Article Text

Abstract
Objective Inflammatory bowel diseases (IBD) have been intrinsically linked to a deregulated cytokine network, but novel therapeutic principles are urgently needed. Here we identify the interleukin (IL)-33 and its receptor ST2 as key negative regulators of wound healing and permeability in the colon of mice.
Design Expression of IL-33 and ST2 was determined by qRT-PCR, ELISA, immunohistochemistry and western-blot analysis. Wild-type and St2−/− mice were used in wound healing experiments and in two experimental models of IBD triggered by 2,4,6-trinitrobenzene sulphonic acid or dextran sodium sulphate (DSS). Neutralisation of ST2 was performed by using a specific blocking antibody.
Results Nuclear localisation and enhanced expression of IL-33 in myofibroblasts and enterocytes was linked to disease involvement independently of inflammation, while the expression of ST2 was primarily restricted to the colonic epithelia. In two experimental models of IBD, genetic ablation of ST2 significantly improved signs of colitis, while a sustained epithelial expression of the cyto-protective factor connexin-43 was observed in DSS-treated St2-deficient mice. Unexpectedly, absence of ST2 in non-hematopoietic cells was sufficient to protect against colitis. Consistently, specific inhibition of endogenous ST2-mediated signalling by treatment with neutralising antibody improved DSS-induced colitis. In addition, IL-33 treatment impaired epithelial barrier permeability in vitro and in vivo, whereas absence of ST2 enhanced wound healing response upon acute mechanical injury in the colon.
Conclusions Our study unveiled a novel non-hematopoietic function of IL-33 in epithelial barrier function and wound healing. Therefore, blocking the IL-33/ST2 axis may represent an efficient therapy in IBD.
- Experimental Colitis
- Epithelial Barrier
- Cytokines
- Antibody Targeted Therapy
- Epithelial Permeability
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Inflammatory bowel diseases (IBD) have been intrinsically linked to a deregulated inflammatory cytokine network.
-
IL-33 has recently been found to be upregulated in human IBD.
-
IL-33 expression is enhanced during wound healing in rats.
-
Intestinal inflammation is reduced in dextran sodium sulphate-treated IL-33-deficient mice when compared with control animals.
What are the new findings?
-
Genetic ablation of the IL-33 receptor (ST2) protected mice in two different experimental models of inflammatory bowel disease.
-
A ST2 blocking antibody significantly reduced signs of colitis upon injury in mice.
-
IL-33 enhances intestinal inflammation upon injury.
-
Non-hematopoietic cells have been identified as key cellular targets of IL33, which promote colitis severity upon injury.
-
IL-33 enhances intestinal permeability in vitro and in vivo.
-
The IL-33/ST2 pathway negatively regulates wound healing in the colon of mice.
How might it impact on clinical practice in the foreseeable future?
-
Neutralisation of the IL-33/ST2 axis may thereby represent an effective therapeutic target in inflammatory bowel disease.
Introduction
Inflammatory Bowel Diseases (IBD) are characterised by relapsing-remitting epithelial barrier dysfunction that is restricted to the colon and/or rectum in ulcerative colitis (UC) or that may affect any part of the gastrointestinal tract in Crohn's disease (CD).1 Novel therapeutic principles are urgently needed to treat and/or prevent the natural history of IBD that affects up to 6 million individuals worldwide. However, the underlying pathogenesis of IBD is not yet well understood. Recent experimental and clinical studies have implicated chronic engagement of stress response of epithelial cells that may account for impaired epithelial regeneration,1 and for enhanced secretion of inflammatory signals,2 including interleukin (IL)-33.3–7
IL-33, previously known as IL1F11 or nuclear factor from high endothelial venules, is the most recently discovered member of the IL-1 cytokine family.8 ,9 Full-length IL-33 is biologically active and is inactivated by apoptotic caspases through proteolytic cleavage.10 ,11 IL-33 functions within the nucleus through its association with chromatin in vivo.12 IL-33 binds to the ST2 chain (also known as IL-33R or IL-1R4) of the IL-33 receptor complex, which confers ligand specificity.8 ST2 is a stable cell surface marker on T(H)2 cells, yet it has become appreciated more recently that ST2 is functionally expressed on epithelial cells as well.13 Interestingly, IL-33 has been shown to drive production of extremely high amounts of the T(H)2 cytokines IL-5 and IL-13 by type-2 innate lymphoid cells after helminth infection in the intestine14 ,15 or influenza virus infection in the lungs.16 ,17 More importantly, ST2 expression has been found to be driven by T(H)2 cytokines,13 suggesting that IL-33 may trigger a key regulatory amplification loop involved in immune homeostasis.
Importantly, IL-33 and ST2 have been linked to several important inflammatory diseases, including asthma, rheumatoid arthritis and atherosclerosis.18 In these different inflammatory conditions, it is worth noting that IL-33 has a dual pathophysiological role that likely depends on the specific immune mechanisms underlying disease pathogenesis.18 However, although the IL-33/ST2 axis is thought to be involved in IBD,3–7 its contribution to disease pathogenesis remains unclear.19 Here our combined in vivo and in vitro approaches unveiled that the IL-33/ST2 axis represents a key regulator of intestinal homeostasis in two experimental models of IBD. More importantly, non-hematopoietic responsiveness to IL-33 promotes intestinal inflammation upon injury. Last but not least, IL-33 controls per se colonic epithelial permeability independently of intestinal inflammation by negatively regulating wound healing in the colon. Collectively, these data identify the IL-33/ST2 axis as a potential therapeutic target in IBD.
