Article Text

Abstract
These guidelines update previous guidance published in 2005. They have been revised by a group who are members of the UK and Ireland Neuroendocrine Tumour Society with endorsement from the clinical committees of the British Society of Gastroenterology, the Society for Endocrinology, the Association of Surgeons of Great Britain and Ireland (and its Surgical Specialty Associations), the British Society of Gastrointestinal and Abdominal Radiology and others. The authorship represents leaders of the various groups in the UK and Ireland Neuroendocrine Tumour Society, but a large amount of work has been carried out by other specialists, many of whom attended a guidelines conference in May 2009. We have attempted to represent this work in the acknowledgements section. Over the past few years, there have been advances in the management of neuroendocrine tumours, which have included clearer characterisation, more specific and therapeutically relevant diagnosis, and improved treatments. However, there remain few randomised trials in the field and the disease is uncommon, hence all evidence must be considered weak in comparison with other more common cancers.
- Clinical decision-making
- decision analysis
- intestinal obstruction
- neuroendocrine cells
- endocrine tumours
- neuroendocrine tumours
- diarrhoea
- infective colitis
- salmonella
- sepsis
- cancer
- pancreatic cancer
- pancreatic endocrine tumour
- pancreatic tumours
- peptide receptors
- gallbladder cancer
- pancreatitis
- pancreatic pathology
- histopathology
- bilary duct carcinoma
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Clinical decision-making
- decision analysis
- intestinal obstruction
- neuroendocrine cells
- endocrine tumours
- neuroendocrine tumours
- diarrhoea
- infective colitis
- salmonella
- sepsis
- cancer
- pancreatic cancer
- pancreatic endocrine tumour
- pancreatic tumours
- peptide receptors
- gallbladder cancer
- pancreatitis
- pancreatic pathology
- histopathology
- bilary duct carcinoma
Recommendations
General recommendations
Multidisciplinary teams (MDTs) at referral centres should give guidance on the definitive management of patients with all varieties of neuroendocrine tumours (NETs). Level of evidence 5, Grade of recommendation D.
MDT representation should normally include specialist physicians in NETs (gastroenterologists, oncologists and/or endocrinologists), surgeons, radiologists, nuclear medicine specialists, histopathologists and clinical nurse specialists. Level of evidence 5, Grade of recommendation D.
Genetics
Clinical examination to exclude complex cancer syndromes (eg, multiple endocrine neoplasia 1 (MEN1)) should be performed in all cases of NETs, and a family history taken. Level of evidence 4, Grade of recommendation C.
In all cases where there is a family history of NETs, or a second endocrine tumour, a familial syndrome should be suspected. Level of evidence 4, Grade of recommendation C.
In all patients, secondary tumours and other gut cancers should be considered. Level of evidence 4, Grade of recommendation C.
Diagnosis (biochemical measurements)
If a patient presents with symptoms suspicious of a gastroenteropancreatic NET:
Baseline tests should include plasma chromogranin A and urinary 5-hydroxyindoleacetic acid. Level of evidence 3, Grade of recommendation C.
Specific biochemical tests should be requested depending on which syndrome is suspected. Level of evidence 3, Grade of recommendation C.
Imaging
For detecting the primary tumour a multi-modality approach is best. CT, MRI and somatostatin receptor scintigraphy (SSRS) are recommended. Gallium-68 (68Ga) positron emission tomography (PET)/CT is recommended for the detection of an unknown primary. Level of evidence 3, Grade of recommendation A/B.
Additional imaging modalities may include endoscopic ultrasound (EUS), endoscopy, digital subtraction angiography (DSA) and venous sampling. Level of evidence 4, Grade of recommendation B/C.
For assessing secondaries, 68Ga PET/CT is the most sensitive modality. Where this is not available, SSRS in combination with CT is the most sensitive modality. Level of evidence 3, Grade of recommendation B.
Histopathology is required to confirm the diagnosis. Level of evidence 3, Grade of recommendation B.
When a primary has been resected, cross-sectional imaging (CT and MRI) using RECIST criteria and SSRS may be indicated for follow-up1 if the patient is involved in a clinical trial. Level of evidence 5, Grade of recommendation D.
Pathology
Pathology is currently the diagnostic gold standard. Level of evidence 5, Grade of recommendation D.
Pathology reporting and reviews should be made by the MDT pathologist. Level of evidence 5, Grade of recommendation D.
Pathological characterisation and classification of NETs should be based on the WHO 2010 classification, the Union for International Cancer Control (UICC) TNM (7th edition), and the European Neuroendocrine Tumour Society (ENETS) site-specific T-staging system. Level of evidence 5, Grade of recommendation D.
Therapy
The aim of treatment should be curative where possible.
The main aim is to keep the patient disease and symptom-free for as long as possible and to maintain a good quality of life (QoL).
The extent of the tumour, its metastases, histological grade and secretory profile should be determined as far as possible before planning treatment. Level of evidence 4, Grade of recommendation C.
The choice of treatment depends on the symptoms, stage of disease, degree of uptake of radionuclide and the histological features of the tumour. Level of evidence 4, Grade of recommendation C.
Surgery should be offered when NETs are resectable and there is curative intent (or when debulking offers palliation) to patients who are fit and have limited disease—primary ± regional lymph nodes. Level of evidence 4, Grade of recommendation C.
Surgery should be considered in those with liver metastases and potentially resectable disease. Level of evidence 4, Grade of recommendation D.
For patients who are not fit for surgery, the aim of treatment is to improve symptoms and maintain an optimal QoL, and where possible to improve survival. Level of evidence 5, Grade of recommendation D.
Treatment choices for non-resectable disease include somatostatin analogues, biotherapy, targeted radionuclide therapy, locoregional treatments including ablation and (chemo)embolisation and chemotherapy. Level of evidence 4, Grade of recommendation C.
External beam radiotherapy may relieve bone pain from metastases. Level of evidence 4, Grade of recommendation C.
Chemotherapy may be used for inoperable or metastatic pancreatic NETs. Level of evidence 1, Grade of recommendation A.
Chemotherapy may be used for poorly differentiated NETs and in selected non-pancreatic NETs of high grade or aggressive clinical course. Level of evidence 2, Grade of recommendation B.
Sunitinib or everolimus may be used as a line of therapy for patients with advanced (inoperable or metastatic), progressive (radiological evidence of disease progression within 12 months), well-differentiated pancreatic NETs. Level of evidence 1, Grade of recommendation A.
If possible, patients with NETs should be entered into formal trials of new drug treatments. Level of evidence 4, Grade of recommendation C.
Ablation
In the setting of metastatic NET, ablation most commonly has a role in small volume tumours, paucilesional disease or in combination with resection. Level of evidence 3, Grade of recommendation C.
Ablation, in common with resection, has been shown to be useful in symptom relief. Level of evidence 3, Grade of recommendation C.
Image-guided ablation can contribute to the cytoreductive approach to metastatic disease. Level of evidence 3, Grade of recommendation C.
Carcinoid heart disease (CHD)
All patients with midgut NETs, with or without hepatic metastasis, and all patients with the carcinoid syndrome, should be screened for CHD: this may include N-terminal pro-brain natriuretic peptide (NT-proBNP) and echocardiography. Level of evidence 1b, Grade of recommendation B.
Patients with elevated NT-proBNP (>260 pg/ml (>30 pmol/l) based on single institution data) should be screened with echocardiography. Level of evidence 1b, Grade of recommendation B.
Referral of patients with confirmed CHD to a cardiology department with expertise in dealing with CHD should be considered. Level of evidence 5, Grade of recommendation D.
Cardiac surgery should be considered in appropriate cases and should be performed by skilled operators in selected centres with experience of dealing with patients with NET. Level of evidence 2c, Grade of recommendation C.
Origin and purpose of these guidelines
These guidelines update previous guidance published in 2005.2 They have been revised by a group who are members of the UK and Ireland Neuroendocrine Tumour Society (UKINETS), with endorsement from the clinical committees of the British Society of Gastroenterology, the Society for Endocrinology, the Association of Surgeons of Great Britain and Ireland (and its Surgical Specialty Associations), the British Society of Gastrointestinal and Abdominal Radiology, and others. The authorship represents leaders of the various groups within UKINETS, but a large amount of work has been carried out by other specialists, many of whom attended a guidelines conference in May 2009. We have attempted to represent this work in the acknowledgements section. Over the past few years, there have been advances in the management of NETs, which have included clearer characterisation, more specific and therapeutically relevant diagnosis, and improved treatments. However, there remain few randomised trials in the field and the disease is uncommon, hence all evidence must be considered weak in comparison with other more common cancers.
It is the unanimous view of the guideline authors that MDTs at referral centres should give guidance on the definitive management of patients with all varieties of NETs, with a particular emphasis on gastroenteric and pancreatic NETs, but also including pulmonary NETs. MDT representation should normally include physicians (gastroenterologists, oncologists and/or endocrinologists), surgeons, radiologists, nuclear medicine specialists, histopathologists and clinical nurse specialists.
The working party that produced these updated guidelines included specialists from these various disciplines who contribute to the management of all NETs, including gastrointestinal NETs. The purpose of these guidelines is to identify and inform the key decisions to be made in the management of gastroenteropancreatic NETs.
The guidelines are not intended to be a rigid protocol, but form a basis upon which to aim for improved standards in the quality of treatment given to affected patients.
The terminology used in this guideline has been chosen to reflect the more modern use of the term ‘NET’ rather than the older but often more familiar term ‘carcinoid tumour’. This decision reflects both the aetiology of NETs and that these tumours are found in diverse locations, not just in the gastrointestinal tract. The term ‘carcinoid tumour’ is still commonly used in clinical practice, but is deemed obsolete by many experts. However, we retain the terms ‘carcinoid syndrome’ and ‘CHD’ and will do so until newer terminology is introduced.
Formulation of the guidelines
Literature search
Members of the guidelines working party were assigned sections of the 2005 guidance to update. A systematic review of the relevant literature was performed, with synthesis of the available evidence; this was followed by peer group appraisal and then expert review.
Categories of evidence
The Oxford Centre for Evidence-based Medicine's Levels of Evidence (May 2001) were used to evaluate the evidence cited in these guidelines.3
Aetiology, epidemiology, clinical features, prognosis and genetics
Aetiology
The aetiology of NETs is poorly understood. Most NETs are sporadic but there is a small familial risk (see under Genetics). NETs constitute a heterogeneous group of neoplasms that share certain characteristic biological features, and can therefore be considered a common entity. The origin of the cells in the gut is debated, but they may arise from pluripotential progenitor cells that develop neuroendocrine characteristics. It seems unlikely that these cells migrate in from the neural crest, as previously thought. Such tumours originate from pancreatic islet cells, gastroenteric tissue (from diffuse neuroendocrine cells distributed throughout the gut), neuroendocrine cells within the respiratory epithelium, and parafollicullar cells distributed within the thyroid (these tumours being referred to as medullary carcinomas of the thyroid). Pituitary, parathyroid and adrenomedullary neoplasms share certain characteristics with these tumours, but are not considered here. These guidelines apply to all NETs arising from the gut, including the pancreas and liver (gastroenteropancreatic), as well as those arising from the lung that have metastasised to the liver or abdominal lymph nodes. Small intestinal and pancreatic NETs have different signatures, as do benign and malignant tumours. The general term of NET is preferred and encouraged for describing gastrointestinal and pancreatic NETs (often denoted pNET), although the term carcinoid is still in common usage and usually denotes a well-differentiated serotonin (5-hydroxytryptamine)-secreting midgut tumour.
Historically, gut-derived NETs were classified according to their embryological origin, into tumours of the foregut (bronchi, stomach, pancreas, gallbladder, duodenum), midgut (jejunum, ileum, appendix, right colon) and hindgut (left colon, rectum).4 The WHO has issued guidance on the classification of NETs according to histopathological characteristics (see under Pathology). The molecular biology of NETs is still poorly understood but there are emerging data to suggest that molecular profiling and identification of common genetic elements may enhance tumour classification and identify potential targets that may be involved in tumour progression.5–7
Epidemiology
The incidence of NETs is reported to be rising. Early data from the UK, Sweden and Switzerland8–11 suggested that the incidence of gastrointestinal NETs was between 2 and 3 per 100 000 persons per year with an overall slight preponderance in women. The largest and most recent analyses of the epidemiology of NETs have examined data from the USA (the Surveillance, Epidemiology and End Results (SEER) Programme) and Norway (the Norwegian Registry of Cancer (NRC)).12–14 The USA data cover nearly five decades and demonstrate a steady increase in the incidence, or reporting, of stomach and rectal tumours and a decrease in that of appendiceal NETs.12 14 There are reported ethnic differences in NET incidence, with African–Americans having the highest overall value at 6.5 per 100 000 per year.12 The most common site for a primary NET in Caucasians in the USA is the lung, accounting for about 30% of all new cases, whereas in Asian/Pacific, American Indian and African–Americans the rectum is the most common site, with about 27% of new cases having the primary here.12 In Norway, the most common primary site is the small intestine (25%).13 The overall incidence of NETs in Caucasians is 4.44 per 100 000 persons per year in the USA and 3.24 per 100 000 persons per year in Norway. This rate of occurrence is higher than previously thought, but is in keeping with data from autopsy studies in Sweden from 30 years ago.15
Other analyses suggest an even higher incidence of NETs, reporting a fourfold increase between 1973 and 2004, from 2.1 to 9.3 new cases per 100 000 persons per year.16 This report concluded that NETs are the most common small bowel tumour (37.4%), followed by adenocarcinoma (36.9%), lymphomas (17.3%) and stromal tumours (8.4%). However, another analysis of the SEER dataset from the USA suggests that the rate of increase in the incidence of NETs has been from 1.09 to 5.25 per 100 000 persons per year over the same time period.14 The incidence of tumours in the appendix, caecum and pancreas almost doubled between 1975 and 2005, but collectively these tumours make up only a small percentage of the total number of cases, and for each the incidence is about 0.1–0.2 per 100 000 persons per year. The incidence of rectal, small intestinal and pulmonary primaries increased by 4–5-fold over the same period with incidences of between 0.9 and 1.3 per 100 000 persons per year.12 14 17
Whatever the precise incidence of NETs, it appears that the number of patients presenting with these tumours has been steadily increasing.18 Indeed, since many NETs are slow-growing or of uncertain malignant potential, with even malignant NETs associated with prolonged survival, the prevalence of NETs is relatively high.14 It is reported that, despite the rising number of cases, there is still a delay of up to 7 years between the appearance of first symptoms and a diagnosis of NET.18
It is not possible to predict who might develop a NET. There is an increased risk in patients with atrophic gastritis and persons of Afro-Caribbean origin.19 In the USA, there is evidence that a long-term history of diabetes mellitus, especially when combined with a family history of cancer, also increases the risk of developing a gastric NET.20 This work also reported that a history of smoking or alcohol use has no apparent effect on the risk of developing a NET. Overall, men have a greater risk than women of developing small bowel cancer, and small intestinal NETs are 30% more common in men than in women.13
The sites and overall frequencies of primary NETs, as reported in the USA and Norway, are shown in table 1.13 A number of other national databases are in the process of reporting and publication, although most suffer from selective reporting and accrual from specialist centres. In general, however, the rates and tumour types in table 1 are in agreement with these series.
