absorptive effects.
How might it impact on clinical practice in the foreseeable future?
Teduglutide has the potential to reduce the burden often seen with parenteral support in patients with short bowel syndrome with intestinal failure, and could add to the limited clinical treatment armamentarium in treating patients with short bowel syndrome.
Introduction
Short bowel syndrome (SBS) is characterised by large heterogeneity where patients with intestinal insufficiency are able to compensate for their malabsorption of fluids, electrolytes, trace elements, vitamins or nutrients by increasing oral intake and adapt metabolically,1 2 whereas patients with intestinal failure depend on parenteral support (fluids, electrolytes or nutrients).3–5 A large part of this heterogeneity is explained by differences in the anatomy of the remnant bowel.6 7 Patients with mild intestinal failure with a jejunostomy or ileostomy need approximately 1000 ml of fluid and electrolytes taken over a few hours 3–7 times per week. Patients with SBS with jejunostomies or ileostomies frequently have complications such as dehydration and electrolyte deficiencies due to stomal losses. In severe cases, significant protein and energy malabsorption can occur and may require supplementary hypertonic nutrients and electrolyte infusions administered both daytime and nocturnally. Patients with SBS and intestinal failure who have a preserved colon in continuity often suffer from large amounts of rectal fluid loss, fear of incontinence and the consequences of colonic fermentation such as gaseous distension and flatulence, whereas fluid and electrolyte deficiencies are less prominent.7 Since some of these patients do not imminently suffer from dehydration, days off parenteral nutrients are possible. However, on those nights when nutrient infusions are required, both the infusion and the accompanying excessive urine production may disturb the sleep pattern of the patients. In the most severe cases, nocturnal nutrients as well as daytime fluid and electrolytes are required.
Although frequently life-saving in patients with SBS with intestinal failure, the parenteral administration of fluids, electrolytes, trace elements, vitamins and nutrients has been associated with potentially life-threatening complications. Poor catheter care technique, insertion site, tunnel and catheter-related blood stream infections may lead to bacteraemia and even septicaemia, and the presence of a central catheter may lead to central venous thrombosis and even embolism.5 In addition, parenteral constituents and chronic dehydration may contribute to progressive intestinal failure-associated liver and renal disease and eventually failure.8 9 Mutually, the symptoms of SBS with intestinal failure and the inconveniences and complications in relation to parenteral support may cause potential restrictions in the lifestyle of these patients and may lead to significant impairment of their quality of life.10 11
In the past, the clinical care of patients with SBS has mainly focused on ‘making the most of what the SBS patient still had’ by optimising remnant intestinal function through dietary interventions, oral rehydration solutions, antidiarrhoeal and antisecretory agents. Furthermore, anastomosis of excluded bowel has been advocated, when it is possible, and experimental surgical procedures have also been employed. Intestinal transplantation is currently only recommended in patients failing parenteral support due to recurrent life-threatening sepsis, loss of venous access and end stage intestinal failure-associated liver disease.5 Treatments focused on improving the structural and functional integrity of the remaining intestine by so-called intestinal rehabilitation which minimise or eliminate the need for parenteral support are therefore needed.
In recent years hormonal stimulation to augment remnant bowel adaptation has been suggested, with glucagon-like peptide 2 (GLP-2)—a peptide which is secreted from the intestinal L-cells following ingestion of a meal—as a key factor. Repeated administration of GLP-2 promotes the expansion of the intestinal mucosa via the stimulation of crypt cell growth and the reduction of enterocyte apoptosis.12 Exogenous GLP-2 administration inhibits gastric acid secretion and gastric emptying,13 14 stimulates intestinal blood flow,15 increases intestinal barrier function16 and enhances nutrient and fluid absorption in both preclinical and clinical models.17 In addition, GLP-2 may decrease bone resorption and it has been suggested as a potential therapy in osteoporosis.18
Open uncontrolled clinical studies have suggested positive effects of exogenously administered GLP-2 and the di-peptidyl peptidase IV degradation resistant analogue, teduglutide, in patients with SBS.19–22 In an open-label 3-week study where the oral intake and parenteral support were intentionally kept constant during 72 h balances, teduglutide reduced faecal wet weight excretions by 711±734 g/day (p=0.001) and increased wet weight absorption by 743±477 g/day causing increases in urine volumes of 555±485 g/day (p<0.001). In addition, faecal energy losses decreased by 808±1453 kJ/day (p=0.04) in relation to teduglutide treatment, but all effects reverted 3 weeks after treatment.22
In the present study, the largest randomised placebo-controlled trial ever performed in patients with SBS with intestinal failure, the objective was to determine whether teduglutide could reduce the burden of parenteral nutrition and intravenous fluid requirements. Secondary end points included the ability to obtain additional days off or eliminate the need for parenteral support in this SBS population with demonstrated intestinal failure. In detail, during 24 weeks of treatment with placebo or teduglutide, adjustments in the parenteral support were performed when the urine volume was increased to a certain threshold. This algorithm-based approach considered the changes in urine volumes to be based on changes in intestinal wet weight absorption. In addition to safety evaluations, DEXA scanning, histological evaluation of bowel morphology in biopsies, plasma citrulline and quality of life questionnaires described changes in body composition, structural intestinal adaptation and quality of life, respectively, in relation to placebo and teduglutide treatment.