Results
IL-33/ST2 expression in human IBD and in experimental models of IBD
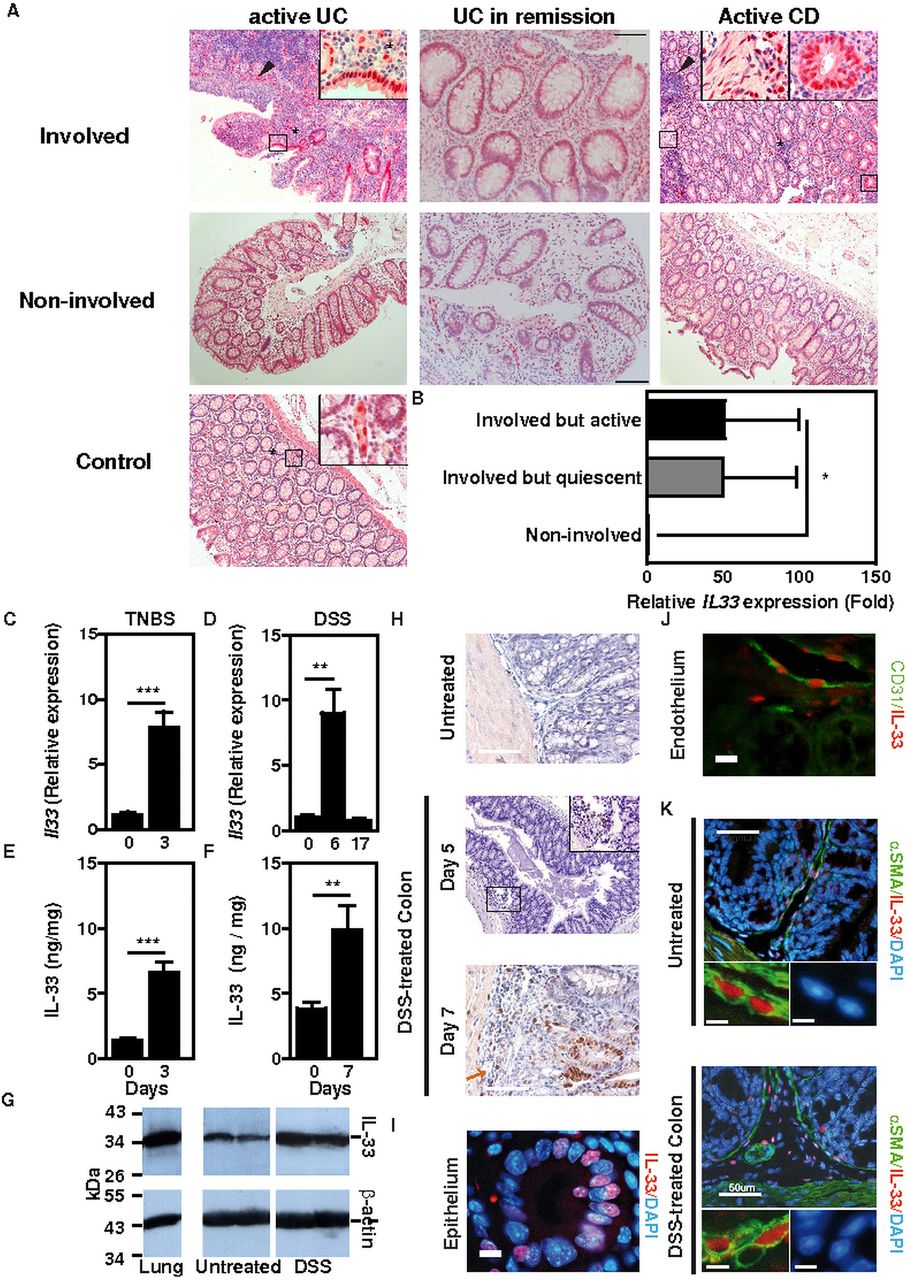
IL-33 has recently been found to be upregulated in human IBD.3–6 In colon biopsies from CD patients, inflammatory aggregates were found surrounding IL-33+ cells, while the latter frequently formed ‘shield-like’ clusters underneath ulcerated areas in UC (figure 1A). Furthermore, certain IL-33+ cells were located within the muscularis in some IBD patients (figure 1A). IL-33 expression was found upregulated within the inflamed mucosa of resection specimens collected from IBD patients with active disease when compared with the expression in non-involved patient biopsies or in colon tissue from non-IBD controls (figure 1A and online supplementary tables S1 and S2). Likewise, transcript level of IL-33 was enhanced within the involved colon mucosa during remission independently of the Mayo score when compared with non-involved colonic areas (figure 1A,B and online supplementary table S3). Similar to what is observed in human IBD (figure 1A,B), we found that both mRNA and protein level of IL-33 was significantly elevated in damaged colons of mice undergoing two well-established acute models of colitis in mice, namely trinitrobenzene sulphonic acid (TNBS)—(figure 1C,E) and dextran sodium sulphate (DSS)-induced (figure 1D,F) colitis. Western-blot analysis of colon extracts further confirmed these findings by showing upregulation of the active, full-length form of IL-33 in colons at the peak of disease severity following exposure to DSS (figure 1G).10 ,20 Likewise, IL-33 was already detectable in submucosal connective tissue fibroblasts on day 7 of DSS treatment, whereas it was barely detectable in naive mice (figure 1H).

IL-33 in human inflammatory bowel diseases (IBD) and in two experimental models of colitis. (A) Immunohistochemical analysis of IL-33 expression in non-IBD controls and in involved and non-involved mucosal areas of IBD patients in remission or with active disease. Inserts represent enlargements of the boxed areas. Size bars, 100 μm. (B) Relative IL-33 mRNA expression in ulcerative colitis patients in remission (n=5). (C) Relative IL-33 mRNA expression in trinitrobenzene sulphonic acid (TNBS)-induced colitis (n=5). (D) Relative IL-33 mRNA expression in dextran sodium sulphate (DSS)-induced colitis. C57BL/6J mice were left untreated (n=4) or were given 2% DSS in the drinking water for 6 days (n=9), or for 7 days then returned on normal drinking water for the following 10 days (n=7). (E) IL-33 protein levels in TNBS-induced colitis (n=5). (F) IL-33 protein expression in DSS-induced colitis. C57BL/6 mice were given 3% DSS (n=4) or not (n=5) in the drinking water for 7 days. (G) Western-blot analysis of control or DSS-treated colon homogenates using specific antibodies. Lung tissue is shown as control. (H) Immunohistochemical analysis of IL-33 expression in DSS-induced colitis. Size bars, 50 μm. (I–K) Immunofluorescence staining for IL-33 (red) and α-smooth muscle actin (SMA) or CD31 (green) in DSS-induced colitis. DNA was counterstained with DAPI (blue). Representative images of IL-33 staining in colonic epithelium (I), CD31+ endothelium (J) and a-SMA+ myofibroblasts (K).
In healthy colonic mucosa, nuclear staining of IL-33 was primarily restricted to endothelial cells, whereas within the involved mucosa nuclear IL-33 immunoreactivity was predominately in colonic epithelial cells most often in UC during either remission or relapse rather than in CD (figure 1A). We next further examined which are cellular sources of IL-33. We thereby performed specific double immunofluorescence staining of DSS-treated colonic sections in mice. At the peak of disease severity on days 7 and 8, nuclear IL-33 was detectable in few enterocytes (figure 1I) and in cells that express either a cognate marker of endothelial cells (CD31) (figure 1J) or of myofibroblasts (α-smooth muscle actin, αSMA) (figure 1K). We next aimed at determining which cells are targeted by IL-33 within the colonic mucosa upon injury. Our screening approach revealed that ST2 is primarily expressed by colonocytes in both mice (figure 2A) and humans (figure 2B), while its expression was barely detectable among leukocytes from the lamina propria if any. Subepithelial infiltrates contained many ST2-positive cells in either relapsing or remitting IBD (figure 2B), as well as in experimental model of IBD (figure 2C), further supporting a key role of the IL-33/ST2 axis in the pathogenesis of IBD.