Sites and overall frequencies of primary NETs in the USA (from the SEER Programme) and Norway (from the NRC)12–14
The frequencies of primary NETs reported as occurring in the liver or biliary tract were <1%. Unknown primary sites or sites other than those listed in table 1 accounted for 11–14% of cases.13
For patients with a small bowel primary NET, the likelihood of nodal metastases being present at the time of diagnosis is about 60%, irrespective of the precise site of origin within the small bowel; with a colonic primary, the likelihood is about 40–70%, with a greater risk within this range if the primary is in the ascending colon, and a lower risk if the primary is in the rectum.12 Nodal metastases are present in 5% of patients with an appendiceal primary and 15% of those with a pulmonary primary.12 The chance of liver metastases being present at the time of diagnosis is about half the risk of nodal spread having occurred.12
The chance of metastases being present at the time of diagnosis and the likelihood of MEN1 is shown in table 2.
Clinical features
Gastroenteropancreatic NETs may be classified into non-functioning tumours, which have no hormone-related clinical features, and functioning tumours, which cause symptoms due to peptide and hormone release. In all NETs, presenting features may include non-specific symptoms such as pain (which may be intermittent and present for many years), nausea and vomiting, and, in some cases, anaemia due to intestinal blood loss (table 3).18 25–27 Pain may be due to local tumour invasion, bowel obstruction or mesenteric ischaemia. Most gastroenteropancreatic NETs are non-functioning and present with mass effects of the primary tumour or metastases (usually liver). A high index of suspicion is needed to identify patients, and diagnosis is often delayed for several years, often made as an incidental finding at surgery or during radiological assessment.28–32 Gastroenteropancreatic NETs are also associated with other primary malignancies.
Clinical features of pancreatic NETs
Non-functioning gastroenteropancreatic NETs
Gastric and rectal NETs are often diagnosed coincidentally at endoscopy, or may be the source of anaemia. Type I and II gastric NETs are related to hypergastrinaemia, and are usually small (measuring several millimetres) and multiple. Type I tumours are associated with achlorhydria, while type II are associated with Zollinger–Ellison syndrome and MEN1; neither are likely to metastasise when small. In contrast, type III gastric NETs are often larger (measuring several centimetres) and solitary, are not associated with elevated gastrin, and usually have malignant potential. Appendiceal NETs may be identified during surgery for appendicitis, or during colonic surgery, and are less commonly associated with a secretory syndrome. Approximately 60% of all pancreatic NETs are non-functioning. The primary tumour may be large at presentation, when approximately 50% have metastasised. UK data show that bronchial NETs may present with cough and recurrent pneumonia (22%), incidentally on chest radiography (18%), with haemoptysis (13%), or with shortness of breath (9%).33 Rarely, these tumours may be associated with ectopic adrenocorticotropic hormone (ACTH) secretion and Cushing's syndrome.
Functioning tumours
Functioning pancreatic NETs cause symptoms dependent on the peptide hormone released (table 3).
Carcinoid syndrome
Carcinoid syndrome occurs in ∼20% of cases of well-differentiated endocrine tumours of the jejunum or ileum (midgut NET) and consists of (usually) dry flushing (without sweating; 70% of cases34) with or without palpitations, diarrhoea (50% of cases) and intermittent abdominal pain (40% of cases)35; in some patients, there is also lacrimation and rhinorrhoea. Carcinoid syndrome occurs less often with NETs of other origins and is very rare in association with rectal NETs. It is usually due to metastasis to the liver, with the release of vasoactive compounds, including biogenic amines (eg, serotonin and tachykinins), into the systemic circulation. However, it may also occur in the absence of liver metastases if there is direct retroperitoneal involvement, with venous drainage bypassing the liver. Pain due to hepatic enlargement may also be a presenting feature, as may upper right abdominal pain (similar to that of pulmonary infarction) secondary to either haemorrhage into, or necrosis of, a hepatic secondary tumour. Wheezing and pellagra are less common presenting features. CHD is present in ∼20% of patients at presentation and usually indicates that the syndrome has been present for several years (see also under Carcinoid heart disease).36
Carcinoid crisis
Carcinoid crisis is characterised by profound flushing, bronchospasm, tachycardia and widely and rapidly fluctuating blood pressure. It is thought to be due to the release of mediators, which lead to the production of high levels of serotonin and other vasoactive peptides. It is usually precipitated by anaesthetic induction for any operation, intraoperative handling of the tumour, or other invasive therapeutic procedures such as embolisation and radiofrequency ablation.
Prognosis
NETs are slow-growing tumours and survival depends on a number of factors. The highest 5-year survival rates in both the SEER and NRC epidemiological studies were for patients with rectal primary tumours (74–88%), while the lowest were found among patients with pancreatic primaries (27–43%; table 4).12 13 However, for patients with a benign insulinoma, the 5-year survival rate may be over 95%.38 It had been thought that appendiceal NETs were relatively benign in their behaviour, yet the SEER data have reported 5-year survival rates of 95% for patients with localised disease and 35% for patients with distant spread at the time of diagnosis.12 The most recent SEER and NRC data show that, overall, patients with appendiceal primaries have 5-year survival rates between 70% and 80%.13
The SEER data show that the 5-year survival of all patients with NETs, regardless of the primary site and degree of spread, did not change between 1973 and 2002 and remained at 60–65%. Over this period there may have been a slight improvement in outcome for patients with a small bowel primary, with an overall 5-year survival of ∼65% in 2002.18 The 5-year survival among just over 4000 patients from England and Wales presenting between 1986 and 1999 was 57% for those with a well-differentiated tumour, and 5% for those with a ‘small-cell’ tumour.37
Length of survival is directly related to both the extent of the disease at the time of diagnosis and the degree of differentiation of the tumour. According to the most recent SEER data, the 5-year survival of patients with well or moderately well-differentiated tumours was:
82% for local spread.
68% for regional spread.
35% for distant spread.
For poorly differentiated tumours these values were lower:
38% for local spread.
21% for regional spread.
4% for distant spread.14
The histopathological type of the tumour, its Ki-67 or MIB-I proliferation index, size and location, as well as the age of the patient, also affect survival.13 14 39–45 It is no longer appropriate to quote survival figures based on overall survival of all patients with a tumour at a particular site. Such information, when discussed with patients, should be based on a consideration of the impact of the above factors.
Using the newer pathological classification aids in the prognostication of survival. Thus, the 5-year survival rates for grades 1, 2 and 3 tumours are 96%, 73% and 28%, respectively. Similarly, using the recommended TNM staging system, 5-year survival rates for stages I, II, III and IV are 100%, 90%, 79% and 55%, respectively, demonstrating the utility of such newer classifications.46
Genetics
NETs may occur as part of complex familial endocrine cancer syndromes such as MEN1, MEN2,47 neurofibromatosis type 1 (NF1),48 49 Von Hippel Lindau (VHL) and Carney complex, although the majority occur as non-familial (ie, sporadic) isolated tumours. The incidence of MEN1 in gastroenteropancreatic NETs varies from virtually nil in gut NETs, to 5% in insulinomas, to 25–30% in gastrinomas.28 However, it is important to search thoroughly for MEN1, MEN2 and NF1 syndromes in all patients with NETs by obtaining a detailed family history and undertaking a clinical examination with appropriate biochemical and radiological investigations. The diagnosis can now be confirmed by genetic testing. In addition, mutations involving the succinate dehydrogenase subunit D, which are usually associated with paragangliomas and phaeochromocytomas, have been reported in patients with midgut NETs.50
A diagnosis of MEN1, MEN2, NF1 or a paraganglioma syndrome not only has important implications for the patient but also for the patient's relatives, who should be considered for screening for the associated tumours and genetic testing. Gastrointestinal NETs may, on rare occasions, occur as an isolated familial cancer, without evidence of a MEN syndrome.51 This is consistent with results from epidemiological studies, which show a small increased familial risk, with standardised incidence rates of 4.35 (n=4; 95% CI 1.86 to 7.89) for small intestinal and 4.65 (n=4; 95% CI 1.21 to 10.32) for colon NETs in offspring of parents affected by NETs. This familial clustering was seen to be more pronounced with midgut and hindgut tumours, and very few patients had obvious MEN1, indicating that much of this association is independent of MEN1.10 Patients with tuberous sclerosis complex may also be at increased risk of NETs.52
A family history of cancer is also a significant risk factor for all types of NETs. Risk is elevated more often in women than in men. In a USA-based case–control study, the adjusted ORs (95% CI) of positive family history of cancer for small bowel, gastric, lung, pancreatic and rectal NETs were 1.4 (0.9 to 2.2), 0.8 (0.3 to 2.6), 2.7 (1.2 to 6.1), 1.5 (0.8 to 2.9) and 1.8 (0.5 to 5.8) among men and 2.3 (1.2 to 4.3), 5.2 (1.4 to 19.6), 2.1 (1.1 to 4.3), 3.7 (1.4 to 9.9) and 1.3 (0.4 to 3.0) among women, respectively.20 Moreover, it appears that up to ∼20% of patients with gastric NETs may also develop another synchronous cancer, typically affecting the gastrointestinal tract.12 There are insufficient data to know whether this is a feature in patients with other NETs. Risks for second cancers in men were increased during the first year of follow-up. Slightly lower risks were noted in women.
Diagnosis
Diagnosis of NETs is based on clinical manifestations, peptide and amine secretion,53 and specialised radiological and nuclear imaging. Diagnosis is secured by detailed histology, which should be obtained whenever possible.
Biochemistry
The measurement of the secretory products of NETs is helpful in three respects:
To assist with initial diagnosis.
To assess the efficacy of treatment.
To assess changing prognosis.
The life of patients with NET may be under more immediate threat from the syndrome than from the underlying malignancy. As the syndromes are driven by circulating secretory products, it is usually beneficial to reduce these.
Chromogranin A (CgA) is the only general marker for NETs as it is usually found in high concentrations regardless of whether the tumour is accompanied by hormone-related clinical features.29 54 55 However, chromogranin B (CgB) may be elevated when CgA is in the reference range.56–58 Two chromogranin B assays are commercially available, from Euro Diagnostic UK and Byorbyt Ltd. Pancreastatin, one of the post-translational products of CgA, is a useful additional marker,59 60 as it is only raised in metastatic NETs and results are not confounded by conditions that raise CgA (eg, treatment with proton pump inhibitors, or atrophic gastritis).
Pancreatic polypeptide (PP), a product of the normal pancreas, is secreted in high concentrations from a significant proportion of NETs throughout the gastroenteropancreatic tract (50–80% of pancreatic NETs and >30% of all gut NETs). Therefore PP is a useful additional or alternative general marker in some circumstances, particularly when CgA or CgB are within the reference range.26 61
The measurement of specific markers is useful for the diagnosis and monitoring of specific tumours.53
The majority of tumours of the jejunum, ileum, proximal colon and appendix (>70%) and a significant number of NETs of the stomach and respiratory system (10–35%) secrete serotonin. Reliable assay of serotonin in blood has been problematic, but is performed in a few centres.62 The breakdown product of serotonin, 5-hydroxyindoleacetic acid (5-HIAA), has been used as an alternative and may be readily measured in a 24 h urine collection. However, there are many dietary restrictions and drug interference problems associated with the measurement of serotonin and 5-HIAA, and the laboratory should be contacted to ensure that inappropriate foods and drugs are excluded for 3 days before and during the urine collection (online appendix 1).
As many tumours in the ileum and colon present with bowel obstruction, and will have been resected surgically before diagnosis of tumour type, laboratory specimens are often collected postoperatively. Urinary 5-HIAA may then be within the reference range. CgA and neurokinin A measurement,63 together with measurement of 5-HIAA, will indicate residual disease in more than 90% of patients.64 65 As surgical cure is rare, all of these patients should be followed-up indefinitely using laboratory testing (and imaging), with the exception of those with some small tumours of the appendix (see under Surgery).
A range of peptide markers specific to the tumour site may also be measured. These are detailed in table 5.
Peptide markers specific to the tumour site
Laboratory diagnosis of some tumour types is not straightforward.