Methods
Patients, study design, efficacy and safety
After receiving approval from local IRBs and medical ethics committees, centres screened patients of both sexes aged ≥18 years with a history of SBS due to intestinal resection and dependent on parenteral support (fluids, electrolytes or nutrients) at least three times per week for a period of at least 12 months prior to the start of the study. Exclusion criteria are shown in box 1.
Exclusion criteria
Pregnancy or lactation.
Body mass index <18 or >27 kg/m2.
Active Crohn's disease as evaluated by standard procedures employed by the investigator.
Radiation enteritis, scleroderma, coeliac disease, refractory or tropical sprue, diabetes.
Alcohol or drug abuse within the last year.
Previous use of teduglutide or potential allergies to teduglutide or its constituents.
Inadequate hepatic function: ALT and AST both >2.0× upper limit of normal (ULN), total bilirubin >1.25× ULN or alkaline phosphatases >2.5× ULN.
Inadequate renal function: serum creatinine or blood urea nitrogen >1.5× ULN.
Urine sodium <20 mmol/day.
Any hospitalisation within 1 month before screening.
Use of infliximab, growth hormone or growth factors such as native GLP-2 or other biological therapy within the last 12 weeks.
Use of systemic corticosteroids, methotrexate, cyclosporine, tacrolimus, sirolimus, octreotide, intravenous glutamine or any investigational drug within last 30 days.
The use of antimotility and antidiarrhoeal agents (loperamide, difenoxylate, codeine and other opiates), H2-receptor antagonists, proton pump inhibitors, bile sequestering agents, oral glutamine, diuretics and oral rehydration solutions were required to be stable for ≥4 weeks prior to baseline evaluations and remain stable during the study.
The basic study design is presented in figure 1.
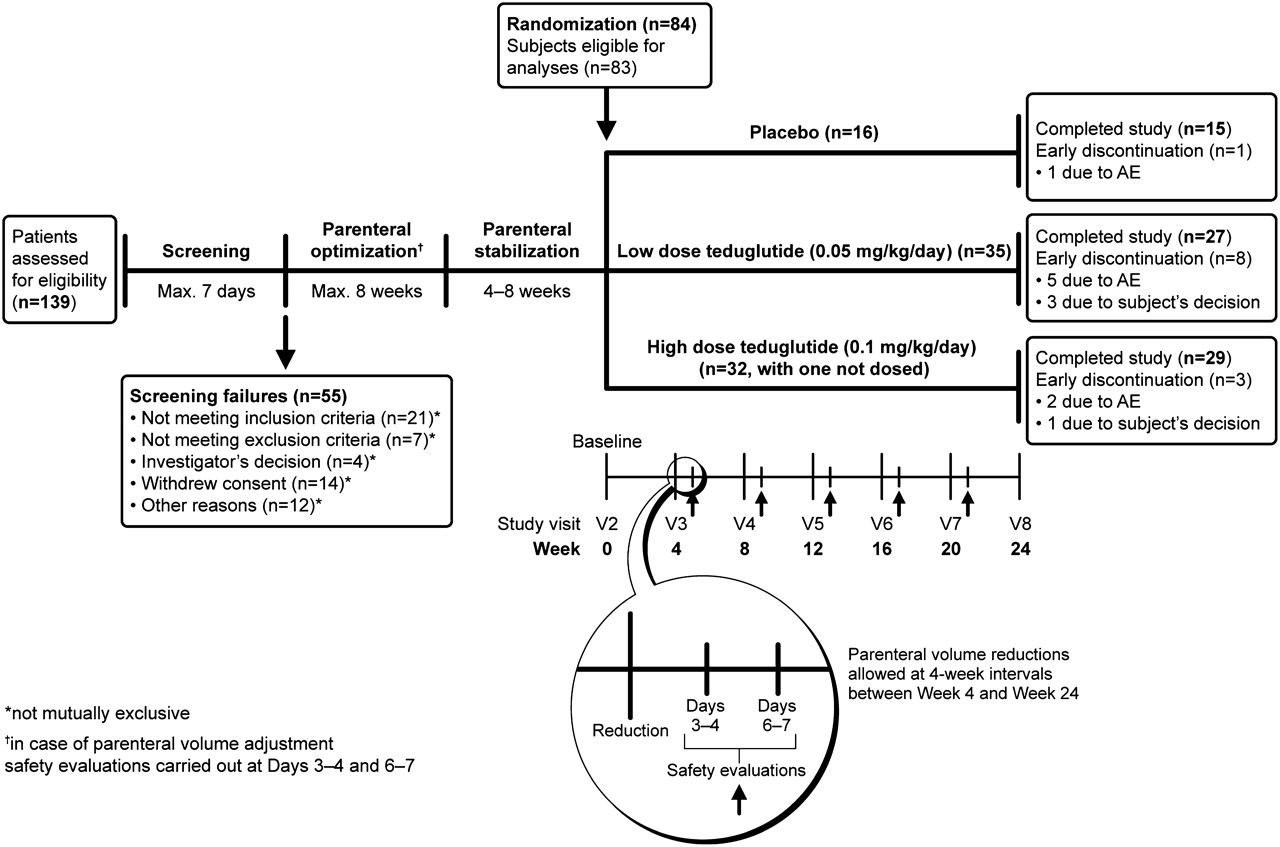
Basic study design. AE, adverse event.