ST2 in human inflammatory bowel diseases and in experimental models of colitis. (A) Relative mouse ST2 mRNA expression was determined by qRT-PCR analysis for different cells types and normalised using Actb (n=4). (B) Immunohistochemical analysis of human ST2 expression in controls, active Crohn's disease, active ulcerative colitis (UC) and UC in remission patients. Size bar, 100 μm. (C) Immunohistochemical analysis of mouse ST2 expression on day 5 and on day 8 of DSS-induced colitis. Inserts represent enlargements of the boxed areas. Lower edges of the large panels: 689 μm length, of the inserts: 143 μm. Data show means±SEM. A second independent experiment gave similar results.
ST2 deficiency results in decreased disease severity in two experimental models of IBD
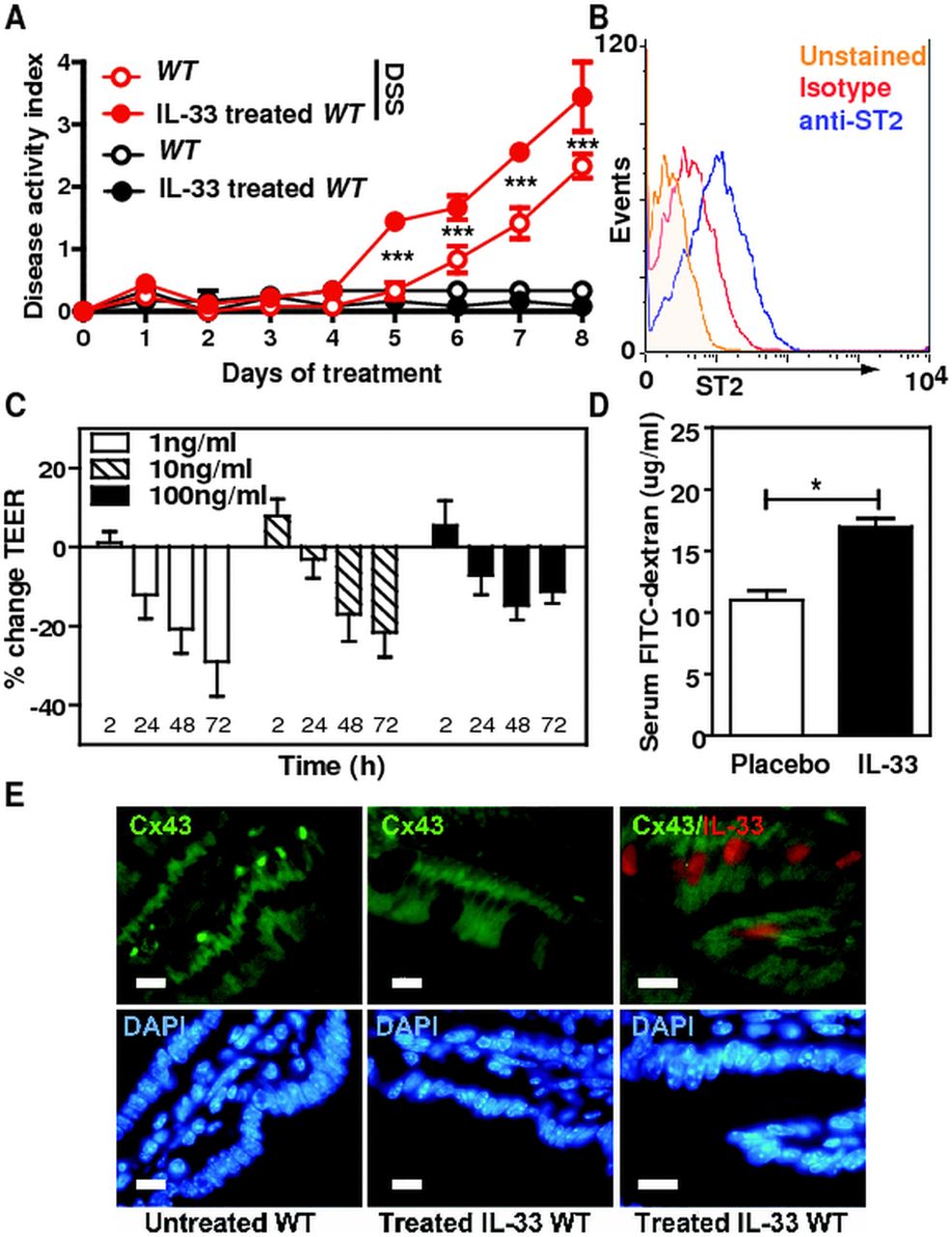
To formally assess the role of the IL-33/ST2 pathway in intestinal homeostasis, we asked whether absence of IL-33-mediated signalling pathway would abrogate disease severity in two experimental models of acute ulceration/intestinal inflammation. Mice deficient or not for the IL-33 receptor (St2–/–) were first exposed to TNBS by the intrarectal route. Under this experimental condition, St2-deficient mice had less inflammation as assessed by histological scoring (figure 3A,B) and colon length (figure 3C) as compared with their WT littermates. Consistently, the colonic mucosa of St2-deficient mice showed lowered production of KC (figure 3D) as compared with their WT littermates. We next determined whether absence of the ST2-mediated signalling pathway might protect in another model of colitis that is induced by DSS. Mice deficient or not for St2 were exposed ad libitum to DSS for 7 days. The disease activity index (DAI) was assessed daily as an average of loss-of-body weight and signs of rectal bleeding and diarrhoea. No weight loss was observed in DSS-treated St2–/– mice whereas DSS-treated WT mice had significant progressive weight loss over time; up to 16% of their initial body weight at day 7 of exposure to DSS (figure 4A). Consistently, DSS-treated St2–/– mice experienced reduced signs of morbidity over the course of the disease when compared with similarly DSS-treated WT animals (figure 4A). At necropsy, DSS-treated St2−/− mice limited overt epithelial ulceration, infiltration by neutrophils and lymphocytes in the colon (figure 4B) that coincided with reduced secretion of IL-6 (figure 4C) and KC (figure 4D). Consistently, reduced colon shortening was found in St2–/– mice upon injury when compared with control animals (figure 4E). We therefore demonstrated that engagement of the ST2 pathway contributes to the exacerbation of colitis in two experimental models of IBD, suggesting that pharmacological inhibition of ST2 may represent a novel therapeutic principle in IBD. Thus, we assessed whether neutralisation of ST2 with a specific monoclonal antibody injected intraperitoneally on days 1, 3, 5 and 7 following DSS challenge would protect mice from colitis. In line with our previous findings, WT mice treated with a blocking anti-ST2 antibody displayed significantly reduced signs of disease compared with DSS-treated mice that received isotype control (figure 4F). Consistently, histological score (figure 4G) and secretion of IL-6 (figure 4H) was improved upon treatment with ST2 blocking antibody when compared with placebo. In addition, mice deficient for IL-33 showed a reduced histological score (figure 4I) and colon shortening (figure 4J) upon DSS challenge when compared with similarly treated control animals. Collectively, we provided experimental evidence that therapeutic neutralisation of the IL-33/ST2 axis may represent a novel effective therapeutic principle for the treatment of IBD.