In the stomach, type I NETs are associated with atrophic gastritis, while in type II NETs the gastrin results from a gastrin-secreting tumour. Circulating gastrin is raised in both types I and II, and causes enterochromaffin-like (ECL) cell hyperplasia and, ultimately, may cause ECL NETs. In type III tumours, gastrin is not raised. CgA is raised in all three types.66 67
The laboratory diagnosis of gastrinoma may be difficult. Both gastrin and CgA are raised in gastrinoma. However, both gastrin and CgA are also raised in many common conditions, particularly when gastric acid is reduced or absent—for example, in patients with atrophic gastritis or in those receiving proton pump inhibitor (PPI) therapy. Upper gastrointestinal endoscopy and gastric biopsy is always required to differentiate gastrinoma from atrophic gastritis; secretin stimulation and intragastric pH may also be needed in some cases.
When circulating gastrin and CgA are raised, a fasting specimen is required; autoimmune atrophic gastritis must be excluded, and Helicobactor pylori should be eradicated. Recurrent peptic ulcer disease, especially with gastrointestinal bleeding in the absence of H pylori, gives a strong suspicion of gastrinoma. PPIs and H2 antagonists raise both circulating CgA and gastrin (online appendix 2). PPIs should be withdrawn with great caution and ideally stopped 10 days to 2 weeks before any planned estimation of circulating fasting gastrin. In patients with suspected gastrinoma, oral H2 antagonists may be used instead for this period, but it is advisable for these to also be interrupted 48 h before the test.68 Patients with a gastrinoma should be advised that it is dangerous to stop PPIs without supervision. Where a question regarding the diagnosis remains, a secretin test may be performed, with or without gastric acid studies.
Patients with gastrinoma may present with circulating gastrin <10% above the reference range.69 The majority of gastrinomas are located in the duodenum rather than in the pancreas.
All patients with gastrinoma should be considered as candidates for MEN1 syndrome. Fasting calcium, parathyroid hormone and prolactin measurements should be made.
Insulinoma may be difficult to diagnose, as circulating insulin concentrations are often within the reference range in these patients. The insulin level, however, will be inappropriate to the blood glucose. Measurement of C peptide or pro-insulin is helpful. The majority of insulinomas are benign, and CgA will not be raised unless the tumour is metastatic. A 48–72 h fast under hospital supervision with serial blood glucose analysis is the gold standard diagnostic tool and will usually trigger hypoglycaemia within 24 h. In ∼5% of patients, however, the hypoglycaemia may only be revealed postprandially.70
Non-functioning tumours of the pancreas often secrete PP, and this may be a helpful marker in patients with these tumours.61
The majority of rectal tumours do not secrete CgA. Many secrete PP and some may secrete enteroglucagon, human chorionic gonadotropin-β or acid phosphatase. When a marker is identified, this is helpful. However, the absence of a marker does not equate to the absence of a tumour.
A range of peptide markers can be measured in two laboratories in the UK, and specimens may be sent to these laboratories through local hospitals. Some other NHS laboratories measure CgA.
Some peptides show a significant rise postprandially, particularly insulin, gastrin and PP. These peptides may remain elevated for more than 6 h71; therefore it is ideal that all specimens should be collected after an overnight fast. For some of the markers (eg, CgA), a fasting specimen is not required. If specimens at clinics are not always collected after a fast, the fasting or random status of the specimen should be recorded on the accompanying form to enable appropriate interpretation by the laboratory. With the exception of insulin, peptide markers for NETs are all raised in the circulation of patients with renal failure.72 73 Interpretation of results from these patients is difficult.
A number of circulating markers have been reported to be of prognostic value: CgA for the majority of tumours,74 pancreastatin for hepatic tumour bulk,59 and neurokinin A for serotonin-secreting tumours of the small bowel.64
In a significant number of sporadic NETs, cell type may change and tumours will produce different peptides in addition to the original ones. This indicates worsening prognosis; in particular, ACTH is associated with poor prognosis.75–77 Circulating prognostic indicators are of value in progressive disease and in assessing treatment response since they may be repeated frequently.
Imaging
Imaging is indicated at different stages in the patient's care, including:
Screening of at-risk populations
Primary lesion detection
Assessing extent of disease
Follow-up and assessing response to treatment.
Screening of at-risk populations
Patients with a family history of MEN1 syndromes should be considered for screening according to the established MEN syndrome guidelines.78 In principle, screening of asymptomatic family members should, where possible, be undertaken without exposure to radiation, and thus ideally with MRI.
Primary tumour detection
Precise localisation and measurement of the primary tumour is helpful for surgical planning. It is unclear whether locating and resecting the primary tumour changes prognosis. However, primary tumour resection may reduce the likelihood of local complications, such as bleeding and obstruction.
Gastrointestinal and pulmonary NETs
Endoscopy is the investigation of choice for suspected gastric, duodenal and colorectal NETs. EUS, where available, is a useful adjunct for the assessment of depth of invasion and for biopsy when a mass is detected.79–81 CT and/or bronchoscopy are recommended for the investigation of suspected bronchial NETs.82 SSRS and CT83 provide additional staging information.
CT is the most widely used initial imaging investigation for patients with a suspected gastrointestinal NET involving the small bowel. The sensitivity for detection of the primary tumour is limited (table 6), but mesenteric disease, lymphadenopathy and liver disease are well demonstrated.93 94 The technique of CT enteroclysis combines the cross-sectional display of solid organs with the luminal and mural display of the small bowel and is more sensitive than routine CT in demonstrating an occult primary tumour in patients strongly suspected of having a gastrointestinal NET of the small bowel. Multiplanar reformatting facilitates viewing of the small bowel loops. The reported sensitivity and specificity for detection of small bowel lesions by CT enteroclysis is 85% and 97%, respectively, and midgut NETs are well demonstrated.84 95 Magnetic resonance (MR) enteroclysis is currently under investigation as a radiation-free alternative for small bowel assessment.85 96 Transabdominal ultrasound is no longer generally used for the initial detection of gastrointestinal NETs.
Capsule endoscopy
Small-scale studies have reported the successful detection of occult small bowel NETs using capsule endoscopy where other techniques have failed.96 97 It is advisable to use a dissolvable ‘patency’ capsule to safeguard against the capsule being retained within strictures. A disadvantage of capsule endoscopy is that precise localisation of the tumour within the small bowel is not usually possible.
Pancreatic NETs
Functioning pancreatic NETs (ie, those associated with a hormonal syndrome) may be picked up at an earlier stage than non-functioning tumours. The potential for surgical cure necessitates accurate localisation, ideally using a combination of CT, MRI and EUS, often together with SSRS and, in some centres, DSA with intra-arterial calcium stimulation. Meticulous technique is required to optimise detection rate (online appendix 3).
CT
With the development of multidetector CT (MDCT) and the use of thin reformats, there has been a reported increase in sensitivity for the detection of insulinomas to 94%.86 When the results of MDCT are combined with experienced EUS, a sensitivity of 100% can be achieved.86 Small functioning tumours are usually isodense to pancreas before contrast and enhance strongly after contrast, although the vascular blush is often transient. The best visualisation is usually with arterial phase imaging but portal venous phase imaging is complementary. Large tumours are more likely to be non-functioning than small tumours. Signs of malignancy include large size, necrosis, calcification and invasion/infiltration of surrounding structures.
MRI
Marked improvements in MR technology have occurred in the past decade. The diagnostic performance of MRI has improved and has been shown, in several studies, to exceed or equal that of CT.87 98 99 MRI has a sensitivity of 94% for pancreatic lesions, but this is lower for extrapancreatic lesions.88 98
Pancreatic NETs are typically of high signal intensity on T2-weighted and T2 fat-saturated images and of low signal intensity on T1-weighted and T1 fat-saturated images. Enhancement of a lesion following intravenous contrast administration may render it isointense to the surrounding pancreatic parenchyma.
EUS
EUS is invasive, operator-dependent and is not available at all centres. It greatly improves the sensitivity for the detection of small tumours and multiple pancreatic NETs in MEN1 or VHL syndromes compared with cross-sectional imaging.100 101 The primary aims of EUS are to obtain a tissue sample, and to decide whether the patient should have an enucleation or a Whipple's procedure.
Although the diagnostic performance of EUS is operator-dependent, reports indicate that overall the technique is highly sensitive in experienced hands, with sensitivities as high as 79–100%.101 102 There is a close correlation between aspiration cytology and the final histology after resection, and it has a low complication rate.103–106
Intraoperative ultrasound (IOUS)
This technique has similar advantages to EUS. IOUS may improve the intraoperative sensitivity for identifying small lesions in the pancreatic head and show multiple lesions in up to 92−97% in patients with the MEN1 syndrome; it may be a useful adjunct to palpation of the gland.107 108 IOUS has the advantage over EUS of being able to assess the liver. However, it is not as sensitive as surgical palpation in detecting extrapancreatic lesions.
Intra-arterial calcium with DSA
This technique may be particularly important for localising occult gastrinomas.109 Intra-arterial calcium stimulation combined with hepatic venous sampling has been reported to achieve a success rate of up to 90% in the localisation of insulinomas. Results should be interpreted in combination with those from other imaging modalities.110 111
Scintigraphy (in all gastroenteropancreatic NETs)
Scintigraphy, including PET/CT, may be used to locate the primary tumour in cases where endoscopic or CT findings are inconclusive. These techniques are considered in the next section.
Assessing extent of disease
Many patients with NET present with metastatic disease without a known primary site. Investigations for localising the primary site have been described above. In cases where CT has not identified the primary lesion, and in cases where the primary lesion has been detected but whole-body imaging for the detection of metastatic disease is indicated, whole-body scintigraphy is indicated, using SSRS with single-photon emission CT (SPECT) or PET/CT (using gallium-labelled somatostatin analogues).112 Imaging with SSRS or meta-iodobenzylguanidine (mIBG) will also identify patients with inoperable or metastatic disease who might be candidates for high-activity targeted radiotherapy. Krenning et al113 reported that SSRS before surgery revised the staging and changed management in 33% of patients with NET. In patients with poorly differentiated NET, [18F]fluorodeoxyglucose (18F-FDG) PET/CT may be helpful for staging.
111Indium (111I)-octreotide
The observation that NETs overexpress somatostatin receptors (SSTRs) has led to the development of radiolabelled somatostatin analogues for diagnostic imaging. Of the five recognised receptor subtypes, SSTRs 2 and 5 have been targeted for imaging purposes. SSRS is widely available and well established for the localisation of primary NETs.113–115 The reported sensitivity of SSRS is 61–96%.1 116–124 Sensitivity and specificity are enhanced by topographic image acquisition (SPECT) and by the recent development of SPECT/CT image fusion. In pancreatic NETs, diagnostic performance varies by tumour type. Sensitivities approaching 75% have been reported for gastrinomas, VIPomas, glucagonomas and non-functioning tumours, compared with 50–60% for primary insulinomas.89
Limitations of SSRS include: (1) reduced sensitivity in smaller (sub-centimetre) lesions and in lesions exhibiting low receptor density; (2) 2-day imaging protocol; and (3) potential interference by co-adminstration of therapeutic somatostatin analogues (online appendix 4). The demonstration of somatostatin receptor status by 111In-octreotide or 68Ga-labelled peptide PET/CT imaging positively predicts response to somatostatin analogue therapy.
Meta-iodobenzylguanidine
NETs of pancreatic origin are rarely mIBG avid. In gastrointestinal NETs of the small bowel, 123I-mIBG scintigraphy is less sensitive than 111In scintigraphy.125 126 The main indication for mIBG imaging lies in selecting patients for high activity 131I-mIBG-targeted radionuclide therapy. Between 40% and 85% of NETs accumulate mIBG depending on primary tumour origin.
PET/CT
68Ga somatostatin analogues
Several 68Ga-labelled somatostatin analogues have been developed for diagnostic imaging. DOTA octreotide (DOTATOC) and DOTA octreotate (DOTATATE) bind to SSTR2 and SSTR5, whereas DOTA-NaI-octreotide (DOTANOC) binds to SSTR2, SSTR3 and SSTR5. Rapid tumour accumulation and background clearance facilitate imaging within 100 min of administration.90 In a study of 84 patients, the sensitivity (96%) and specificity (92%) of 68Ga-DOTATOC proved superior to that of CT or SSRS for detection of unknown primary, initial tumour staging or follow-up after therapy. There was an increase in the detection of unrecognised bone metastases, and additional sites of metastatic disease were identified.90 However, CT is required to ensure detection of liver and lung metastases.90 127 A retrospective study of 68Ga-DOTATOC PET in duodenopancreatic NETs reported a sensitivity of 87% and a specificity of 83%.91 68Ga-DOTANOC PET/CT has recently been reported to have led to either staging or treatment change in 50 of 90 patients (56%) assessed prospectively.128 The major practical disadvantage of 68Ga-peptide PET/CT in the UK remains its limited availability.
18F-FDG PET/CT
Although well-differentiated NETs are not typically 18F-FDG avid, 18F-FDG PET/CT is useful in staging primary bronchial and poorly differentiated aggressive NETs.129
Emerging tools
18F-DOPA PET/CT (18F-DOPA)
The sensitivity of 18F-DOPA in metastatic NETs approaches 100%, demonstrating more lesions than SSRS, CT, or SSRS and CT combined. Carbidopa pretreatment is used to reduce artefacts related to physiological activity in the peripancreatic tissues.130
Follow-up and assessing response to treatment
The role of follow-up imaging and optimal imaging frequency depend on clinical circumstances and tumour grade.133–135 The modality of choice should be that which best demonstrated the tumour at diagnosis. Thus, SSRS imaging is recommended for tumours known to be SSTR-positive, supplemented by CT and MRI where necessary. Follow-up for SSTR-negative tumours relies on MDCT or MRI.