Parenteral optimisation
To establish that the patients minimally tolerated baseline parenteral support resulted in a urine output of 1.0–2.0 l/day, a period of optimisation was used. The patients were instructed how to perform home collections of their 48 h urine output and completed these 2 days before each visit. Although the osmolarity and oral intake were not kept strictly controlled, study participants were asked to try to keep the timing, quantity and quality of beverages as constant as possible during the 48 h collection periods. The study subjects were seen in outpatient clinics at 2-week intervals. Adjustment to the study subjects' baseline parenteral volume was performed when urine volume fell below 1.0 l/day or exceeded 2.0 l/day. At all times, interim safety evaluations were performed within 1 week after adjusting parenteral volumes by repeating 48 h urine collections, again recording and keeping oral beverages the same as during previous balances. Blood samples were taken (including haematocrit, blood urea nitrogen and creatinine), urine sodium was measured and a clinical evaluation was performed to check for clinical signs of dehydration. If tolerated, the new parenteral volume was maintained stable until the next visit and, if not, the original parenteral volume was resumed. The patient was excluded from the study if parenteral optimisation was not achieved, defined as stable urine output volume of 1.0–2.0 l/day after 8 weeks.
Parenteral stabilisation
After optimisation, the patients were maintained for 4–8 weeks on the stabilised tolerated parenteral volume. If the patients still had a urine volume of 1.0–2.0 l/day while keeping oral beverages constant, the patients were eligible for randomisation.
Randomisation
Eligible patients were randomly assigned to one of three groups according to a computer-generated interactive response system (Fisher Automated Clinical Trial Services). Randomisation was stratified for the three groups and the parenteral volume at three levels of consumption: (1) parenteral volume consisting of intravenous fluids and electrolytes only (3–7 times weekly); (2) parenteral volume consisting of fluids and nutrients 3–5 times weekly; and (3) parenteral volume consisting of fluids and nutrients (6–7 times weekly). These patients with SBS, depending on their parenteral support (nutrients and/or fluids), were randomised to receive teduglutide (Cangene, Winnipeg, Manitoba, Canada) at doses of 0.05 or 0.10 mg/kg/day or placebo (2:2:1) given subcutaneously into one of the four quadrants of the abdomen or either thigh once daily in the morning for 24 weeks. The placebo consisted of a lyophilised powder containing L-histidine, mannitol, monobasic and dibasic sodium phosphate, which were also contained in the active treatment.
Parenteral adjustments after randomisation
A strict parenteral weaning algorithm was used in the protocol that allowed for no more than 10% reductions in parenteral volumes at 4-week intervals (figure 2). Weaning was performed if the 48 h urinary volumes exceeded the baseline values by more than 10%, regardless of the absolute amount. Comparisons for subsequent reductions were always made to baseline urinary volumes. Greater reductions were allowed if urinary volumes exceeded 2.0 l/day. A maximum of five reductions in parenteral support were allowed from baseline to week 24. The physician responsible for adjusting the parenteral support was expert in the management of intestinal failure and parenteral infusion weaning. The physicians were educated in the algorithm and instructed to follow it. This physician was required to be different from the person conducting the physical examinations and assessing safety because the observation of stomal swelling, known to occur in relation to GLP-2 and teduglutide treatment, may have unblinded the observer.22 The mode of parenteral adjustments, either by adjusting daily parenteral provisions or providing days off parenteral support, was entrusted to this person and was not specified in the protocol. As previously described, interim safety evaluations were performed within the week following adjustment to parenteral volumes. This was done by repeating 48 h urine collections, attaining weights and by reviewing the recorded oral fluid intakes as per the previous balance studies leading to a change in parenteral support in the algorithm. Blood samples were taken (including haematocrit, creatinine, blood urea nitrogen), urine sodium was measured and a clinical evaluation was performed checking for clinical signs of dehydration. Only if this assessment led to a conclusion that the new parenteral volume was tolerated would the weekly parenteral support volume be maintained until the next visit. Otherwise, the original parenteral volume was resumed.

Algorithm for parenteral volume adjustment during dosing.
The primary efficacy variable in the study was initially the responder rate—that is, the percentage of patients who had a reduction from baseline in parenteral volume of ≥20% at week 20 of treatment and again at week 24. A decrease of at least 20% in parenteral fluid was considered to result in a clinical benefit to the patients. After an independent review of the protocol by a statistical and regulatory panel prior to database lock, an expanded graded primary end point was introduced to compare the patients treated with teduglutide versus placebo with respect to a graded response score (GRS) criterion that accounted for both intensity and duration of a response at the end of the 24-week period. The intensity of the response relied on a reduction from baseline in weekly parenteral volume (from 20% to 100%). The duration of the response considered the responses at weeks 16 and 20, as well as weeks 20 and 24. The analysis of this expanded end point took into account higher levels of response and earlier onset of response coupled with a longer duration of response as shown in table 1. Thus, the score arose from the concept that, optimally, a graded change could be seen at the earlier time point and still observed at the later time point.
Criterion values for the graded response score
The statistical analysis of the GRS score compared the effects of placebo and teduglutide, starting with the 0.10 mg/kg/day dose according to a pre-specified step-down procedure.