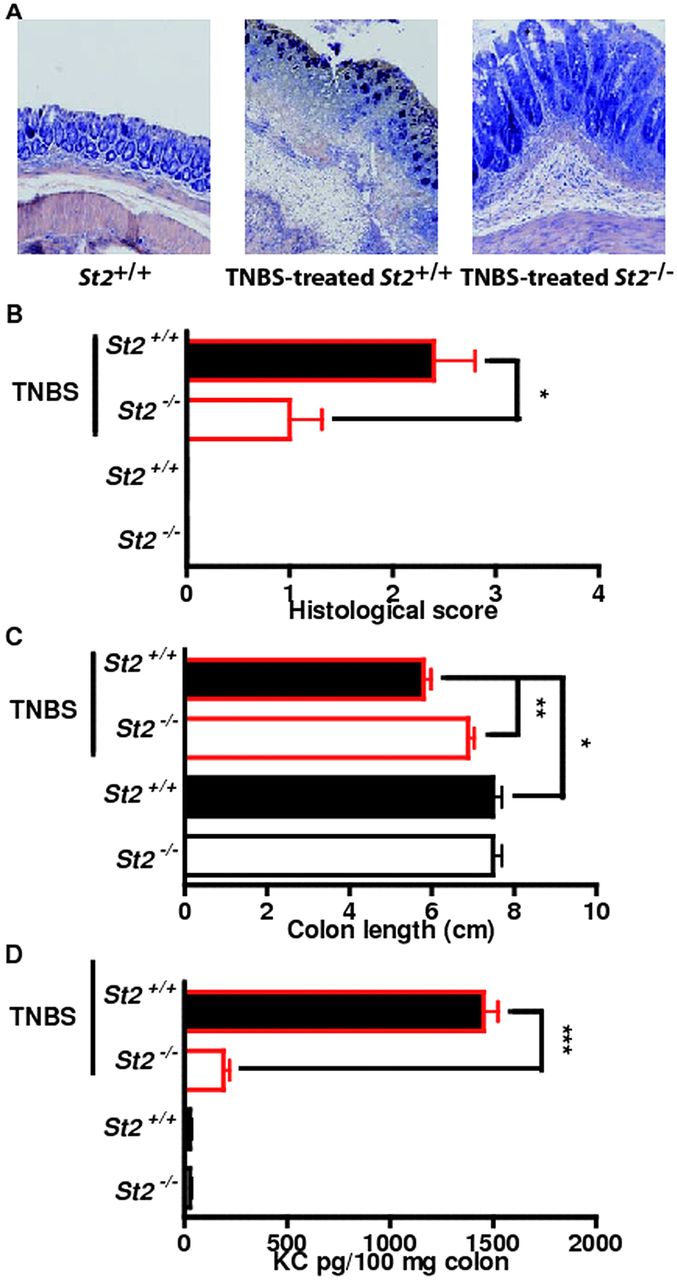
Genetic ablation of ST2 reduced colonic inflammation in a hapten-mediated model of acute ulceration/intestinal inflammation. (A–D) trinitrobenzene sulphonic acid-induced colitis. Mean values of six animals per group±SEM out of two independent experiments. Representative May-Grünwald & Giemsa stainings (A) and histological score in colons (B), colon length (C) and KC expression (D) are depicted.

Genetic and/or pharmacological inhibition of the IL-33/ST2 axis reduced colonic inflammation in a chemical-induced model of acute colitis. (A) Disease activity index (DAI), (B) Histological Score, (C) IL-6 and (D) KC ELISA from colon homogenates and (E) Colon length of dextran sodium sulphate (DSS)-treated wild-type and St2−/− animals. Mean values of DAI±SEM out of four independent experiments are shown. (F) DAI (G) Histological Score, (H) IL-6 ELISA from colon homogenates of DSS-treated mice administered 50 µg neutralising anti-ST2 antibody (rat IgG1) or isotype control intraperitoneally on days 1, 3, 5 and 7. (I) Histological Score and (J) Colon length of DSS-treated wild-type and IL-33-deficient mice. Mean values of four animals per group±SEM out of two independent experiments.
Absence of ST2-mediated signalling in non-hematopoietic cells protects against DSS-induced colitis
To further compartmentalise how the ST2-mediated signalling pathway may drive intestinal inflammation upon injury, bone marrow chimaera experiments were performed. Lethally irradiated wild-type and St2−/− mice were reconstituted with bone marrow cells from either wild-type or St2−/− mice. Three months later, chimaeric animals were challenged with DSS 3% for 7 days. In line with previous findings of reduced colon inflammation in St2−/− mice, shortening of colon from DSS-treated irradiated St2−/− mice that were reconstituted with bone marrow cells from St2−/− (KO→KO) mice was reduced when compared with similarly treated control (WT→WT) animals (figure 5A). Consistently, colon levels of MPO enzymatic activity (figure 5B) and histological score (figure 5C) were also reduced in mutant (KO→KO) chimaeric mice relative to controls. More importantly, the enhanced resistance against DSS-mediated colitis in mutant animals was also recapitulated in St2−/− recipients that were reconstituted with bone marrow cells from wild-type (WT→KO) donors. In contrast, disease severity in irradiated wild-type mice that were reconstituted with bone marrow cells from St2−/− (KO→WT) animals was similar to that observed in control (WT→WT) recipients. Given that both IL-33 and ST2 are highly expressed in the intestinal epithelium upon injury, we therefore hypothesised that protection from colitis in St2−/− mice might reflect sustained intestinal barrier function. In response to epithelial insults, enterocytes need to migrate, a process which depends on gap junction intercellular communication through the engagement of Connexin 43 (Cx43).21 ,22 Importantly, Cx43 expression was almost completely lost in DSS-treated WT mice, but not in similarly treated St2−/− mice (figure 5D). In addition, it is worth noting that the colonic epithelium of St2−/− mice, but not of wild-type animals, was never positive for IL-33 (figure 5E). Collectively, we demonstrated that the absence of ST2-mediated signalling pathway in non-hematopoietic cells protects mice against DSS-mediated colitis with maintenance of gap junctions.