The follow-up interval depends on the rate of growth of the tumour. Initially, follow-up imaging may be taken at 3–6-month intervals. If the disease is relatively slow growing, the interval can be increased to 9–12 months. In slow-growing tumours, particularly in younger patients, MRI may be used for follow-up in order to to reduce radiation exposure to the patient. In the context of clinical trials, standardised criteria may be used to assess response, although these remain imperfect for the evaluation of NETs. Functional MRI techniques, including diffusion-weighted MRI and dynamic contrast-enhanced MRI, are currently being evaluated.
Assessment of QoL
Metastatic disease is a common presentation in patients with NETs; therefore, often the aim of treatment is to improve QoL rather than achieve a cure. It is therefore recommended that QoL should be assessed regularly throughout management.
Patients with NETs, in spite of long-term and metastatic disease, often perceive their health-related QoL as good,136 but the treatment options available to these patients are expensive and are not without side effects. The data derived from QoL measurement may be helpful to both patients and clinicians in decisions about treatment options.137 It can be used to help inform economic analyses and resource allocation and to influence healthcare policy.138 QoL data may also be useful in highlighting those areas where we need to develop interventions for the amelioration or prevention of treatment-related problems.139 A specific QoL score questionnaire for patients with NETs is in phase IV validation.140 At present, the best tool to assess health-related QoL in patients with NETs is the European Organization for the Research and Treatment of Cancer's Quality of Life Questionnaire (EORTC QLQ) C-30.141
Pathology
Pathological reporting of NETs
Pathologists dealing with NETs should have a special interest in endocrine or gastrointestinal pathology, or participate in a network with the opportunity for pathology review. Tumours should be classified according to the WHO 2010 classification.142 This classification is fundamentally different from the WHO 2000 classification scheme,143 as it no longer combines stage-related information with the two-tiered system of well and poorly differentiated NETs. The WHO 2010 classification is based on the concept that all NETs have malignant potential, and has therefore abandoned the division into benign and malignant NETs and tumours of uncertain malignant potential. Instead, it organises the classification of NETs according to grade and stage. The classification scheme uses the terms NET (or neuroendocrine neoplasm) and neuroendocrine carcinoma as outlined in table 1 of online appendix 5.
Grading is performed on the basis of morphological criteria and the proliferative activity of the tumour (online appendix 5). Tumour staging is performed according to a system of site-specific criteria. While the WHO 2010 classification recommends using the Union for International Cancer Control (UICC) TNM (7th edition) staging system,144 it acknowledges the existence of the ENETS staging system,145 146 and the fact that grading and staging definitions may need adjustment in the future as the schemes evolve.
The ENETS TNM staging system for gastrointestinal and pancreatic endocrine tumours has been described previously.145 146 The T-staging criteria differ from those proposed by UICC TNM (7th edition)144 for gastric and pancreatic NETs, and in particular, for NETs of the appendix.
The recommendations are therefore:
To grade the tumour according to the WHO 2010 grading system142
To stage the tumour according to the UICC TNM (7th edition)144 site-specific system
To also stage the tumour according to the ENETS site-specific T-staging system when different from the UICC TNM (7th edition)—that is, for NETs of the stomach, pancreas and appendix145 146
To state which TNM classification is used
To document the underlying features that contribute to the T-stage classification (eg, tumour size, extent of invasion, etc) to allow translation between different current and future classification systems.147
In addition, pathologists should refer to the WHO 2000 classification143 for those NETs that, according to this system, fall into the category of benign tumours. This is applicable to NETs that are small and locally confined without distant spread, and relates in particular to a proportion of pancreatic insulinomas, gastric NETs in the context of chronic atrophic gastritis, and incidental appendiceal NETs. The benign behaviour of these NETs is widely accepted and established in clinical practice, and it is therefore deemed clinically useful to report on this additional information at a time of transition between both classification systems.
The prognostic validity of the TNM system as proposed by ENETS has been established,46 148–152 but similar validation studies are still awaited for the recently introduced WHO 2010 and UICC TNM (7th edition) classification and staging schemes.
Specimen handling, and gross and microscopic assessment should be carried out according to a standard protocol that is based on the Royal College of Pathologists dataset for NETs of the gastrointestinal tract and pancreas153 and that follows the ENETS consensus guidelines.154 Further detailed recommendations are given in online appendix 5.
Treatment
Objectives
The aim of treatment should be curative where possible but it is palliative in the majority of cases. Patients often maintain a good QoL for a long period despite having metastases. Although rate of growth and malignancy are variable, the aim should always be to maintain a good QoL for as long as possible. For all patients, the aim is to keep the patient free from disease and symptoms for as long as possible.
Surgery
General approach
Surgery is the only curative treatment for NETs. As with all gastrointestinal tumours, the conduct of surgery with intent to cure depends on the method of presentation and the stage of disease. Specific issues with NETs include determining the extent of local and distant tumours, identification of synchronous non-NETs, recognition of fluid and electrolyte depletion from diarrhoea, and, in advanced cases, detection of less obvious cases of carcinoid syndrome as well as detection of cardiac abnormalities. The treatment plan should be modified accordingly, whether to meet immediate or long-term objectives, within a multidisciplinary framework. With NETs, if the primary lesion is <2 cm in diameter (depending on the site of origin), the incidence of metastasis is low.30 155 However, nodal or liver metastases are found at presentation in 40–70% of patients.30 155–157
Perioperative preparation of patients with functional NETs
When major surgery or hepatic artery embolisation are planned in patients with carcinoid syndrome, prophylactic administration of somatostatin analogues should be considered to prevent a potential carcinoid crisis, even in patients who are receiving long-acting formulations of these agents. Short-acting octreotide is preferably used by constant intravenous infusion at a dose of 50 μg per hour, initiated 12 h before, and given for 24–48 h after, surgical intervention.158–163 It is also important to avoid drugs that release histamine or activate the sympathetic nervous system.164 Despite octreotide therapy, patients may still develop life-threatening cardiorespiratory complications that can tax even the most experienced anaesthetist, who may have to use α- and β-adrenoreceptor blocking drugs to avoid severe complications.165
In addition, short-acting octreotide should always be available, even when a non-syndromic patient with a small bowel NET undergoes an interventional procedure. In cases of unexpected carcinoid crisis, bolus intravenous doses of 100–500 μg octreotide should be given, followed by continuous infusion (see doses above). Antihistamines and corticosteroids may also be beneficial.35
Similar prophylactic measures may be required for pancreatic and periampullary NETs, for example glucose infusion for insulinoma, PPI (oral or infusion) and intravenous octreotide for gastrinoma.
Lung
The treatment of choice is a major lung resection or wedge resection plus node dissection.166 Direct bronchial ultrasound may assist in determining the resection margin.167 Five-year survival after such surgery is between 67% and 96%, depending on the histology of the tumour168–171 and the extent of lymph node involvement.172–178
Emergency abdominal presentations
Patients presenting with suspected appendicitis, intestinal obstruction or other gastrointestinal emergencies are likely to require resections sufficient to correct the immediate problem. Once definitive histopathology is obtained, a further, more radical resection may have to be considered. The most common circumstance is when a NET arising in the appendix 2 cm or more in diameter has been removed. Under these circumstances, a right hemicolectomy is usually indicated, despite the common absence of obvious malignant features characterising the NET.179–181 Goblet cell appendix tumours always require right hemicolectomy because they behave in a much more aggressive way.182–184
Well-differentiated tumours that are <2 cm may require further resection if they:
Breach the serosal surface
Invade the mesoappendix by more than 3 mm
Are located at the base of the appendix.185
There is no definite evidence for the requirement of further surgery if histology shows vascular, neural or lymphatic invasion, but close follow-up is advisable, and further surgery could be discussed with the patient.
Most patients should be followed-up for 10 years.186 Complete resection by appendicectomy of lesions smaller than 1 cm in diameter with no other adverse features is likely to be curative.180 Extended follow-up in this case does not appear to be necessary.
If a perforation has occurred at the site of the tumour, some authorities would recommend right hemicolectomy, although there is no direct evidence relating to this.
Practice point
Although not underpinned by any evidence from prospective studies, the guideline development group holds the view that, particularly in women, there is an increased risk of pelvic peritoneal metastases (especially bilateral ovarian) with goblet cell NETs of the appendix. Such patients should therefore be counselled about this risk and the provision of ‘prophylactic’ bilateral oophorectomy should be discussed.185 187 188
Level of evidence 5, Grade of recommendation C.
A limited emergency small bowel resection for an obstructing NET can be followed at a later date by elective surgery to remove further small bowel or to undertake mesenteric lymphadenectomy. This is particularly appropriate if a second tumour has been identified. A substantial minority of patients with midgut NETs have multiple tumours,189 190 so a search should be made after removal of an obstructing lesion before any further surgery. Furthermore, it is not uncommon to encounter significant desmoplastic reaction in the mesentery (occasionally with varices), which renders resection extremely difficult and dangerous in inexperienced hands. In such cases it is not unreasonable to refer intervention on to a surgeon with greater experience of midgut NETs.
Level of evidence 3–5, Grade of recommendation C.
Stomach
In patients with gastric NETs, the surgical approach depends on the type of tumour, of which there are three types.
Type 1 gastric NETs are associated with hypergastrinaemia and chronic atrophic gastritis. They originate from ECL cells, and can synthesise and store histamine. The frequency of direct invasion into muscularis and metastasis is extremely low, and in most cases only annual endoscopic surveillance is appropriate.191–195 Limited surgery with endoscopic polypectomy and/or antrectomy may be preferable, especially when B12 deficiency anaemia is compounded by iron-deficiency anaemia due to bleeding from the gastric NETs,191 196–198 although achlorhydria also contributes to iron deficiency due to iron malabsorption.
Type 2 gastric NETs occur in patients with hypergastrinaemia due to Zollinger–Ellison syndrome in combination with MEN1 syndrome.199 Small type 1 and 2 tumours with no extension into muscle on EUS or CT can be resected endoscopically, and a combined laparoscopic and endoscopic technique has been used.
Type 3 gastric NETs are sporadic and have a more malignant course.192 200 They are not associated with hypergastrinaemia. These tumours have often metastasised at the time of diagnosis. Most lesions will need resection and clearance of regional lymph nodes and are effectively treated as for gastric adenocarcinoma.191 201–203
Level of evidence 3–5, Grade of recommendation C.
Small intestinal NETs
By far the majority of small intestinal NETs are malignant in nature. Whether liver metastases are present or not, resection of the primary tumour and extensive resection of associated mesenteric lymph nodes is appropriate to cure or to delay progression that would otherwise endanger the small bowel. Occasionally, nodal metastases cause sclerosis with vascular compromise of the associated small bowel, which can lead to pain, malabsorption and even death. Patients who are discovered to have small intestinal NETs only after laparotomy and histological examination may be candidates for further surgery, notably for extensive mesenteric lymphadenectomy. Resection of mesenteric metastases may alleviate symptoms dramatically and, in large series, is associated with prolonged survival. It is recommended that such surgery is undertaken in centres where surgeons have experience of operating on patients with midgut NET disease.
Level of evidence 2, Grade of recommendation B.
As yet there is no clear guidance on the role of resection of asymptomatic primary NETs in the presence of unresectable liver metastases; ideally this question should be answered by a randomised controlled trial. Data from a UKINETS audit204 and other authors205 suggest that there may be a survival benefit for such practice.
Level of evidence 4, Grade of recommendation C.
Colorectum
Standard resection with locoregional lymphadenectomy is appropriate.206 Clearance of metastatic lymph nodes is a worthwhile objective that may contribute to long-term survival, and nodal clearance does not add significantly to the risk of mortality, which should in any case be <2% when conducted by specialist colorectal teams. Small lesions that are <1 cm in diameter and that have a well-differentiated histology (particularly those in the rectum, which tend to have a less aggressive course) may be considered adequately treated by complete endoscopic removal or transanal mucosal resection; however, the patient will require follow-up endoscopy to ensure this has been accomplished. Lesions that are more than 2 cm in diameter should be managed as per adenocarcinoma; there is debate concerning lesions of 1–2 cm, but some of these will invade locally and metastasise.
Level of evidence 3, Grade of recommendation C.
Pancreas
Pancreatic and periampullary NETs form a special group that requires particular consideration.207–212 As with all other neoplasms at these sites, surgery should only be undertaken in specialist hepatopancreatobiliary units. It is recognised that pancreatic resectional surgery is increasingly being performed via laparoscopic access.213–216 For the moment, the decision as to whether this surgery is performed by the traditional open or laparoscopic route should be left to the discretion of each designated specialist pancreatic surgery centre.
According to the WHO classification, tumour size correlates with malignant potential. Localised tumours larger than 2 cm in diameter warrant aggressive resection. Surgical management remains to be proven effective for non-functioning pancreatic NETs in MEN1 syndrome. Often the diagnosis is established biochemically before surgery and, although preoperative localisation can be difficult, the biochemical diagnosis provides some indication of the likelihood of malignancy (eg, low with insulinoma), and localisation can be aided by hepatic venous sampling after calcium stimulation. Thus, for insulinoma, if the lesion is clearly localised before surgery, and is near or at the surface of the pancreas and easily defined at surgery, enucleation may be sufficient, provided histopathology demonstrates complete excision and benign features.217 However, this may not be possible, and Kausch–Whipple pancreatoduodenectomy, left pancreatectomy or even total pancreatectomy may be justified in selected cases.218 These operations are also applied to selected cases with localised disease arising from other functioning, as well as non-functioning, NETs of the pancreas.219
In patients with Zollinger–Ellison syndrome who do not have MEN1 syndrome, surgical exploration should be offered for a possible cure of the disease. There is controversy concerning patients with this syndrome who have MEN1, however, since older data suggest poorer survival in patients treated surgically.220 Nevertheless, the majority of these patients die from malignant spread of their gastrinomas, suggesting that resection is preferable for tumours 2 cm or larger to prevent metastatic spread.221
Level of evidence 3, Grade of recommendation B.