Secondary efficacy end points included the number and percentage of subjects who responded (defined as a parenteral volume reduction of ≥20% from baseline at week 20 and maintained at week 24); the absolute reduction from baseline in parenteral volume and parenteral kilojoules; achievement of at least one day reduction in weekly parenteral administration or total weaning from parenteral support. Further exploratory end points included the change from baseline in oral fluid intake and urine production, body composition (evaluated by DEXA),22 plasma citrulline (an amino acid produced by enterocytes as a biomarker of a reduced enterocyte mass),23 bowel morphology (histopathological evaluation and villus height and crypt depth morphometrics, optionally taken via stomas or by colonoscopy)22 and health-related quality of life questionnaires (SF-36,24 the EuroQol EQ-5D25 and the IBDQ26).
Safety evaluations were conducted throughout the study, which included all reports of adverse events (AEs) and clinical laboratory tests. An independent data and safety monitoring committee oversaw the study.
All patients completing the randomised 24-week placebo-controlled trial were offered active treatment in a 28-week extension trial. The results from this study will be presented in a separate publication.
Statistical analysis
Based on the previous findings in a phase 2 study,22 the prospective primary hypothesis was that subcutaneous injections of teduglutide would result in a higher GRS than placebo in patients with SBS dependent on parenteral support. The statistical analysis plan specified a step-down procedure which required teduglutide at a dose of 0.10 mg/kg/day to be statistically significantly greater than placebo before evaluation of the 0.05 mg/kg/day dose. Statistical analyses were performed on the intent-to-treat population. The analysis of AEs included all 83 patients who received at least one dose of the assigned treatment. Results are expressed as mean±SD.
All statistical tests were two-sided with an α level of 0.05. A sample size of 80 randomised subjects (32 subjects in each of the two teduglutide treatment groups and 16 subjects in the placebo group) was required to provide at least 90% power to detect an increase in the percentage of subjects who had the protocol-defined minimum response defined as a parenteral volume decrease of ≥20% for week 20 and maintained at week 24 which, on average, was estimated to correspond to one day off parenteral support (from 5% in the placebo treatment group to 50% in the teduglutide treatment groups in the study). The power calculations were based on two-sided tests of significance using the Fisher exact test.
For the analysis of the primary efficacy end point (the GRS), pairwise treatment comparisons were made using a rank analysis of covariance (an extension of the Wilcoxon rank sum test) with strata for the baseline parenteral consumption level used for the stratification of the randomisation and treatment groups, with the baseline weekly parenteral volume as a covariate and a step-down procedure for multiple comparisons. For the main secondary end point (responses maintained from week 20 and week 24 and defined as a ≥20% reduction from baseline in weekly parenteral volume), pairwise comparisons between treatment groups were made using the Fisher exact test.
Results
Patients
In the period from May 2004 to November 2007, 139 patients signed informed consent forms and were screened at 32 centres in the USA, Canada, Denmark, France, Poland, Germany, the Netherlands, the UK and Belgium. Eighty-four patients with SBS were randomised and 83 were dosed (figure 1). The study data were gathered by the investigators and by a contract research organisation, and the data were held and analysed by NPS Pharmaceuticals in collaboration with the principal investigators on the writing team who had access to all data.
There were no significant differences in the demographic characteristics and medications among the groups at baseline compared with the placebo group, although parenteral volume and energy infusions tended to be higher in the 0.10 mg/kg/day teduglutide group (table 2). The numbers of patients completing the 24-week study were 15 (94%), 29 (88%) and 27 (77%) in the placebo, 0.10 mg/kg/day teduglutide and 0.05 mg/kg/day teduglutide groups, respectively.
Demographic characteristics and medication at baseline
Efficacy
Primary efficacy end point: GRS
The primary efficacy end point of the study, the GRS, was not significantly different from placebo in the teduglutide 0.10 mg/kg/day group (p=0.16), although a greater frequency of higher category responses was seen (table 3).
Summary of results for the graded response score (GRS)
Ad hoc analysis of the primary efficacy end point
The prespecified statistical analysis plan required the 0.10 mg/kg/day dose to be significantly greater than placebo before further analyses. To gain further understanding about the effect of teduglutide, it was decided to explore the effect of the 0.05 mg/kg/day dose on the primary end point. These results showed a statistically significant improvement compared with placebo in the GRS for the 0.05 mg/kg/day teduglutide dose group (p=0.007).
Secondary and exploratory efficacy end points
The binary response end point represented the proportion of patients that responded to treatment which was defined as the achievement of a ≥20% reduction from baseline in weekly parenteral volume at week 20 and maintained at week 24. The responder rate was not significantly different between the teduglutide 0.10 mg/kg/day dose group and the placebo group (25% (8/32) vs 6% (1/16), p=0.17), but the responder rate was significantly higher in the teduglutide 0.05 mg/kg/day dose group compared with placebo (46% (16/35) vs 6% (1/16), p=0.005).
Three subjects were completely weaned from parenteral support; two patients in the 0.05 mg/kg/day teduglutide treatment group became completely independent of parenteral support after 25 and 6.5 years on this treatment, receiving 5.4 l and 3.5 l parenteral support per week at baseline, respectively. Another patient receiving the 0.10 mg/kg/day teduglutide dose, who had been receiving parenteral support for 3.7 years and received 4.5 l parenteral support at baseline, was also completely weaned from parenteral support at the end of treatment at week 24. Neither active treatment arm resulted in a significant reduction in the number of days on parenteral support.
As shown in figure 3, a minor but statistically significant reduction in parenteral volume was observed in patients in the placebo group at weeks 12 and 24 compared with baseline (106±167 ml/day, p=0.02 and 128±202 ml/day, p=0.03, respectively). No significant changes were seen in the oral fluid intake or urine volume in the placebo group.