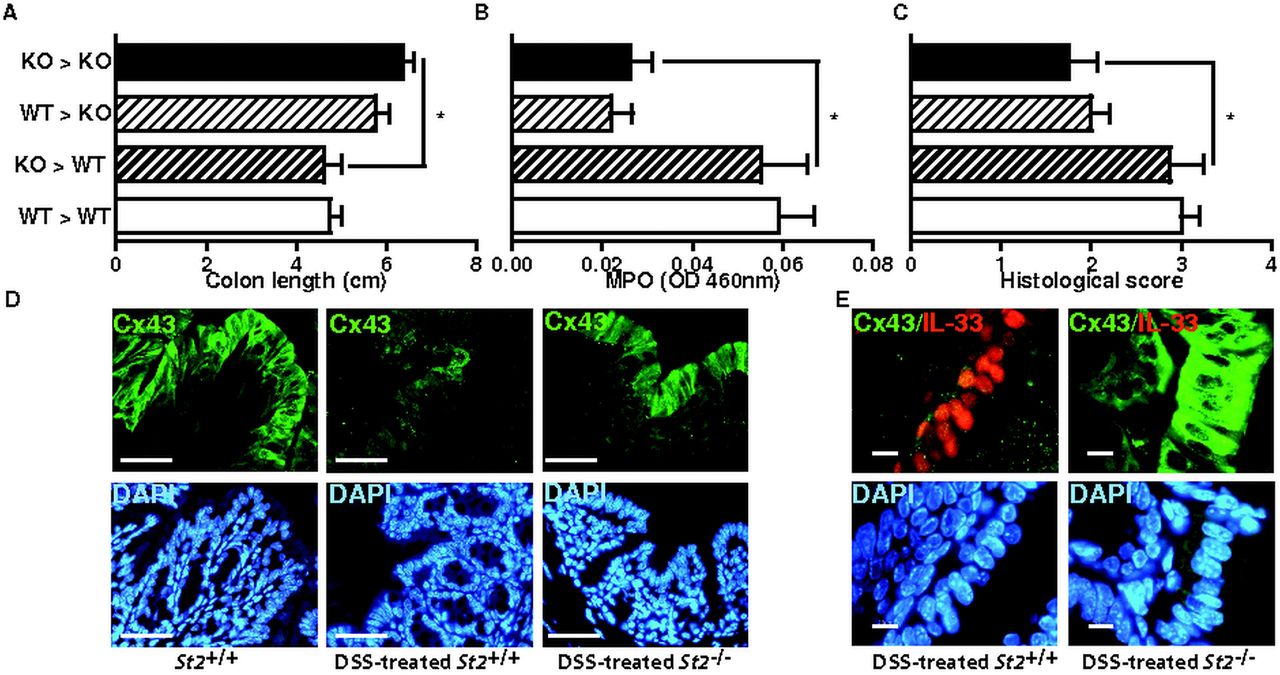
Loss of responsiveness to IL-33 within the radio-resistant compartment is sufficient to protect mice from dextran sodium sulphate (DSS)-induced colitis. (A) Colon lengths (B) MPO levels and (C) histological scores of DSS-treated chimaeric animals at autopsy. Mean values±SEM are given (n=4 per group). (D, E) Expression of endogenous Cx43 and IL-33 was analysed by double immunofluorescence staining with specific antibodies. Size bar, 50 μm.
IL-33 impairs epithelial barrier function and exacerbates DSS-induced colitis
To further assess the role of IL-33 on intestinal inflammation, recombinant IL-33 was injected intraperitoneally on days 1, 3 and 5 to DSS-treated WT mice and DAI was followed over time. Importantly, IL-33 treatment significantly augmented the morbidity of DSS-induced colitis, as judged by the more severe DAI, but not in mice that were not challenged by DSS (figure 6A). Given the proinflammatory role of IL-33 in the colon, we hypothesised that IL-33 may directly influence per se intestinal permeability that may enhance the inflammatory response of the colonic mucosa upon injury. We first assessed whether IL-33 impaired the integrity of polarised monolayers of the human colonic epithelial Caco-2 cell line that expresses ST2 (figure 6B). Through transepithelial electrical resistance measurements across polarised Caco2 monolayers, exogenous IL-33 was found to negatively regulate epithelial integrity (figure 6C). To further confirm our in vitro findings, we next evaluated permeability of the intestinal tract in groups of mice that were not challenged with DSS but that either was treated or not with exogenous IL-33. Remarkably, an enhanced intestinal translocation of FITC-dextran was observed upon systemic IL-33 treatment independently of colitis when compared with placebo (figure 6D). No Cx43 expression change was observed in response to exogenous administration of IL-33, suggesting that the sustained expression of Cx43 in DSS-treated St2−/− mice was a surrogate marker of the enhanced resistance of the aforementioned animals to colitis (figure 6E). Collectively, our findings support the hypothesis that the IL-33/ST2 axis plays a key role in controlling epithelial barrier integrity and permeability independently of the colonic inflammatory milieu.

IL-33 negatively regulates epithelial barrier function in the colon. (A) Disease activity index of 3% dextran sodium sulphate-treated mice that received intraperitoneal injections of 0.5 µg recombinant IL-33 on days 1, 3 and 5 is depicted. Mean values±SEM are given (n=4 per group). (B) ST2 expression on Caco-2 cells. (C) Transepithelial electrical resistance of Caco-2 monolayers treated with indicated doses of recombinant IL-33 at the indicated time points. Pooled data of three independent experiments are shown, bars represent means±SD. (D) Intestinal permeability in placebo and rmIL-33-treated mice assessed by measuring serum FITC-Dextran levels 4 h after administration. Data are displayed as mean±SEM with n=4 per group. One representative of two independent experiments is shown. *p<0.05, unpaired t test. (E) Colonic expression of endogenousCx43 and IL-33 is shown in (E) untreated and (F) IL-33-treated mice.