With advances in diagnostic imaging, small non-functioning (<2 cm) pancreatic NETs are increasingly being diagnosed in asymptomatic, otherwise well patients.222 223 While, intuitively, such patients should benefit from surgical resection of such tumours, the management of these patients remains controversial. In the absence of consensus, these patients should be managed by an expert MDT experienced in the management of pancreatic NETs.
Level of evidence 4, Grade of recommendation C.
While there is evidence to support hepatectomy for resectable NET liver metastases,224 the role of resection of asymptomatic primary pancreatic NETs in the presence of unresectable liver metastases remains controversial.225 In the absence of adequate evidence, such decisions should be made by an expert MDT experienced in the management of pancreatic NETs.
Level of evidence 4, Grade of recommendation C.
Appendix
The appendix is a common primary site, with tumours often found incidentally at appendicectomy. Some 75% of these NETs are located near the tip. Classical NETs <1 cm in size can usually be managed by appendicectomy, while for tumours >2 cm in size or for goblet cell tumours of any size, right hemicolectomy is the treatment of choice.184 226–228 For tumours 1–2 cm in size, any of the following mandates right hemicolectomy: serosal breach by tumour, cellular atypia, invasion of mesoappendix by more than 3 mm, or involvement of the base of the appendix.229 230 Some centres recommend right hemicolectomy for perforation of the appendix in the presence of a NET and for lymphovascular and perineural invasion, but no definite evidence exists. Long-term (10-year) follow-up is required for any high-risk features.
Level of evidence 3–4, Grade of recommendation C.
Liver
In the presence of liver metastases ‘curative’ liver resection is possible in about 10% of cases,224 if the lesion(s) is confined to one lobe. With bilobar metastases and one very dominant lesion causing symptoms, a debulking operation may be carried out for palliation, particularly if there is resistance to medical therapy. The 5-year survival after resection of the primary and/or liver secondary is up to 87%, and postoperative mortality is 6%.161–163 231–236 Several series have shown low morbidity and excellent medium-term survival after liver resection, with better outcomes than for patients whose tumours are not resected (although this may partly reflect stage of disease).231 237 238 A very small number of patients with no obvious primary tumour may have primary hepatic neuroendocrine malignancy, and surgery can be curative239; for such patients, surgery is the treatment of choice, with a recurrence rate of 18% and 5-year survival of 74% reported in one series.240 Many patients will need somatostatin analogues, which predispose patients to gallstones, hence the gallbladder is usually removed at the time of liver surgery.
Level of evidence 3, Grade of recommendation B.
Orthotopic liver transplantation (OLT)
Patients with end-stage NET and uncontrollable symptoms that are unresponsive to any other therapy have been considered for liver transplantation.241–248 The highest disease-free survival reported to date at 1 year is 77%.247
An analysis of UK transplants for NETs249 reported actuarial disease-free survival of 62% at 1 year and 23% at 5 years, with similar data in a series from France.250 The UK and French series both included patients from many years ago, when survival rates would be expected to be lower, and many patients in these series predated modern imaging techniques. More recent data from the French group251 252 show 5-year survival at 47%, which is close to the acceptable limit for transplantation in the UK.
OLT is currently outside the routine remit of UK Transplant Agency guidelines in view of the disease-free survival of <50% at 5 years. However, survival has progressively increased245–247 such that in the future, in selected patients and using new drug treatments, OLT may be a management option. At present, only exceptional cases that cannot be treated by other means should be considered, and this should preferably be part of a national trial so that data are standardised and examined regularly. OLT should be avoided in patients who have undergone multiple organ transplants, and those who have pancreatic primary sites and high Ki-67-expressing tumours.
At present, organ shortage combined with the low survival data suggest that liver transplantation should only be considered in exceptional circumstances. Further research is needed to try to assess pretransplant prognostic factors.
Symptomatic treatment
Symptoms associated with NET hormonal hypersecretion may impair patients' QoL and in some instances can be life-threatening (eg, severe diarrhoea and hypokalaemia in VIPomas). Administration of specific medications to treat symptoms should therefore start as soon as clinical and biochemical signs indicate the presence of hypersecretory NETs, even before the precise localisation of primary and metastatic lesions is confirmed. Treatments include somatostatin analogues, and the use of PPIs for gastrinomas and diazoxide for insulinomas. Additional medications such as loperamide, cholestyramine and corticosteroids may be used as required.
Somatostatin analogues
The only proven hormonal management of NETs is by the administration of somatostatin analogues. SSTRs are present in the vast majority (75–95%) of NETs, but are identified in only 50–60% of insulinomas, and are less evident in poorly differentiated NETs and somatostatinomas. Since natural somatostatin has a very short half-life (2–3 min), analogues with longer half-lives have been developed for clinical use. Somatostatin analogues bind principally to SSTR subtypes 2 (with high affinity) and 5 (with lower affinity),253 thus inhibiting the release of various peptide hormones in the gut, pancreas and pituitary; they also antagonise growth factor effects on tumour cells, and, at very high dosage, may induce apoptosis. The effects of somatostatin analogues are demonstrable as biochemical response rates (inhibition of hormone production) in 30–70% of patients and as symptomatic control in the majority of patients.
There are two commercially available somatostatin analogues: octreotide and lanreotide. The immediate-release form of octreotide (half-life of 1.5–2 h) has to be administered subcutaneously two to three times per day or by continuous intravenous infusion. Longer-acting (slow-release and depot) formulations of somatostatin analogues include octreotide long-acting release (LAR), lanreotide Autogel and lanreotide LA.254 The long-acting agents have produced significant improvement in the QoL of patients with NET and have comparable or better efficacy than short-acting octreotide.255–258 Long-acting formulations of somatostatin analogues should therefore now be considered the standard of care for symptomatic treatment of NETs. Short-acting formulations may still have a role in patient stabilisation (used over short periods) and in the management of carcinoid crisis.
The licensed dosage of octreotide LAR is 10, 20 or 30 mg every 4 weeks, and for lanreotide Autogel the recommended dose is 60, 90 or 120 mg every 4 weeks. Patients should generally be started on lower doses with treatment up-titrated to achieve stabilisation. In cases of breakthrough symptoms, rescue doses of subcutaneous octreotide can be used two or three times per day up to a maximum daily dose of around 1 mg. Alternatively, if the breakthrough symptoms occur mainly during the week before the next long-acting injection, a reduction of administration intervals from 4 to 3 weeks may be considered.259
Role in prevention of carcinoid crisis
See under ‘Perioperative preparation of patients with functional NETs’ for an overview of the use of somatostatin analogues in the prevention of carcinoid crisis.
Nuclear medicine imaging and treatment with radiolabelled somatostatin analogues during treatment with somatostatin analogues
In theory, co-administration of somatostatin analogues may reduce the sensitivity of SSRS imaging. Where possible, most UK centres recommend withdrawal of short-acting somatostatin analogues for 24–48 h before111In-octreotide or 68Ga-peptide injection until imaging is completed.
In patients receiving long-acting analogues, SSRS or 68Ga PET/CT, studies should ideally be scheduled towards the end of the dosing interval and just before the next planned injection.
ENETs guidelines suggest withdrawing long-acting somatostatin analogues for 6 weeks before planned radiopeptide treatment, and substituting short-acting somatostatin analogues for symptom control until 24 h before treatment.260 In the absence of evidence from randomised controlled trials, most UK centres avoid the potential morbidity of this approach by adhering to the simpler regimen described above for diagnostic imaging.
Somatostatin analogue treatment does not interfere with mIBG or 18F-FDG PET/CT imaging.
Adverse effects
The most common adverse effects of somatostatin analogues are usually mild and resolve with time.8 261 262 They include local reactions (pain and erythema) at the injection site, abdominal cramps, nausea, flatulence, diarrhoea and steatorrhoea. There is also a risk of cholelithiasis (10–50%),263 264 which may be asymptomatic; in such cases, cholecystectomy is not necessary. However, prophylactic cholecystectomy during abdominal surgery is recommended in patients who are already receiving, or are due to start, long-term treatment with somatostatin analogues.265 Rare adverse effects include bradycardia, abnormal metabolism of glucose, malabsorption of vitamins A, B12 and D, and alopecia.266
Efficacy and indications in various syndromes
Functioning small bowel NETs (carcinoid syndrome)
Somatostatin analogues are effective in the management of the symptoms of carcinoid syndrome. Most studies report improvements in diarrhoea and flushing in 60–70% and 70–80% of patients, respectively, and a significant reduction (>50%) in biochemical markers (especially 5-HIAA) in 40–60% of patients.123 256 267
VIPoma (watery diarrhoea, hypokalaemia, achlorhydria (WDHA) syndrome, Werner–Morrison syndrome)
Rehydration and electrolyte replacement are always indicated and may improve the clinical condition considerably. In patients with this rare life-threatening syndrome, the administration of somatostatin analogues results in significant improvement of symptoms in 80–90% of patients and in reduction of vasoactive intestinal peptide levels in 60–80%.268 269 However, biochemical improvement does not always correlate with symptomatic improvement, and thus the drug dose should be titrated mainly against symptoms.
Glucagonomas
Somatostatin analogues have been reported to result in improvements in patients with the glucagonoma syndrome (migratory necrolytic erythema rash, diabetes, weight loss, etc). The characteristic rash of necrolytic migratory erythema can be life-threatening. After the initiation of somatostatin analogue treatment, 80–90% of patients with glucagonomas show a significant improvement in migratory necrolytic erythema rash. Treatment is less effective in terms of controlling diabetes and weight loss, which are common symptoms in these patients. Treatment with somatostatin analogues may result in a reduction in circulating glucagon levels in about 60% of patients, although in many cases these levels are very high, so normalisation is unlikely.258 270
Gastrinomas
PPIs are currently the treatment of choice for the control of gastric acid hypersecretion-associated symptoms (see under Proton pump inhibitors). Somatostatin analogues are not considered as first choice agents by the majority of clinicians and should only be used in refractory cases.
Insulinomas
Somatostatin analogues are often not effective in the control of hypoglycaemia in patients without SSTR 2-positive tumours (50–60% of insulinomas), and their use may lead to variable effects on blood glucose levels, possibly because of inhibition of glucagon secretion.271 272 Diazoxide has been shown to be effective in controlling hypoglycaemic symptoms in many patients with insulinoma (see under Other medications).273
Other clinical syndromes
Somatostatin analogues have been reported to improve parathyroid hormone related peptide (PTHrP)-related hypercalcaemia in rare PTHrP-secreting pancreatic NETs.274 They may also be beneficial in NETs patients with paraneoplastic Cushing's syndrome or acromegaly, associated with ectopic secretion of ACTH or growth hormone-releasing hormone (GHRH), respectively.275 276
Non-functioning NETs
The role of somatostatin analogues in patients with non-functioning NETs is unclear and the routine use of these agents in such NETs can only be recommended once further evidence is available.
Antiproliferative potential of somatostatin analogues
It has been postulated that somatostatin analogues may have an antiproliferative effect. Several studies suggest that, in pancreatic NETs, partial/complete tumour response is achieved in fewer than 10% of patients, although stabilisation of radiologically documented tumour progression has been demonstrated in 24–57% of patients.277–284
Similar results have been demonstrated for small bowel NETs.256 257 267 277–283 In addition, recent data from one double-blind, placebo-controlled study (PROMID; Placebo-controlled, double-blind, prospective, Randomised study on the effect of Octreotide LAR in the control of tumour growth in patients with metastatic neuroendocrine MIDgut tumours) demonstrated prolonged progression-free survival (PFS) in patients with metastatic NETs of midgut origin receiving octreotide LAR with a median time to progression of 14.3 months compared with 6 months in the placebo group. In this study, the best results were demonstrated in patients with low hepatic tumour load and a resected primary lesion.285
Finally, there is an ongoing study assessing the antiproliferative effects of lanreotide Autogel in patients with non-functioning intestinal and pancreatic NETs.
While the evidence is thus not wholly conclusive, and further studies in a wider range of tumours and using different doses will be valuable, at present there is a rationale for the use of somatostatin analogues in attempting to moderate the tumoural progression of NETs.
New somatostatin analogues
New somatostatin analogues are in development, with the agent SOM-230 (pasireotide) in phase II at the time of writing. These studies may extend the use of somatostatin analogues for symptomatic therapy, and may offer treatment for patients resistant to conventional octreotide and lanreotide.
Proton pump inhibitors
PPIs are currently the treatment of choice for the control of gastric acid hypersecretion-associated symptoms and prevention of peptic complications in gastrinoma patients. Higher doses of PPIs may be required in patients with complicated disease.