{kind=link}
{kind=link}
{kind=link}
Overall effects of placebo, teduglutide 0.10 mg/kg/day and teduglutide 0.05 mg/kg/day at baseline and weeks 4, 8, 12, 16, 20 and 24 on the reduction in oral fluid intake, increase in urine volume and reductions in daily parenteral volume based on the 48 h measurements at home. The figure also shows the composite effect, which is the sum of these beneficial combined effects.
Despite not meeting the a priori end point of a minimum reduction of 20% in parenteral fluid volume at weeks 20 and 24, patients receiving the teduglutide 0.10 mg/kg/day dose reduced their oral fluid intake by 342±599, 250±624, 365±575, 307±525, 359±638 and 392±647 ml/day at weeks 4, 8, 12, 16, 20 and 24, respectively, compared with baseline (all p<0.05). Oral fluid intake was significantly lower than placebo at weeks 12 and 20 (447±526 ml/day and 667±738 ml/day, p<0.05). Oral fluid intake in this group (teduglutide 0.10 mg/kg/day) initially increased urine production by 210±537 ml/day at week 4 compared with baseline (p=0.04), but urine production subsequently decreased to baseline levels. In addition, parenteral volume was gradually decreased by 159±342, 242±361, 275±367, 309±431 and 353±475 ml/day at weeks 8, 12, 16, 20 and 24, respectively (all p<0.05) compared with baseline. These parenteral volume reductions were not significant when compared with placebo (p=0.08 at week 24).
Demonstrating a consistent effect, patients receiving the teduglutide 0.05 mg/kg/day dose produced significantly more urine at all time points (260±521, 207±485, 288±477, 213±261, 306±567 and 367±485 ml/day at weeks 4, 8, 12, 16, 20 and 24, all p<0.05 vs baseline), in spite of maintaining a constant oral fluid intake and having parenteral volume significantly decreased by 109±230, 193±223, 246±277, 319±295 and 354±334 ml/day at weeks 8, 12, 16, 20 and 24 (all p<0.05), respectively, compared with baseline (figure 3). At week 24 both active treatment arms exhibited a mean weekly reduction in parenteral volume of −2.5 l compared with −0.9 l for placebo (p=0.08 for each comparison of active versus placebo).
Reductions in parenteral energy at week 24 of 243±450 kJ/day (p=0.056), 447±1051 kJ/day (p=0.030) and 912±1333 kJ/day (p=0.001) were seen in the placebo group, teduglutide 0.10 mg/kg/day group and the teduglutide 0.05 mg/kg/day group, respectively, compared with baseline. However, the reductions in parenteral energy were not significant between active teduglutide (0.10 mg/kg/day and 0.05 mg/kg/day) and placebo (p=0.11). Oral energy intake and faecal excretion were not measured.
Body weight was measured at all time points, while DEXA scans were only performed at baseline and at week 24. As shown in table 4, statistically significant increases in body weight were seen in the two teduglutide dose groups at various time points compared with placebo. These changes were mainly confined to the lean body mass, as illustrated in table 5.
Changes in mean±SD body weight (kg)
Changes in body composition by DEXA
A statistically significant increase in the total body bone mineral content was observed after 24 weeks of treatment with teduglutide 0.10 mg/kg/day (p=0.046), and a numerical increase was also seen in the teduglutide 0.05 mg/kg/day dose group (p=0.06, table 5) versus placebo. However, no significant changes in regional Z- or T-scores, reflecting bone mineral density, were demonstrated in this study (data not shown).
To demonstrate direct restoration of structural integrity of the intestine, changes in bowel morphology were evaluated in a subset of patients who were willing to have a biopsy taken (table 6). Significant increases in small bowel villus height were seen in both teduglutide dose groups compared with baseline and placebo. Significantly increased colonic crypt depths compared with placebo (p=0.016) were seen at the 0.10 mg/kg/day dose. At baseline, plasma citrulline, a biomarker of reduced enterocyte mass in patients with SBS, was lower in the 0.10 mg/kg/day teduglutide group than in the placebo group (p=0.051). Plasma citrulline increased significantly in both teduglutide groups compared with baseline and placebo. These increments were higher in the 0.10 mg/kg/day teduglutide group.
Short bowel (SB) villus height and crypt depth, colonic crypt depth and plasma citrulline levels
The overall results from three quality of life assessments (SF-36, EuroQol EQ-5D and IBDQ) indicated no major effect on quality of life parameters (data not shown).
Safety and tolerability
Both doses of teduglutide were well tolerated throughout the 24 weeks of observation. The overall compliance was >85% in all treatment groups. Of the 83 patients included in the study, 79 (95%) experienced at least one AE. The number of patients with AEs and serious adverse events (SAEs) and treatment-emergent AEs and SAEs leading to discontinuation of treatment is given in table 7.
Treatment-emerging adverse events (AEs) and serious adverse events (SAEs)
The most common AEs in the teduglutide treatment groups were abdominal pain (24%), headache (24%), nausea (22%), nasopharyngitis (16%) and vomiting (15%). The most frequently reported SAEs were catheter-related complications, catheter sepsis, catheter site infection, small intestinal obstruction and fever. No deaths were reported during the active treatment phase of the study. One patient died from a bleeding ulcer during the screening phase of the study. No major findings were reported in the laboratory/chemistry or haematology tests of the teduglutide-treated patients compared with placebo.