IL-33 delays wound healing of the injured colonic epithelia
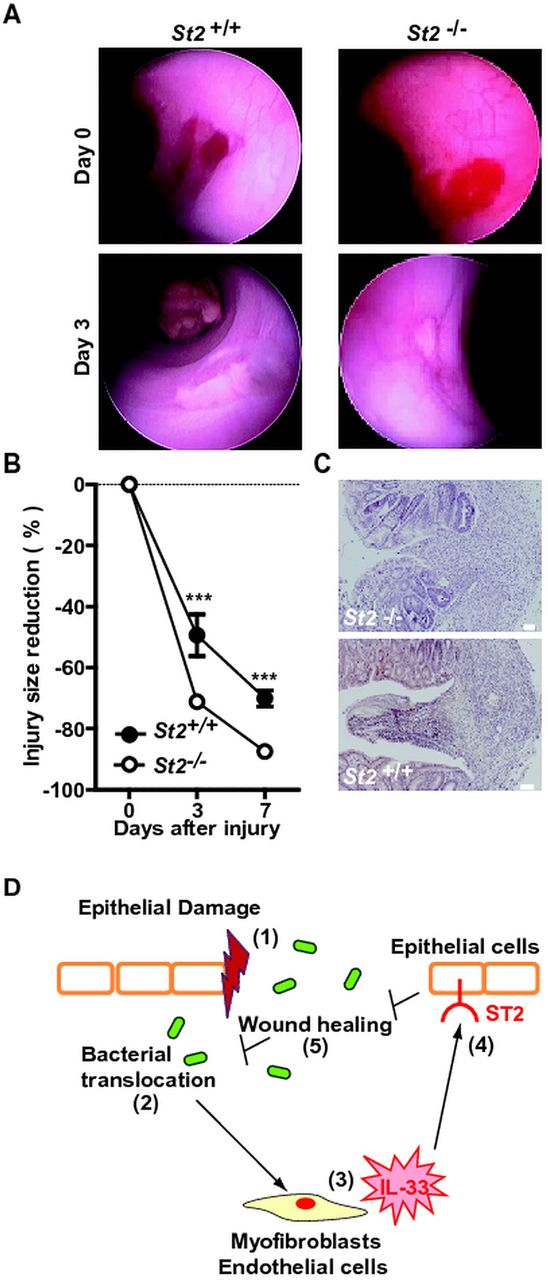
Recently, administration of IL-33 was found to regulate the Notch-mediated signalling pathway23 that is involved in wound healing. To assess whether the IL-33/ST2 axis affects re-epithelialisation following colonic damage, mucosal regeneration was monitored after biopsy injury of the descending colon in St2-deficient and control animals. Oval-shaped lesions of the mucosa of the distal colon had recovered by 49% and by 70% in control wild-type animals at 3 and 7 days post injury, respectively. In contrast, a significant accelerated regeneration of the average surface lesion area was observed in St2-deficient mice (figure 7A,B). Consistently, the histological aspect of colon wounds appeared less severe in the absence of St2 (figure 7C). Therefore, we concluded that the IL-33-ST2 axis promotes leakiness of the colonic mucosa by delaying wound healing in mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Absence of ST2 improved wound healing in the colon. The mucosal regeneration was assessed after biopsy-induced injury of the descending colon of St2-deficient (n=9) and control (n=6) animals by using a straight-type rigid miniature endoscope and 3-French biopsy forceps. (A) Representative endoscopy pictures. (B) Wound diameter was assessed just after biopsy at day 0, 3 and 7. (C) Representative photographs of H&E staining of paraformaldehyde-fixed tissue at day 4 post injury. Size bars, 50 μm. (D) Schematic overview of the physiological role of the IL-33/ST2 axis in intestinal homeostasis.
Discussion
The function of IL-33 is influenced by underlying disease condition.18 Notably, the anti-infective properties of IL-33 against Trichuris muris infection were linked to enhanced T(H)2-type cytokine expression together with reduced secretion of T(H)1 and T(H)17 cytokines.24 IL-33 also conferred protection in a mouse model of sepsis by increasing neutrophil recruitment and bacterial clearance at the site of infection.25 Our data unveiled for the first time, to our knowledge, a previously unrecognised non-hematopoietic mediated mechanism of IL-33 in the colon by linking epithelial permeability and wound healing in IBD. Our data demonstrated that IL-33 impairs intestinal barrier function independently of inflammation, providing a working model whereby enhanced IL-33 may favour microbial translocation that perpetuates a vicious circle of colonic inflammation.26 In addition, we report a key pathogenic role for IL-33/ST2 axis in two experimental models of IBD. In line with previous findings using IL-33−/− mice26 including ours, we demonstrated that St2−/− mice have reduced clinical signs and inflammatory lesions upon injury, which coincided with sustained expression of molecules involved in gap junction intercellular communication. Consistently, neutralisation of responsiveness to any isoforms of IL-33 by a very specific anti-ST2 blocking antibody improved the course of DSS-induced colitis. In addition, administration of exogenous recombinant IL-33 impaired intestinal barrier function and enhanced the severity of DSS-induced colitis by increasing intestinal permeability, strongly suggesting that bioactive IL-33 is released locally upon injury. More importantly, an upregulated secretion of IL-33 was observed in involved areas of the colonic mucosa in mice and human IBD independently of inflammation. Our immunofluorescence and Western blot analyses indicate that the full length biologically active form of IL-33 is found within the involved colonic area in both mice and human IBD and that the number of cells showing nuclear expression of IL-33 is increased in both the submucosal and epithelial compartment of the diseased colon. Noteworthy, IL-33 may directly act on epithelial barrier function upon injury since ST2 was primarily expressed by the intestinal epithelium of UC patients. In line with our hypothesis, absence of ST2-mediated signalling pathway in non-hematopoietic cells was sufficient to protect animals against DSS-induced colitis as determined by bone-marrow chimaera experiments. Collectively, we propose that the IL-33/ST2 axis acts as a colonic alarm in responding to epithelia injury. It remains now to be determined how non-hematopoietic response to IL-33 may regulate epithelial barrier function and wound healing effects by investigating several mechanisms, such as TGFβ.27 It is worth noting that it may interfere with activation of the innate immune system,26 and may limit microbial translocation by promoting wound healing. Enhanced wound healing is a promising therapeutic approach for maintenance of remission and we thereby propose that neutralisation of the IL-33/ST2 pathway will be a novel two-pronged approach in IBD.
Materials and methods
Patients
Clinical diagnosis of CD or UC was based on established clinical, endoscopic and histological criteria. Paraffin-embedded blocks from colectomy samples of the involved and non-involved regions from both therapy-resistant active CD or UC patients (see online supplementary tables S1 and S2) and from biopsies samples of UC patients in remission (see online supplementary table S3) were analysed. Tissues from 20 CD patients and 19 UC patients were included in this study. Normal colons from six patients were either healthy tissue from resection edges of tumour biopsies (n=4) or diagnostic biopsies from health monitoring studies (n=2) that appeared healthy at the histological level. Approval of the experiments was obtained for KM and GC from the Ethics Committee of Both Basels (EKBB: 264/11 ‘Die Rolle von Interleukin-33 in chronisch-entzündlichen Darmerkrankungen’). Information about the cohort of UC patients in remission is reported to the Commission Nationale de l'Informatique et des Libertés (no.1404720), which supervises the implementation of the act regarding data processing, data files and individual liberties that came into effect on 6 January 1978, and was amended on 6 August 2004, to protect the personal data of individuals.