Other medications
Diazoxide is used as short-term treatment in patients with insulinoma scheduled for surgery, or as a long-term therapy in patients with unresectable disease. Diazoxide is a hypertensive medication with hyperglycaemic effects, and is usually effective in controlling hypoglycaemia symptoms in patients with insulinoma. The recommended daily dose is 200–600 mg orally. Adverse effects, including oedema, weight gain, hirsutism and renal dysfunction, are common but are not usually troublesome.273 There are two small case series of patients with malignant insulinoma refractory to conventional agents that report significant improvement in glycaemic control with the novel mammalian target of rapamycin (mTOR) inhibitor, everolimus,286 287 and also a case report of a similar patient showing control of hypoglycaemia with oral rapamycin.287
In patients with small bowel NETs, loperamide and ondansetron can be used for the management of secretory diarrhoea, cholestyramine for bile salt malabsorption-related diarrhoea, and oral pancreatic supplements for steatorrhoea following treatment with somatostatin analogues. Oral antibiotics may be used for control of small bowel bacterial overgrowth in patients with extensive mesenteric fibrosis. Patients may also benefit from oral vitamin B compounds to prevent clinical features of vitamin B deficiency.
In some patients with insulinoma, verapamil and phenytoin can be used as alternatives to diazoxide. Corticosteroids can be used in patients with insulinoma with refractory hypoglycaemia and in those with VIPoma with life-threatening diarrhoea that does not respond to maximum doses of somatostatin analogues.288
In patients with glucagonoma, zinc salts can be used to prevent further skin lesions.
Appropriate prophylaxis should be considered in all patients with NETs with increased risk of thromboembolic episodes (eg, those with glucagonoma).
Bisphosphonates may be used for symptom control in patients with bone metastases, although they are not specifically indicated for NETs.289
Interferon α
Interferons are immune modulators necessary to combat viral infections. Interferon α acts via specific cell-surface receptors to activate downstream cytoplasmic kinases. In addition, it induces arrest in the G1 and G0 phases of the cell cycle, inhibits production of growth factors, induces class 1 antigens, and has antiangiogenic properties.290 It is used for the treatment of both functional and other NETs, either on its own or added to long-acting somatostatin analogues if the patient is not responding to maximum dosage of somatostatin analogues. Interferon α 3–5 mega units three times per week subcutaneously is the usual dose used. However, there is conflicting evidence as to its efficacy, with only one major group supporting its widespread use. However, there is some evidence that it may have a greater effect in tumours with low mitotic rate.266 Biochemical response has been demonstrated in 40–60% of patients, while symptomatic improvement and significant tumour shrinkage have been reported in 40–70% and a median of 10–15% of patients, respectively.291–293
Somatostatin analogues have been added to interferon α with the aim of enhancing its antitumour effect,294 but studies thus far have failed to demonstrate this. An increase in 5-year survival from 37% with interferon α alone to 57% in combination with octreotide was seen in one study (HR 0.62 (95% CI 0.3 to 1.1); p=0.132), although the results were not statistically significant because of small patient numbers (n=68).295 Two other small randomised studies have shown this combination to increase toxicity without any additional survival benefit (although both studies were underpowered to do so).296 297 Although sometimes useful for control of refractory hormonal symptoms, the combination of interferon α and somatostatin analogues for the prolongation of survival should still be considered investigational.
Emerging evidence suggests that pegylated interferon is better tolerated and also demonstrates some activity298 299; however, current data do not support its use outside of clinical trials.
Chemotherapy
There are only four published randomised trials of chemotherapy for NETs, and none have compared chemotherapy with best supportive care.300 301 There are numerous small, uncontrolled, retrospective studies in heterogeneous or poorly characterised patient populations. Many older studies report response rates in terms of clinical, radiological and biochemical response rather than according to WHO or RECIST criteria.302 303
For pancreatic NETs the seminal randomised trials performed by Moertel et al304 305 established streptozotocin (STZ)-based regimens as the standard of care. The first trial demonstrated a superior, but non-significant, survival benefit for patients treated with the combination of STZ and 5-fluorouracil (5-FU) compared with STZ alone,304 while the second showed better survival for the combination of STZ and doxorubicin (DOX; 26 months) over single-agent chlorozotocin (18 months) or the 5-FU/STZ combination (17 months).305 In these trials, the best response rates were 63% and 69%, respectively, but these rates were based on clinical and biochemical responses as well as radiological response.
A more realistic response rate is provided by three recent single-arm studies, which applied WHO or RECIST criteria in patient cohorts of at least 45 patients. Using STZ/DOX,306 STZ/5-FU/DOX307 or STZ/5-FU/cisplatin (FCiSt),308 response rates were remarkably consistent at 36%, 39% and 38%, and were associated with median overall survival of 24, 37 and 32 months, respectively. The results of the recently completed randomised phase II NET01 trial comparing STZ/capecitabine with or without cisplatin (FCiSt; http://Clinicaltrials.gov identifier NCT00602082) are awaited.
Temozolomide (TMZ), like STZ, is an alkylating agent but is orally bioavailable, and has been reported to have a 70% response rate in a recent small (n=30) single-arm study when used in combination with capecitabine.309 Previous results using TMZ as a single agent or in combination with thalidomide or bevacizumab have been less impressive,310 311 and randomised comparisons with STZ regimens are required. Of interest, response to TMZ may be related to expression of the DNA repair enzyme methylguanine DNA methyltransferase (MGMT), and two small retrospective studies have shown increased responses to TMZ by MGMT-deficient tumours.311 312 The value of MGMT expression as a potential predictive marker of response of NETs to alkylating agents needs to be investigated further before changing clinical practice.
For non-pancreatic NETs, there are two randomised trials. The first compared 5-FU/STZ versus DOX in 172 patients and, using clinical, biochemical and radiological criteria, reported respective response rates of 22% and 21%, with no significant difference in survival.301 The second trial compared 5-FU/DOX versus 5-FU/STZ in 176 patients and reported a response rate (WHO criteria) of 16% in both arms, but a superior survival of 24 months for 5-FU/STZ compared with 16 months for 5-FU/DOX (p=0.027).300 Small, single-arm, single-agent studies exploring newer cytotoxic agents including the taxanes,313 gemcitabine,314 pemetrexed315 and topotecan316 have not been encouraging, with response rates of <10%.
Tumour grade (WHO criteria based on Ki-67 and mitotic index) and differentiation status also influence response to chemotherapy. A recent analysis of 82 patients treated with FCiSt demonstrated responses ranging from 14% for low-grade tumours to 33% for intermediate-grade tumours, and 60% for high-grade tumours (p=0.025). Both Ki-67 (p=0.019) and mitotic index (p=0.008) correlated independently with response.308 For Ki-67, a minimum cut-off of 10% was associated with a response rate of over 38%, but further studies are required to define the optimum value to direct therapy. For poorly differentiated carcinomas, response rates of 42–67%, lasting a median of 8–9 months, are reported in historical studies using cisplatin and etoposide.317–319 Increasing the intensity of treatment by adding paclitaxel to carboplatin and etoposide, although feasible, had no obvious advantage over doublet therapy.320
Given the paucity of adequately powered definitive phase III studies incorporating modern response assessments and up-to-date histological subtyping aimed at reducing patient heterogeneity, patients and clinicians should be encouraged to participate in well-designed, prospective clinical trials of chemotherapy or novel targeted therapies. See under Emerging therapies below for a discussion of newer agents.
At the time of writing, the use of chemotherapy should be based on the following principles:
Where appropriate, patients should be invited to participate in clinical trials (eg, chemotherapy or novel targeted therapy trials—see under Emerging therapies below).
For poorly differentiated NETs, a platinum-based regimen should be considered.
Level of evidence 2, Grade of recommendation B.
A STZ-based combination should be considered for moderately and well-differentiated tumours, particularly when the following apply:
The tumour is pancreatic in origin.
The tumour is of intermediate or high grade, based on mitotic index or Ki-67.
There is rapid clinical or radiological progression.
Level of evidence 1, Grade of recommendation A.
Although there are limited data in terms of efficacy, systemic chemotherapy could also be considered in patients with recurrent and/or metastatic goblet cell appendiceal NETs. The optimal regimen needs to be defined; however, colorectal cancer regimens have been used in view of the aggressive nature of this tumour.
Ablation therapies
Radiofrequency ablation: image-guided ablation
Radiofrequency ablation has been used with some effect in stabilising or reducing tumour size, but randomised trials are lacking.321 It may be indicated in patients with inoperable bilobar metastases.322 Ablation therapies are rapidly evolving. Treatment by ablation can be performed percutaneously or laparoscopically. Most devices are now able to deliver 3–5 cm spherical ablation zones, and large expandable probes and multipolar devices can yield larger treatments. The percutaneous approach is the most commonly used, as it is least invasive, cheapest and has the additional benefit of CT or MRI guidance. The laparoscopic approach has the benefit of intraoperative ultrasound scanning, which is ideal for the detection of tiny tumours, but it does require considerable skill.323
Ablation can be used to reduce hormone secretion and/or to reduce tumour burden. Most patients with neuroendocrine metastases have a large number of small metastases that are hormonally active. Parallels should not be drawn with the ablation experience seen in metastatic colorectal disease. The Cleveland Clinic group assessed ablation outcomes following laparoscopic radiofrequency ablation, noting a 34% local recurrence rate for colorectal metastases, but only 6% recurrence for neuroendocrine metastases.324
The Essen group has usefully stratified metastatic NETs into three groups: (1) solitary or paucilesional disease; (2) isolated metastatic bulk with smaller deposits (often contralobar); and (3) disseminated metastatic spread.325 Ablation is usually most appropriate in groups 1 or 2 alongside resection or as an adjunct to somatostatin analogue radionuclide therapy. As with surgical resection or embolisation in the setting of a functioning NET (usually metastatic midgut NETs), the procedure should be carried out in conjunction with a prophylactic continuous octreotide infusion (see under Surgery above).
The main limitation for radiofrequency ablation is the size and number of tumours. Neuroendocrine metastases are small, numerous and very slow growing. Therefore, it is possible to treat patients with indolent disease, with as many as 20 small (<3 cm) tumours, at multiple treatment sessions over a period of years. Complete ablation is, however, still limited to small volume (<4 cm) tumours.
Destroying the largest lesion may not necessarily switch off hormone production. To achieve a reduction in hormone secretion, it is usually necessary to ablate at least 90% of the visible tumour.326–331 Tumour location is not as important as for liver resection. Recent case series suggest that after laparoscopic radiofrequency ablation for NET liver metastases, local recurrence is around 6% and symptomatic relief is achieved in 70–95% of patients, with a 5-year survival of 57% and a median survival of 3.9 years after the first radiofrequency ablation.324 332 Patients who have biliary–enteric anastomoses after pancreatic surgery are at significant risk of secondary infection in the ablated area of the liver and require 3 months of rotating oral antibiotics after the procedure. All cases considered for cytoreductive ablation should be discussed at a specialist hepatobiliary MDT meeting.
Targeted radionuclide therapy
Systemic targeted radionuclide therapy
This is a useful approach for patients with inoperable or symptomatic NETs and has become a standard of care for these patients in the UK and Europe. Significant responses have been observed in patients who would otherwise not be treatable.
Indications for treatment include symptom palliation after maximal medical therapy and tumour progression. Patient selection criteria include demonstration of superior radiopharmaceutical uptake at all known tumour sites on diagnostic imaging by comparison with normal tissues, reasonable bone marrow reserve, and adequate renal function. Contraindications include pregnancy and breast feeding. Patients should be continent and self-caring in order to minimise risk to nursing staff. Several different therapeutic radiopharmaceuticals are available. No randomised controlled trials have been performed.
131I-mIBG
Up to 80% of functioning malignant NETs concentrate mIBG,333 although the intensity of tumour uptake by comparison with normal tissues is often too low to justify high-activity radionuclide therapy. Treatment protocols vary between different centres. 131I-mIBG therapy is administered in a dedicated, shielded isolation facility for radiation protection reasons. Thyroidal uptake of free radioiodide is prevented by potassium iodide/iodate blockade.
The usual prescribed 131I-mIBG activities in the UK range between 7.4 and 11.2 GBq administered at 3–6-month intervals. mIBG therapy is the only licensed radionuclide therapy for NETs. Treatment is well tolerated, and toxicity limited to temporary myelosuppression 4–6 weeks after therapy. Myelotoxicity is more severe in patients who have bone marrow infiltration by tumour at the time of treatment or who have undergone previous chemotherapy or radionuclide therapy. Myelosuppression is cumulative and may be dose-limiting after repeated treatment cycles.
Response rates of 40–60% have been reported after repeated cycles of 131I-mIBG therapy.334–336 Treatment offers symptom palliation, improved QoL and reduced requirements for somatostatin analogue therapy. Partial objective responses (by the WHO criteria) of 10–15% have been reported,335 but complete radiological response is rare. Survival benefit appears to be related to symptom response and initial administered activity, with a reported actuarial survival improvement of 22% at 5 years.335 Hormone response is not associated with survival gain.
Radiolabelled peptide therapy
Experience using different radiolabelled peptides has been published but no randomised controlled comparison between individual radiopharmaceuticals has been undertaken. The radiolabels of choice are Yttrium-90 (90Y) and Lutetium-177 (177Lu). Available peptides include DOTATOC, DOTATATE, DOTA-lanreotide and DOTANOC, which exhibit different affinities for individual SSTR populations. Optimal peptide selection in an individual patient is determined by diagnostic tracer imaging. The main toxicities of radiopeptide treatment are temporary myelosuppression and radiation nephritis. Nausea and vomiting during and immediately after treatment are partly attributed to co-administration of amino acids for renal protection (see below) and are mitigated by prophylactic antiemetics. Pain due to temporary radiation oedema may occur in patients with bulky tumours and is managed by corticosteroids and analgesics.
90Y-DOTATOC and 90YDOTATATE
Activities in the range 3–6 GBq administered at 6–8-week intervals, to a cumulative activity of 12–18 GBq, are recommended. Most patients report subjective benefit within two treatment cycles, often in association with a reduction in biochemical tumour markers.