Histopathological evaluation of the intestinal tissue samples confirmed no dysplastic transformation in any of the biopsies.
Discussion
This study is the largest randomised placebo-controlled clinical trial ever performed on treatment in patients with SBS. The results of this study show that further exploration of teduglutide as a therapeutic option in restoring structural and functional intestinal integrity in patients with SBS with demonstrated intestinal failure is warranted. Teduglutide at 0.05 mg/kg/day significantly increased both the GRS (p=0.007) and the responder rate (p=0.005) compared with placebo. Many of the secondary end points were positive using either of the teduglutide doses. Contrary to expectation of a dose response, teduglutide at 0.10 mg/kg/day did not meet statistical significance in increasing the GRS or responder rate compared with placebo. Although the primary hypothesis was therefore not supported, the findings from the ad hoc analysis with the 0.05 mg/kg/day group suggest that the non-significance of the 0.10 mg/kg/day dose versus placebo could be explained by limitations imposed by the protocol and a trend towards larger baseline parenteral volume requirements in the 0.10 mg/kg/day group.
When considering the trial design and end points for this study, it is paramount to address both the numerator and the denominator in the equation used to determine parenteral volume and support while searching for a logical explanation for the discrepancy in effect between the two teduglutide doses. The numerator is best illustrated in figure 3 which shows in detail the overall effects of placebo, teduglutide 0.10 mg/kg/day and teduglutide 0.05 mg/kg/d dose at baseline and weeks 4, 8, 12, 16, 20 and 24 on the reduction in oral fluid intake, the increase in urine volume and reductions in daily parenteral volume based on 48 h measurements at home. The figure also illustrates the composite effect, which is the sum of these positive combined effects.
This figure confirms that patients receiving the 0.05 mg/kg/day dose had a constant oral fluid intake throughout the study. However, since urine output increased steadily during the study, this is in contravention to the objective of the study protocol which was to maintain a constant urine output by progressively reducing the parenteral volumes. Thus, the absolute effect of teduglutide 0.05 mg/kg/day on the reduction in parenteral volume (the numerator) appears to be underestimated. This may be explained by the restrictions in the weaning algorithm (ie, the maximally allowed reductions in parenteral support of 10% per month) or varying degrees of protocol adherence of personnel responsible for adjustments in parenteral support used in 32 centres around the globe. It can be hypothesised that a more liberal protocol allowing for more aggressive adjustments in the parenteral regimen or a better adherence to the parenteral weaning algorithm may have yielded a similar result for the two active treatment groups. Highlighting the total effect of the 0.05 mg/kg/day teduglutide dose, the composite end point increased significantly by 816±982 ml/day compared with placebo (p=0.03) at week 20.
When addressing the absolute effects of the 0.10 mg/kg/day teduglutide dose, it is obvious that this dose led to a reduction in oral fluid intake which was evident throughout the study (figure 3). According to the protocol, the parenteral volume could not be reduced until after 4 weeks of teduglutide treatment and, unless urine output exceeded 2 l/day, only monthly decrements of 10% of baseline parenteral volumes were allowed. It is likely that the patients with SBS receiving the high dose of teduglutide adjusted their oral fluid intake in order to reduce excessive urine production (figure 3) and prevent fluid retention, as also demonstrated by increases in body weight and lean body mass (tables 4 and 5). These findings are encouraging in that they demonstrate consistency with previous investigations showing that increases in intestinal wet weight absorption led to a reduction in oral fluid intake in patients with SBS if the parenteral volume was kept constant.20 It is possible that an earlier and more aggressive intervention would have enabled more substantial parenteral weaning, precluding the reduction in oral fluid intake. However, it is also possible that the high dose of teduglutide may have influenced gastric motility which may have led to decreased oral fluid intake, but this would not lead to increases in urine production at the same time (week 4, figure 3). Highlighting the total absolute effect of the 0.10 mg/kg/d teduglutide dose, the composite effect increased significantly by 489±619, 700±723 and 953±830 ml/day at weeks 12, 16 and 20, respectively (all p values <0.05) compared with placebo.
Summarising the effects on the composite effect of the two teduglutide doses, the urine volume increased by ∼350 ml/day and the parenteral volume was decreased by ∼350 ml/day in the 0.05 mg/kg/day teduglutide dose group, whereas the oral fluid intake decreased by ∼350 ml/day and the parenteral volume decreased by ∼350 ml/day in the 0.10 mg/kg/day teduglutide dose group. Thus, the effects on the reduction on the parenteral volume, the numerator, is equivalent with the two teduglutide doses and the true effect of either teduglutide dose on intestinal wet weight absorption is probably around 700 ml/day (ie, 4.9 l/week), without taking into account the positive effects on the fluid balance evaluated by the lean body mass in DEXA scans and body weight. This is highly consistent with findings in the phase 2 study where true metabolic and fluid balance studies suggested an effect of teduglutide in doses ranging from 0.03 to 0.15 mg/kg/day on wet weight absorption equivalent to 743±477 g/day.22
Turning to the denominator of the equation, it is noteworthy that, although not consistently statistically significant, imbalances between patients with SBS receiving 0.10 mg/kg/day and 0.05 mg/kg/day teduglutide were evident. Remnant small bowel length and plasma citrulline tended to be lower in the patients with SBS receiving the 0.10 mg/kg/day dose than in those receiving the 0.05 mg/kg/day dose at baseline and, more importantly, the parenteral energy and specifically baseline parenteral volume tended to be higher in the 0.10 mg/kg/day teduglutide group than in the 0.05 mg/kg/day teduglutide group (1.82 l/day vs 1.37 l/day, table 1). In addition, the number of patients who received >21 l parenteral fluid per week was higher in the 0.10 mg/kg/day teduglutide group than in the 0.05 mg/kg/day teduglutide dose group (13% vs 0%). The relative parenteral volume reductions (18±26%) in the 0.10 mg/kg/day teduglutide group were therefore numerically lower than in the 0.05 mg/kg/day teduglutide group (28±28%), while the absolute parenteral volume reductions at week 24 were comparable in the two groups (353±475 ml/day and 354±334 ml/day, respectively, figure 3). This may have contributed to the counterintuitive finding that the low dose of teduglutide had a higher responder rate than the high dose. When considering absolute parenteral volume reductions along with the results of the secondary end points including the bowel mucosa and plasma citrulline concentrations, it seems likely that the two active teduglutide doses exhibit similar efficacy.