Mice
Mice deficient for St2 (St2–/–)28 and for IL-33 (Il33−/−)26 were back-crossed eight times on C57BL/6J genetic background and bred with wild-type mice in our Specific-Pathogen Free animal facility at the Transgenose Institute (CNRS, TAAM, Orleans, France).
Experimental models of colitis
Experimental model of acute ulceration/intestinal inflammation was induced in 8–10 weeks old male St2–/–, Il33−/− and St2+/+ mice by either intrarectal administration of TNBS (TNBS, 150 mg/Kg, as described before29) and/or by administering ad libitum 3% DSS (DSS; MW: 30 000–40 000, TdB Consultancy AB) in the drinking water for 7 days. Age-matched and gender-matched animals were housed five per cages and had free access to a standard laboratory chow diet in a half-day light cycle exposure and temperature-controlled Specified-Pathogen Free environment as determined by the FELASA recommendations. All animal studies were approved by the local investigational review board and were performed in two accredited establishments according to the governmental guidelines N°86/609/CEE. DAI was determined daily by combining the average score of loss-of-body weight, rectal bleeding and stool consistency as described before.29
Bone marrow transplantation experiments
Recipient mice underwent a lethal total-body irradiation as reported before.30 Twenty-four hours postirradiation, mice received 2×106 fresh bone marrow cells. Blood was collected in EDTA-containing tubes at regular intervals, and the haematological parameters were determined with a Technikon H1E analyser. Two months after bone marrow transplantation, chimaeric mice were next challenged with 3% DSS.
Post mortem and microscopic analysis
Mice were sacrificed by CO2 at day 7 or 8, and the colon was excised and sized. The colon was then rinsed with saline and processed for histopathological analysis (conserved immediately in formaldehyde 10%), and dosage of cytokines. Tissue was fixed in formalin (as mentioned above), paraffin embedded, sectioned, and then stained with either H&E, May-Grünwald & Giemsa or Periodic acid-Schiff staining. Histopathological changes were individually scored by two independent investigators. Each mouse was scored individually for each of the parameters that include inflammatory cell infiltration (score 0–5), tissue damage (score 0–5) and percentage of involvement (score 0–4). The presence of occasional and increased inflammatory cells within the lamina propria was scored as 0 and 1 respectively, whereas transmural extension of the infiltrate was scored 5. For tissue damage, no mucosal damage was scored as 0 and lymphoepithelial lesions were scored from 1 to 5 for extensive mucosal damage and extension into deeper structures of the bowel wall. A score of disease involvement ranges from 0 to 4 that corresponds to either 0%, 1–25%, 26–50%, 51–75% and 76–100% of involvement respectively.30 Infiltrating leukocyte score was done by counting the number of leukocytes per high power field (40×) in the mesenteric border of the colon.
Mouse endoscopy
Mucosal wounding was performed by using a straight-type rigid miniature endoscope and 3-French biopsy forceps. The presence of ulcerations within the colon was monitored by using the Coloview high resolution mouse endoscopic system (Karl-Storz).
Immunoblotting and FACS analysis
Immunoblotting was performed on tissue homogenates that were lysed in RIPA buffer solution supplemented with a protease inhibitor cocktail tablet (Roche) and a phosphatase inhibitor cocktail set II (Merck4Biosciences). A goat anti-mouse IL-33 polyclonal antibody (1/1000; R&D Systems), a donkey anti-goat, HRP conjugated (1/10 000; Promega), an anti-β-actin mouse monoclonal antibody Clone AC-15 (1/3000; Sigma) and a goat anti-mouse, HRP-conjugated (1/10 000; Promega) were used. The immunoreactive proteins were visualised with ECL plus reagents (ECL Western Blotting Detection Reagents, Amersham). For FACS analysis, Caco2 cells were analysed using PE-labelled anti-ST2 antibodies from R&D systems (clone97203) and a matching isotype control.
Histology, immunohistochemistry and immunofluorescence staining
Human sections (10 mm) were preincubated on the BondMax system (Leica, Mannheim, Germany) in Bond Epitope Retrieval Solution 2 (pH 9.0) for 30 min at 95°C, then stained, either for IL-33 using goat antihuman IL-33 IgG (R&D systems, AF3625) at a dilution of 1 : 400, or for ST2 using rabbit polyclonal anti-ST2 (SigmaAldrich, PRS3363) at a dilution of 1 : 400. Colons that were isolated from euthanised animals were embedded in OCT compound (Cellpath) and flash-frozen. Alternatively, colonic sections were fixed in Accustain (Sigma) at 4° for 4 h and embedded in paraffin. 5 μm paraffin-embedded sections were deparaffinised in Histo-clear (National Diagnostics) and rehydrated in graded alcohol series. 6 μm cryosections were postfixed in PFA 4% for 10 min. Staining with H&E (Sigma) was done under standard conditions. Slides were then dehydrated and mounted in Safemount mounting medium (Labonord, France). For immunofluorescence or immunohistochemistry staining, rehydrated paraffin sections were boiled in a microwave oven for epitope retrieval in Sodium Citrate buffer (10 mM pH 6, 20 min). Paraffin or cryosections were equilibrated in PBS and incubated with blocking solution MAXblock (Active Motif) 1 h at room temperature. A polyclonal goat antimouse IL-33 (R&D Systems), a monoclonal rat anti-CD31 (Pharmingen) and a polyclonal rabbit anti-Connexin43 (Zymed) diluted at 1/200, 1/100 and 1/100 in PBS, MAXblock 20% respectively were incubated overnight at 4°C. For IL-33 staining, sections were washed in PBS for 30 min and incubated with a rabbit antigoat biotin polyclonal antibody (1/200; DAKO) followed by SA-HRP (1/100; Vector) and incubated 1 h and 30 min at room temperature respectively. HRP was revealed with DAB (Sigma). Haematoxylin-counterstained sections were dehydrated and mounted with Coversafe medium (Microm). Immunohistochemistry of DSS-induced colitis for mouse IL-33 and ST2 was performed on 5 μm paraffin sections using a goat antimouse IL-33 polyclonal antibody (R&D systems, AF3626) or a rabbit polyclonal anti-ST2 antibody (Abcam, ab25877, 1 : 500). Alternatively, IL-33 immunofluorescence staining sections were incubated with a bovine antigoat Cy3 secondary antibody (Jackson Immunoresearch; 1/200) 1 h at room temperature. For CD31 and Connexin43 staining, sections were respectively incubated with a rabbit antirat IgG biotin polyclonal antibody (1/200; DAKO) and a Donkey antirabbit biotin polyclonal antibody (1/200; Jackson Immunoresearch) followed by SA-FITC (1/200; Vector) and incubated 1 h and 30 min at room temperature respectively. For α-SMA staining, sections were incubated with a mouse anti-αSMA monoclonal antibody (DAKO; 1/100) followed by a donkey antimouse Cy2 polyclonal antibody diluted 1/200 by using a MOM kit (Vector) according to the manufacturer instructions. Sections were counterstained with DAPI and mounted in Mowiol.