A phase II single-arm study337 of 90 patients with refractory NETs treated with three cycles of 90Y-edotreotide reported a response rate of 74.4% (objective response + stable disease), and prolonged overall survival compared with historical controls (26.9 vs 12 months).338 Objective response (>50% partial and complete response) in the range 9–33% by the WHO/RECIST criteria has been reported with 90Y-DOTATOC,339 340 with a median time to progression of 29 months and overall survival of 36.7 months from treatment. A study of 90Y-DOTATATE in patients with progressive disease reported partial responses in 23% of patients, with the remaining 77% having stable disease.341
Toxicity is actively related and includes reversible myelosuppression, which is maximal at 4–6 weeks after treatment, and nephrotoxicity.340 Co-administration of amino acids, particularly d-lysine, reduces tubular peptide binding and is essential to minimise renal toxicity. Patients with pre-existing microangiopathy due to hypertension or diabetes are at increased risk of cumulative nephrotoxicity, which may lead to a decrease in creatine clearance of 7.9% per annum.340
177Lu-DOTATATE
Responses to cumulative activities of 22–29.6 GBq administered in 3.7–7.4 GBq fractions at 6–10-week intervals have been reported: rates of 28% partial response and 54% minor response/stable disease were recorded in a population of patients with refractory, progressive disease at the time of treatment.342 Predictive factors for favourable objective response included high selective tumour uptake on pretreatment imaging and limited hepatic metastatic tumour burden.339 Haematological toxicity is similar to that reported for 90Y-DOTATOC,341 although the risk of nephrotoxicity in a small, unrandomised study was reportedly lower, with a decrease in creatinine clearance of 3.8% per annum. Symptom palliation and QoL improvement as assessed by EORTC QLQ-30 have been demonstrated, although not in a randomised trial.342
Future developments
The therapeutic potential of peptides radiolabelled with α particle emitters343 and combination treatment with radiosensitisers344 is being investigated in preclinical studies. On the basis of emitted β particle range, long-range 90Y may be more suitable for treating bulky metastases, whereas 177Lu would be preferable for small volume disease. Combined 90Y and 177Lu radiolabelled therapy is under consideration. Access to some form of radionuclide therapy should be made available to centres treating patients with NET.
Transhepatic artery embolisation/chemoembolisation
Particle embolisation (polyvinyl alcohol and gel foam powder) of the hepatic artery in patients with liver metastatic NETs has been shown to reduce tumour size and hormone output. It is primarily used for palliation of symptoms.345–348 Chemoembolisation is the regional delivery of chemotherapy (DOX, STZ or cisplatin) in combination with hepatic artery embolisation. The benefits of chemoembolisation have been documented by several investigators,349–351 but there are no completed comparative studies between the two modalities.
There have been several studies since the last guidelines2 looking at outcomes from embolisation and chemoembolisation.204 352–358 The predominant benefit of the procedure is palliation of symptoms, with 70–90% of patients achieving benefit. There may be an additional benefit of radiographic regression of metastasis with a possible improvement in survival, although this has not been demonstrated in a systematic study. Five-year post-embolisation survival rates of 28–44% for NET liver metastases and 18–35% for islet cell metastases have been documented. PFS of 8–22 months and 16 months for NETs and islet cell liver metastasis, respectively, has also been reported. Serious adverse events (sepsis, hepatorenal syndrome and necrotising cholecystitis) have been reported in 7.5–23.8% of patients. Post-embolisation syndrome (fever, abdominal pain and nausea) is common and usually lasts for 24–72 h.
The 30-day mortality from published data is 1.9–9.3%. However, recently the technique of selective hepatic embolisation has developed, whereby the left or right hepatic arterial branches arising from the main hepatic artery are embolised separately. This is thought to reduce the risk of morbidity and mortality. Patient selection is the key to reduce major complications. Patients with >75% liver involvement with or without liver insufficiency, portal vein thrombosis and significant carcinoid heart disease are at increased risk of mortality from the procedure. Other considerations include the presence of systemic infections, significant renal impairment and biliary reconstructive surgery/obstruction. We recommend a multistage technique, aggressive hydration and octreotide infusion (50 μg/h), starting 12 h before the procedure and continued until 48 h after. Intravenous hydrocortisone, prophylactic antibiotics and allopurinol for tumour lysis syndrome may also be considered.
On the basis of published data, there seems no significant advantage of chemoembolisation over embolisation. However, given the better response of islet cell cancers to chemotherapy, these tumours may have a better response to chemoembolisation.
Selective internal radiation microsphere therapy
Embolisation of the hepatic artery with 90Y microspheres for unresectable neuroendocrine liver metastases was first described in 1968.359 360 Three recent studies have investigated the benefits of 90Y radioembolisation.360–362 Early data indicate that it is safe, with no observed deaths within 30 days of treatment in 148 patients. Post-embolisation syndrome was common, and there was a small risk of radiation gastritis and ulceration. Embolisation of the gastroduodenal artery before the procedure may be considered to avoid this. The symptomatic response rate was 55% at 3 months and 50% at 6 months; 2.7% of procedures resulted in complete response, with 60% showing a partial response. Median survival was 70 months.361 Further long-term studies are needed to clarify the role of 90Y microsphere embolisation in the management of patients with unresectable NET liver metastases.
External-beam radiotherapy
NETs have often been regarded as being radioresistant. However, external-beam radiotherapy may provide excellent relief of the pain from bone secondaries, and there has been a suggestion that some secondary deposits in the liver and elsewhere shrink in response to radiotherapy. It is also useful for the occasional patients with brain metastases.363
Emerging therapies
Current systemic anticancer agents (conventional cytotoxics) have limited efficacy in metastatic NETs. Although results are somewhat better for poorly differentiated carcinomas and pancreatic primaries than for other sites, response rates are low and the impact on survival is small. The greatest unmet need in the management of NETs is for new agents and approaches to therapy to improve outcomes. An increasing understanding of the biology of these tumours, together with the ability to synthesise drugs that can interfere with relevant targets, provides an opportunity for the discovery of new therapies.
A major recent development has been the emergence of two new treatment options (sunitinib and everolimus) for patients with advanced (inoperable locally advanced or metastatic), progressive (defined as evidence of radiological disease progression within the previous 12 months by RECIST), well-differentiated pancreatic NETs.364 365
The first of these, sunitinib malate (Sutent; Pfizer), is an oral inhibitor of the tyrosine kinases downstream from key drivers of tumour angiogenesis, including vascular endothelial growth factor (VEGF) types 2 and 3, platelet-derived growth factor and stem-cell factor (c-kit). Pancreatic NETs are highly vascular tumours that have increased VEGF expression; xenograft models treated with VEGF antibodies show significant growth inhibition and reduction of liver metastases. High VEGF expression in tumour biopsy samples correlates with metastatic potential and poorer survival.
On the basis of encouraging results from a phase II study,366 a phase III double-blind study of sunitinib 37.5 mg per day (continuous daily dosing) or placebo was conducted in patients with well-differentiated, progressive pancreatic NETs. The study was closed prematurely (after enrolment of 171 of the planned 340 patients) by the Independent Safety Monitoring Committee following an observed increase in the number of adverse events and deaths in patients receiving placebo. The primary end point (PFS) was statistically superior for patients receiving sunitinib compared with placebo (median 11.4 months vs 5.5 months; p=0.0001), with a HR of 0.42 (95% CI 0.26 to 0.66; p<0.001). The objective response rate was 9.3% for sunitinib, with an additional 63% of patients having stable disease (radiologically, by RECIST); in comparison, 60% of patients receiving placebo had stable disease and none had a response (p=0.007). Toxicities were as expected from the use of sunitinib in other tumour types: the most common major (grade 3–4) toxicities included neutropenia (12% of patients) and hypertension (10% of patients). Other notable toxicities (usually grade 1–2) included diarrhoea, nausea, fatigue, vomiting, hand/foot skin reaction and, rarely, thyroid dysfunction.364
The mTOR is particularly interesting as a potential therapeutic target because genetic abnormalities in the mTOR pathway may be critical to the development of some NETs. A phase II study of the mTOR inhibitor RAD001 (everolimus (Afinitor); Novartis Pharmaceuticals) in 160 heavily pretreated patients with pancreatic NETs revealed an overall response rate of 9.6%, with a further 67.8% of patients experiencing stable disease.367 The subsequent phase III study has confirmed this activity in patients with well or moderately differentiated, progressive pancreatic NETs. Four hundred and ten patients were randomised to everolimus 10 mg once daily (continuous dosing) or placebo. Once again, the study demonstrated a significantly better PFS (the primary end point) of 11.0 months for everolimus and 4.6 months for placebo (HR 0.35 (95% CI 0.27 to 0.45); p<0.001). Treatment was well tolerated, with most toxicities of grade 1–2 severity (stomatitis, rash, diarrhoea, fatigue and infections). More severe toxicities (grade 3–4) included anaemia (6% of patients) and hyperglycaemia (5% of patients). Stable disease (by RECIST) was evident in 73% of patients receiving everolimus, compared with 51% in the placebo group.365
The emergence of sunitinib and everolimus as new treatment options for patients with advanced pancreatic NETs raises a number of additional questions, including the sequencing with other treatment options, activity in higher grade (ie, poorly differentiated) disease, combination with other therapy modalities, activity in non-pancreatic NETs, and use in the different stages of the disease (eg, in the adjuvant setting). Recommendations cannot be made for any of these with the data currently available and these questions remain a focus for ongoing and future research.
Similarly, the effects of targeting other cell signalling pathways (eg, insulin-like growth factor and its receptors, and the epidermal growth factor receptor among others) are subject to ongoing studies. There are many advantages to exploring the potential value of new agents in patients with NETs. Patients are generally fit, despite having extensive disease, and usually have good organ function preservation. Compared with other disseminated malignancies, patients with NETs have a relatively long median survival, which gives new agents a chance to demonstrate their effect. It is clearly essential to include NET patients in programmes of phase I and II trials of new agents, as only in this way will new therapeutic approaches emerge to produce improved outcomes in the future. The networks that now exist for the management of these patients provide large patient numbers and high-quality centres that can undertake early phase trials.
Carcinoid heart disease
The development of CHD leads to dramatic worsening of prognosis in patients with NETs. The 3-year survival of patients with carcinoid syndrome and CHD was found to be 31% compared with 68% in patients with carcinoid syndrome without CHD.368
The reported prevalence of CHD in patients with carcinoid syndrome has decreased in recent decades, from 50% to 70%368 369 to about 20%,36 probably as a result of the introduction of somatostatin analogues and other therapies designed to reduce the tumour load and the production of tumour secretory products.
Up to 20% of patients with carcinoid syndrome present with CHD at diagnosis.369 The most common pathology in CHD is involvement of right-sided valves (tricuspid valve affected more often than pulmonary). Left-sided lesions occur in up to 15% of patients with the carcinoid syndrome, but in 47% of patients with CHD.36 368 370 Almost invariably in patients with involvement of left-sided valves, patency of the foramen ovale is involved.36 Left-sided CHD is very rarely due to a bronchopulmonary NET371 or very severe, poorly controlled carcinoid syndrome.
Clinical examination
Auscultatory and clinical examination is not an accurate predictor of the presence of tricuspid regurgitation.372 Murmurs may be difficult to detect because velocities in the right heart are low. Peripheral oedema, ascites and pulsatile hepatomegaly develop as the disease progresses.
Biomarkers
Urinary 5-HIAA levels have a high sensitivity (100%) but low specificity for CHD and are not suitable as diagnostic biomarkers for this condition. Natriuretic peptides are neurohormones released by the atria and ventricles in response to an increase in wall stress due to both volume and pressure overload.373 NT-proBNP seems to be a very useful biomarker of CHD (cut-off level of 260 pg/ml (30.68 pmol/l)) and could be used to rule out CHD morbidity in patients with carcinoid syndrome.374 375
Electrocardiography
The ECG changes in patients with CHD are non-specific,376 377 but the ECG is very important for detecting arrhythmias.
Echocardiography
Echocardiography is the mainstay tool for diagnosis of CHD.36 The features of CHD are well described368 378 and are pathognomonic in the absence of exposure to the appetite suppressants fenfluramine and phentermine, ergot-derived dopamine agonists, and ergot alkaloid agents such as methysergide and ergotamine.377 379 380 Patients develop thickening and reduction of the mobility and retraction of the leaflets/cusps of cardiac valves. The subvalvular apparatus also becomes involved. Functionally, a combination of valvular regurgitation and stenosis occurs, and the right heart chambers become enlarged. The development of left-sided lesions may be expected in up to half of patients with CHD.36
Contrast echocardiography should be performed in order to detect intracardiac shunts and patency of the foramen ovale. Multiple views of each valve should be obtained for optimal evaluation of right-sided heart valves.381 When necessary, transoesophageal echocardiography (two-dimensional and three-dimensional) should be used.382
Three carcinoid score models have been developed for assessment of the severity of CHD383–385; two of these include right ventricular as well as valvular parameters.
Management
Intuitively, optimising somatostatin analogue therapy should reduce circulating vasoactive substances and carcinoid syndrome and therefore may stabilise CHD. However, to date there is no evidence to support this effect.384 Cytotoxic chemotherapy has been associated with an elevated risk of progressive heart disease.384
Medical management of pure right heart failure is limited and consists of relieving symptoms of right heart failure with diuretic therapy.