Neither active treatment arms resulted in a significant reduction in the number of days on parenteral support. However, the algorithm for weaning parenteral support did not specify for conversion of accumulated effects into days off parenteral support. Many investigators probably found it easier to just reduce daily parenteral volumes. It was noteworthy that two patients in the 0.05 mg/kg/day dose group and one patient in the 0.10 mg/kg/day dose group were completely weaned off parenteral support.
Although the absolute parenteral volume only tended to decrease compared with placebo (p=0.08) in relation to treatment with the two teduglutide doses, the parenteral volume reduction was not associated with detrimental effects on plasma creatinine, body weight or body composition. Unfortunately, renal function was not measured specifically in this study, but a recent study suggests that long-term GLP-2 treatment may have beneficial effects on renal function as evidenced by improvements in creatinine clearance.20 Since it seems that GLP-2 only affects splanchnic and not renal blood flow, improvement in fluid balance and reductions in episodes of dehydration may have accounted for this positive effect.15
It is encouraging that reductions in parenteral energy amounting to 447±1051 kJ/day (p=0.030) and 912±1333 kJ/day (p=0.001) in the 0.10 mg/kg/day and the 0.05 mg/kg/day teduglutide groups, respectively, compared with baseline did not lead to reductions in body weight. Although measurements of dietary energy intake and faecal energy excretions were not measured in this study, it is likely that teduglutide may have decreased faecal energy losses. In the pilot phase 2 study where metabolic balance studies were performed, the reduction in faecal loss was of the same magnitude as the parenteral energy reductions in this study (808±1453 kJ/day).22 However, in this study we cannot exclude the possibility that the patients with SBS may have increased their spontaneous oral energy intake in relation to parenteral energy reductions. The small parenteral volume and energy reduction in the placebo patients emphasises the fact that closely followed patients have the ability to reduce parenteral support and underscores the importance of intestinal rehabilitation programmes. It is noteworthy that, even in dedicated centres managing to recruit patients for this study, the numbers of patients receiving adequate conventional antidiarrhoeal and antisecretory medication seems low (table 2). In general, it is prudent to use these conventional therapies before additional therapies are considered.
Seeking to identify patients especially suitable for teduglutide treatment, no significant correlation was found between residual length of small bowel and the ability to wean from parenteral support in any of the groups (with or without a colon in continuity) or in all the groups put together. When the data from the two teduglutide treatment groups were pooled both the responder rate and GRS were significantly better than placebo (p=0.03 and p=0.04, respectively). However, reductions in parenteral volume and the composite effects were still only of borderline significance compared with placebo (p=0.051 and p=0.07, respectively). A significant correlation was found between the baseline parenteral volume and the absolute parenteral volume reductions at week 24 (r=0.44, p=0.0006), suggesting that the largest absolute effect of teduglutide occurred in patients with the worst remnant bowel function. However, this adds to the argument that a difference in baseline parenteral volume was not the only explanation for the discrepancy between the two teduglutide dose groups with regard to the effect on the primary efficacy end point.
A potential limitation of the current study is that endogenous postprandial meal-stimulated GLP-2 production was not measured. It could be speculated that patients with the largest remnant L-cell mass—that is, patients with residual ileum and colon27—would have the higher endogenous GLP-2 secretions to start with and therefore might be less likely to respond since they already may have been sensitised to stimulation. In fact, 68% of patients with SBS recruited to this study had part of the colon in continuity which would have masked the full potential of teduglutide. In this study we found no significant difference in the percentage reduction in volume of parenteral support between those with and without a colon in continuity. The doses of teduglutide given in this study are known to give rise to supraphysiological teduglutide levels even compared with the elevated endogenously-secreted GLP-2 levels in patients with a preserved colon.28 Previous balance studies suggest that teduglutide was equally beneficial in patients with SBS with a preserved colon and those with a jejunostomy or ileostomy.22
As part of the safety evaluation, all patients with a preserved colon had a screening colonoscopy at baseline and were re-screened at week 24 to exclude the occurrence of premalignant or malignant lesions. Although neither GLP-2 nor teduglutide have been shown to be an independent risk factor for the development of colonic adenomas, dysplasia or malignancy, it was reassuring that no polyps were found on colonoscopy and that histopathological evaluation of the intestinal tissue samples confirmed no dysplasia transformation in any of the 390 individual biopsies taken in relation to this study.