Image acquisition and processing
Epifluorescent images were visualised using an inverted microscope Eclipse TE300 Nikon with 40X/0.75 and 100X/0.5–1.3 objectives at room temperature and captured through a DXM 1200 digital camera using Nikon ACT1 software. Bright field images were visualised using an Eclipse 80i Nikon microscope with 40X/0.75 and 100X/1.30 objectives at room temperature and captured through a Digital Sight DS 5 M L1 Nikon camera using DS 5 M L1 Nikon software. All images were processed using Adobe Photoshop CS4 software.
Cytokine determination and MPO analysis
Protein levels of IL-33 and IL-6 were determined from the supernatants of colon homogenates by specific ELISA according to the manufacturer's instructions (R&D system). For qRT-PCR analysis, colonic biopsies were immediately frozen and stored at −80°C in RNAlater (Ambion, Applied Biosystems). Relative St2 expression was determined in primary cultures of bone marrow derived macrophages and dendritic cells, peritoneal macrophages peripheral blood mononuclear cells, granulocytes, colonic myofibroblasts, and CD4 and CD8 lymphocytes that were collected from C57/Bl6J mice. In addition, the expression of St2 was assessed in the following cell lines, namely L929, 3T3, MC38, Hepa 1.6, P815 and H5V as prototypes of dermal fibroblasts, embryonic fibroblasts, colonocytes, hepatocytes, mast cells and colonic endothelial cells respectively. Briefly, total RNA from cells and colonic specimens was extracted using the RNeasy (Qiagen) and reverse-transcribed with the High-Capacity cDNA Archive kit (Applied Biosystems), according to the manufacturer's instructions. The resulting cDNA (equivalent to 5 ng of total RNA) was amplified using the SYBR Green real-time PCR kit and detected on a Stratagene Mx3005P (Agilent Technologies). qRT-PCR was performed with the forward and reverse primers (sequences available upon request) that were designed using Primer express software, V.1.0 (Applied Biosystems). On completion of the PCR amplification, a DNA melting curve analysis was carried out in order to confirm the presence of a single amplicon. β-actin was used as an internal reference gene in order to normalise the transcript levels. Relative mRNA levels (2-ΔΔCt) were determined by comparing (a) the PCR cycle thresholds (Ct) for the gene of interest and Actb (ΔCt) and (b) ΔCt values for treated and control groups (ΔΔCt). Measurement of MPO was determined as described previously.31
In vitro and in vivo analysis of epithelia permeability
Caco-2 human intestinal colon adenocarcinoma cell line (European Collection of Cell Culture) was maintained in minimum essential medium with Earle's salts and non-essential amino acids, supplemented with 2 mM GlutaMAX, 50 U/ml penicillin-G, 50 μg/ml streptomycin sulphate, and 10% fetal calf serum (Invitrogen) in a humidified atmosphere (95% air-5% CO2) at 37°C. Cells cultured in flasks were used with 70–80% confluence. Caco-2 cells were seeded on transwells for 24-well plates (2.5×105/well, 6.5 mm diameter, 3 μm pore size, Corning B.V. Life Sciences) and cultured for 18–21 days. Transepithelial resistance (TER) was monitored using a Millicell-ERS Volt-Ohm metre (Millipore). Transwells with TER>250Ω*cm2 were used. After baseline TER was measured, recombinant human IL-33 (R&D system) was added to the apical and basolateral sides for 72 h and TER was monitored. IL-33 or PBS-treated mice were orally gavaged with 600 mg/kg of FITC-dextran (at 10 ml/kg H2O; Sigma). After 4 h, serum was collected through terminal exsanguination and the fluorescent intensity of each sample was measured using a spectrophotometer.
Recombinant IL-33 and anti-ST2 blocking antibody
DSS-treated mice were injected intraperitoneally with 0.5 µg of recombinant murine IL-33 on days 1, 3 and 5 (aa 109–266, <1.0 EU/μg as determined by the LAL method; R&D system) or for permeability assays in vivo, with 1 μg/mouse rmIL-33 daily for 7 days (Novartis). ST2 neutralisation was performed by giving 50 µg per mouse of anti-ST2 antibody (kind gift from Dr Dirk Smith, Amgen) or isotype control (rat IgG1) on days 1, 3, 5 and 7 post-treatment by DSS.
Statistical analysis
The non-parametric Kruskal-Wallis test with Dunn's multiple comparison test or the parametric one-way ANOVA test with Bonferroni's multiple comparison test were used (GraphPad Software). All data are presented as mean±SEM. *, p<0.05; **, p<0.01; ***, p<0.001.
Acknowledgments
We are grateful to Sven Bolliger, Céline Cojean, Marc Le Bert, Giulia Ruzzante, Melanie Sachs, Devika Sanmugalingam, Grazyna Wieczorek and Anne Delanoye-Crespin for excellent technical support. We thank Matt Butler for expert advice on experiments and manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
- Data supplement 3 - Online table 3
Footnotes
MAKS, MP and JRM contributed equally.
TJ, MC, J-PG and BR share senior authorship.
-
Correction notice This article has been corrected since it was published Online First. The 4th and 10th author affiliations have been updated.
-
Contributors MAKS, MP, JRM, BF, NO, SN, KM, MB, LB, EL, TG, MLB and MC conducted experiments. All authors were involved in data discussion. LPB, GC and KM were involved in patients’ studies. ST KO mice were provided by PGF. VQ, TJ, MC, JPG, BR designed and supervised the overall study. TJ, MC, JPG, BR prepared and edited the final manuscript and figures.
-
Funding The study was conducted with the support of European grants FEDER (M Chamaillard, V Quesniaux, B Ryffel), Région Nord/Pas-de-Calais (M Chamaillard), Région Centre and FRM allergy (V Quesniaux, B Ryffel), Ligue Nationale contre le Cancer (‘Equipe Labellisée Ligue 2009’ to JP Girard), Associatoin pour la Recherche contre le Cancer Fondation ARC (‘Programme ARC 2011’ to JP Girard) and Fondation pour la Recherche Médicale (‘Equipe FRM 2009’ to M Chamaillard).
-
Competing interests JRM, DS and TJ are employees of Novartis Pharma AG.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Ethics approval Any necessary ethics committee approval was secured for the study reported in mice and humans.