Cardiac surgery
Cardiac surgery offers definitive therapy for symptoms. Marked symptomatic improvement of at least one New York Heart Association class occurs after valve replacement.383 A median survival of 6 years compares favourably with medically treated patients, but perioperative mortality is significant.388–390
Organisation of care
There are many treatment modalities available and most are very expensive with a poor evidence base. Because of the rarity of NETs, not all clinicians and local hospitals will have the full expertise to deal with these patients. It is therefore important that all NET cases are discussed and managed by an MDT or a regional cancer network group that includes at least one recognised member with an interest in NETs. The Department of Health and National Institute for Health and Clinical Excellence (NICE) have developed a comprehensive package of national guidance (the Improving Outcomes Guidance) for different cancers, including rare tumours. The vital role of the MDT has been emphasised in all NICE guidance. In a Department of Health Cancer Plan publication, it was stated that MDTs led to improved communication between various specialists and therefore patients were likely to receive better continuity and coordination of care.391 Although there is no specific guidance for NETs, they are classified as rare cancers; guidance for rare cancers should therefore apply.
For patients with a suspected familial syndrome (eg, MEN1), referral to a genetic centre should be accompanied by appropriate counselling for the patient and their relatives. Even those without a familial syndrome will benefit from psychosocial support and counselling.392
The patient should have information available with which to make rational choices about various treatments. Patients should not only be informed about healthcare and treatment options, but also be given the opportunity to be involved in decisions about their own care at all stages of the cancer journey.391 393 394 The role of the clinical nurse specialist/practitioner or nurse consultant in the MDT is very important in directing the patient through the treatment and decision-making process. It has been shown that nurses are better than doctors in providing information about disease and treatment, and at breaking bad news to patients. Information can also be obtained through centres that regularly treat these patients and from the organisations below.
UKINETS is an organisation composed of specialist doctors and nurses from the UK and Ireland that was set up to discuss the management of NETs. UKINETS can be contacted via their website: http://www.ukinets.org. The patient support group Net Patient Foundation (incorporating ‘Living with Carcinoid’) is open to membership for patients and can be contacted at http://www.netpatientfoundation.com.
It has been reported that cancer patients prefer doctor- or nurse-led support groups to patient-led groups395; therefore the link between UKINETS and the NET Patient Foundation should continue to be strengthened.
Algorithms for overall care
Algorithms for the investigation and treatment of NETs are given in figures 1 and 2.

Algorithm for the investigation of neuroendocrine tumours (NETs). ACP, Acid Phosphatase; BNP, brain natriuretic peptide; CgA, chromogranin A; EUS, endoscopic ultrasound; FDG, fluorodeoxyglucose; GI, gastrointestinal; GPCA, gastric parietal cell autoantibody; HCG, human chorionic gonadotrophin; 5HIAA, 5-hydroxyindoleacetic acid; 5HTP, 5-hydroxytryptophan; Men-1, multiple endocrine neoplasia 1; MIBG, meta iodobenzylguanidine; NF, neurofibromatosis; PET, positron emission tomography; PP, pancreatic polypeptide; PTH, parathyroid hormone; VHL, Von Hippel Lindau.

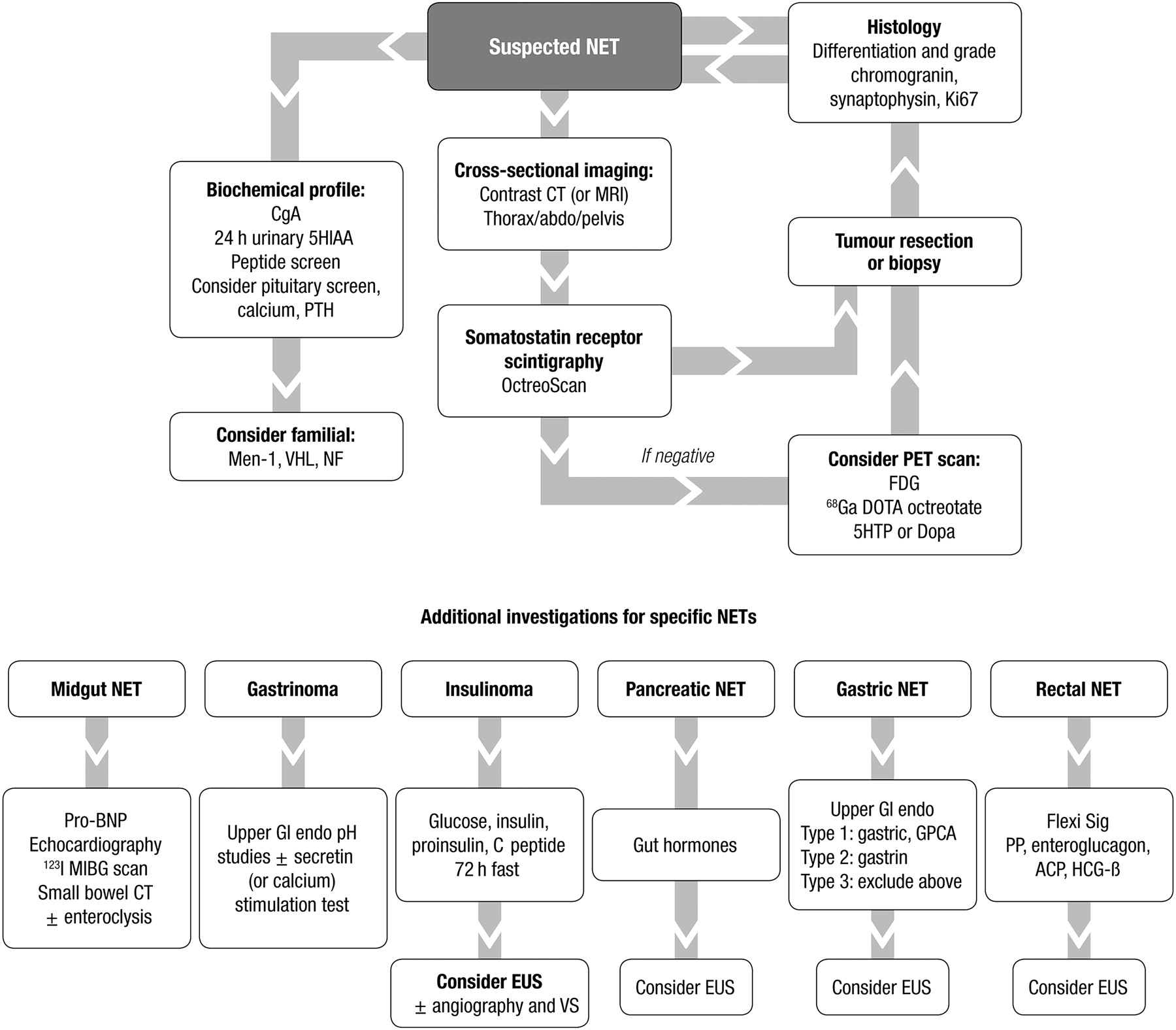{kind=link}
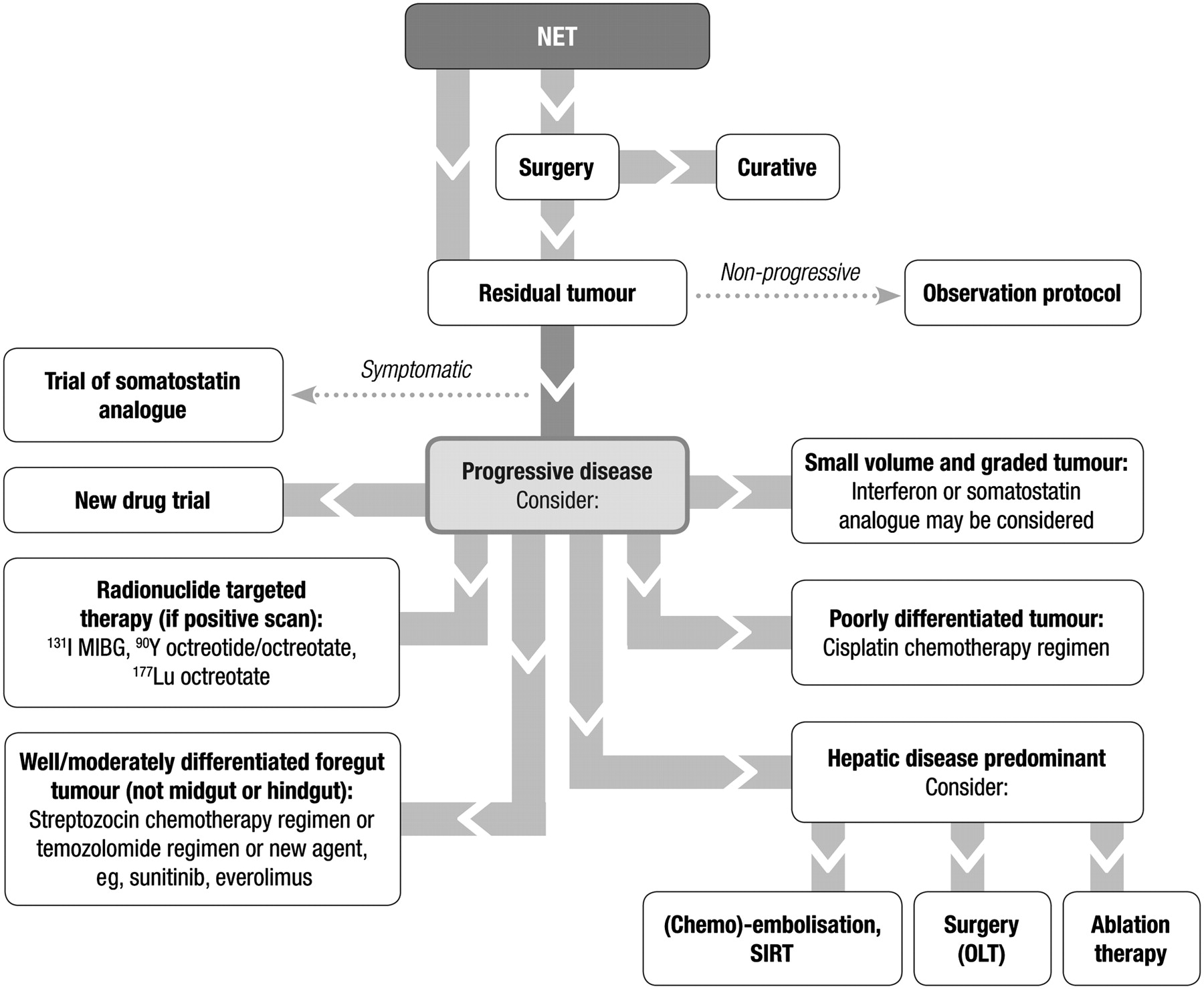{kind=link}
Algorithm for the treatment of neuroendocrine tumours (NETs). MIBG, meta_iodobenzylguanidine; OLT, orthotopic liver transplantation; SIRT, selective internal radiation therapy.
Acknowledgments
In addition to the authors, the following attended a UKINETS guidelines meeting in May 2009 at Basingstoke, Hampshire, UK and contributed to discussion on which these guidelines were revised: Dr Alan Antoney; Dr John Ayuk; Ms Melissa Banks; Ms Andrea Burgess; Dr Muriel Buxton-Thomas; Ms Louise Causer; Dr Joseph Davar; Ms Philippa Davies; Ms Gill Doodson; Mrs Sue Gower; Mrs Barbara King; Professor Derek Manas; Mr Satvinder Mudan; Dr Alberto Quaglia; Dr Aled Rees; Dr Tahir Shah; Dr Christopher Wong. Editorial support was provided by Winnie McFadzean and by Emma Handley and Andrea Carrodus of Porterhouse Medical Ltd.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding Novartis Pharmaceuticals UK Ltd and Ipsen UK provided unrestricted educational grants for the original working party meeting. Novartis Pharmaceuticals UK Ltd also provided financial assistance to allow Porterhouse Medical Ltd, a medical writing agency, to aid the drafting of this manuscript.
Competing interests JR has received honoraria and educational grants from Ipsen and Novartis; AA has received educational support and research grants from Ipsen and Novartis; JA has received research funding, educational grants and travel grants from Ipsen and Novartis; NB has received educational grants and research funding from Ipsen and Novartis; DB has no conflicts of interest; MC has received honoraria from Novartis, Ipsen, Pfizer and Lexicon for advisory boards or invited lectures, and research/educational grants from Novartis and Ipsen; PC has received honoraria for advisory boards and lectures from Novartis and educational travel support grants from Ipsen; AD has received research funding and travel expenses for meetings from Novartis; VL has received honoraria for advisory boards and educational grants from Novartis and Ipsen; TM has sat on an advisory board for Keocyte and received partial funding from Pfizer for attendance at ENETs in 2010; JD has received educational grants from Novartis and Ipsen; JN-P has received honoraria and undertaken consultancy work from Novartis and Ipsen, research funding from Novartis, and educational grants for attending meetings from Novartis and Ipsen; GP has received honoraria for advisory boards and lectures from Novartis and Ipsen, educational travel support grants from Novartis and Ipsen, and support for clinical personnel (NET CNS) from Novartis; NR has received honoraria for advisory boards from Novartis and Ipsen and a travel grant from Pfizer; AR has received honoraria for an advisory board and educational lectures from Novartis as well as travel support to those events; WS has received educational grants and honoraria from Ipsen and Novartis; RT has received honoraria and lecture fees from Ipsen, support for clinical personnel from the MRC and Wellcome Trust Clinical Training Fellowships, and educational grants from MRC, Wellcome Trust, EU FP6 and EU FP7; CT has received honoraria and educational support from Novartis and educational support from Ipsen; JV has received honoraria from Pfizer and Novartis, funding for clinical personnel from Novartis, an educational grant from Novartis and research funding from Pfizer, Novartis and Ipsen; CV has received educational grants for conference attendance from Ipsen; AG has received honoraria and educational grant support from Novartis and Ipsen.
Provenance and peer review Not commissioned; externally peer reviewed.