As a sign of the potency of the drug, small bowel biopsy data showed that treatment with teduglutide increased villus height and crypt depth, although the data rest on only a subset of the patients with SBS who were willing to have small bowel biopsies taken. In the colon the crypt depth only increased significantly in the patients receiving 0.10 mg/kg/day teduglutide. These beneficial effects on bowel morphology are also in accordance with findings in the phase 2 study.22
The statistically significant increases in fasting plasma citrulline concentrations in both groups supported the intestinotrophic effects of teduglutide. Earlier studies have shown that fasting plasma citrulline correlates with residual bowel length in patients with SBS.23 This can be explained by the fact that enterocytes of the small intestine are unique in the body in being able to synthesise and export citrulline to the blood. Thus, the intestinotrophic effects of teduglutide may not account for all the effects on intestinal wet weight absorption. Effects on gastric emptying and secretions are also likely and may need to be further investigated.13 14
The number of subjects reporting an AE and the number of AEs were distributed similarly across all groups. Equally, the distribution of the number of subjects reporting SAEs was similar between groups. Many of the AEs leading to the discontinuation of active treatment in this study have been described previously with native GLP-2 treatment.21 They were mainly related to the pro-absorptive and intestinotrophic effects of teduglutide or restrictions in the parenteral weaning algorithm. It is likely that a more individualised parenteral adjustment algorithm and modification to medications that had narrow therapeutic windows and variable bioavailability may have prevented several of these AEs. However, the occurrence of small intestinal obstruction with teduglutide warrants a degree of caution to be exercised when used in patients with a history of abdominal pain and recurrent small bowel obstructions. Increased intestinal fluid absorption, which is desirable in the majority of patients with SBS, could cause concern in patients with latent or overt cardiac decompensation if the parenteral fluid infusions are not weaned accordingly.
It must be acknowledged that the quality of life tools used in this trial were not specific for patients with SBS. No improvements in health-related quality of life in relation to teduglutide treatment could be demonstrated when employing generic measures such as the SF-36, EQ-5D and IBDQ tools. However, since there is large heterogeneity in the symptoms of patients with SBS, it is likely that the benefits perceived in relation to teduglutide treatments could translate into heterogeneous outcomes not specifically captured by the tools employed. In addition, the study was likely to be underpowered to detect clinically meaningful changes, and the small number of subjects in this study hindered meaningful subgroup analyses. Owing to the large heterogeneity of patients with SBS with intestinal failure, it is difficult to evaluate the clinical meaningfulness of the suggested benefits of teduglutide at present. However, although few in number, the significance of actually getting three patients off all parenteral support should not be underestimated. Furthermore, the concomitant reduction in faecal excretions may also be perceived as a great advantage, especially in patients with SBS with rectum and colon in continuity, large diarrhoeas and fear of faecal incontinence, but also in patients with SBS with jejunostomies or ileostomies and large stomal output which may limit their social interaction. Although teduglutide may not directly decrease the incidence of catheter-related complications, any reduction in the time that patients with SBS are dependent on their central line and parenteral support is beneficial.
In conclusion, teduglutide was safe, well tolerated and demonstrated restoration of structural and functional integrity of the remaining intestine with significant intestinotrophic and proabsorptive effects facilitating reduction in parenteral support in patients with SBS with intestinal failure. Teduglutide has the potential to reduce dependency on parenteral support and its side effects. We suggest that the study design, the parenteral weaning algorithm and imbalance at baseline regarding the need for parenteral support probably jeopardised the effect of the 0.10 mg/kg/day dose of teduglutide on the primary efficacy end point of the study. However, the ad hoc statistically significant effect of the 0.05 mg/kg/day dose on the primary end point and the effects on secondary end points confirmed the overall potency of both doses of teduglutide. A confirmatory phase 3 study will be conducted. Adjustments to the parenteral weaning algorithm—allowing for earlier and more aggressive parenteral weaning—have been made and the 0.05 mg/kg/day teduglutide dose is to be employed. Further studies will determine whether the reductions in parenteral volumes translate into reduced complications in relation to parenteral support and even improvements in quality of life. SBS disease-specific questionnaires are currently in development for this purpose. Studies of longer duration are needed to determine whether patients with milder forms of SBS and intestinal failure can be weaned permanently from parenteral support and be made orally autonomous. The effect of longer dosing in relation to response is unclear, although data from early reports from the short-term teduglutide study suggest that the beneficial effects of teduglutide are reversible within 3 weeks after terminating treatment.22 This suggests that chronic long-term treatment probably will be necessary to maintain the benefits seen in parenteral reductions. An opportunity may exist to study a possible additional effect of teduglutide in the acute adaptive period after intestinal resection. Studies in children with SBS are also warranted as the complications in relation to parenteral support are even more predominant in this group of patients.
Acknowledgments
We thank Kelly Tappenden, Department of Food Science and Human Nutrition, University of Illinois, Urbana, IL, USA, for performing villus height and crypt depth morphometrics.
References
Supplementary materials
online only appendix
Files in this Data Supplement:
Online only appendix
Files in this Data Supplement:
Footnotes
Competing interests The authors have served as consultants for NPS Pharmaceuticals and Nycomed GmbH.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the local IRBs and medical ethics committees in the USA, Canada, Denmark, France, Poland, Germany, the Netherlands, the UK and Belgium.
Provenance and peer review Not commissioned; externally peer reviewed.