Article Text

Abstract
Objective No Crohn’s disease (CD) molecular maker has advanced to clinical use, and independent lines of evidence support a central role of the gut microbial community in CD. Here we explore the feasibility of extracting bacterial protein signals relevant to CD, by interrogating myriads of intestinal bacterial proteomes from a small number of patients and healthy controls.
Design We first developed and validated a workflow—including extraction of microbial communities, two-dimensional difference gel electrophoresis (2D-DIGE), and LC-MS/MS—to discover protein signals from CD-associated gut microbial communities. Then we used selected reaction monitoring (SRM) to confirm a set of candidates. In parallel, we used 16S rRNA gene sequencing for an integrated analysis of gut ecosystem structure and functions.
Results Our 2D-DIGE-based discovery approach revealed an imbalance of intestinal bacterial functions in CD. Many proteins, largely derived from Bacteroides species, were over-represented, while under-represented proteins were mostly from Firmicutes and some Prevotella members. Most overabundant proteins could be confirmed using SRM. They correspond to functions allowing opportunistic pathogens to colonise the mucus layers, breach the host barriers and invade the mucosae, which could still be aggravated by decreased host-derived pancreatic zymogen granule membrane protein GP2 in CD patients. Moreover, although the abundance of most protein groups reflected that of related bacterial populations, we found a specific independent regulation of bacteria-derived cell envelope proteins.
Conclusions This study provides the first evidence that quantifiable bacterial protein signals are associated with CD, which can have a profound impact on future molecular diagnosis.
- Crohn's Disease
- Enteric Bacterial Microflora
- Inflammatory Bowel Disease
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
There are unmet needs for diagnosis, treatment and patient monitoring in Crohn’s disease (CD).
-
No molecular marker has yet advanced to clinical use in CD.
-
The intestinal microbiota is recognised as an essential contributor to disease initiation and perpetuation and, therefore, represents an enormous reservoir for the discovery of novel signatures that could be used as biomarkers and predictors for different disease phenotypes or stages.
What are the new findings?
-
The feasibility of extracting bacterial protein signals relevant to CD by interrogating myriads of intestinal bacteria, even from a small number of subjects.
-
Twelve bacterial protein signals and one human protein signal (glycoprotein 2 of zymogen granule membranes, GP2) were robustly quantified by targeted MS-based proteomics, without the need for antibodies and ELISA testing. All of them make sense in the context of our understanding of CD.
-
Increased IgA at the surface of microbial cells of CD patients coincides with the over-representation of various bacterial proteins with a high immunogenic potential in CD patients.
-
Decreased GP2 at the surface of microbial cells of CD patients may favour adhesion of bacteria to the mucosa and then promote inflammation.
How might it impact on clinical practice in the foreseeable future?
-
Using meta-proteome-wide association studies, we point out new potential biomarkers in CD.
Introduction
Independent lines of evidence converge to suggest a central role of the gut microbial community in Crohn’s disease (CD): microbiota is required for the development of inflammation in genetically predisposed colitis animal models,1 reinfusion of luminal contents after ileal resection rapidly produces recurrent disease in CD patients,2 antibiotics delay postoperative recurrence of CD,3 and the hitherto identified susceptibility polymorphisms contribute or relate to bacterial sensing through innate and adaptive immune pathways.4–6 Finally, a vicious circle of ‘mutualism breakdown’ has been postulated, where the host does not tolerate its own microbiota any longer, and inflammation can favour the selection of aggressive symbionts.7 However, the micro-organisms, or microbial products that signal the disruption of gut homeostasis and may have a critical role in CD, are unknown. Their identification remains a significant challenge due to the high complexity of the intestinal microbiota, forming the most densely populated microbial community in the body. It is composed of 1013–1014 micro-organisms belonging to about a thousand different species, most of them anaerobic,8 and is hitherto largely uncultivable (only 20–30% of enteric bacterial species have been propagated in pure culture). Our group9–11 and others12–15 have used diverse culture-independent approaches based on molecular profiling of 16S rRNA genes to compare the microbial diversity of intestinal or faecal samples from CD patients and healthy people. Despite the biases inherent to different methodological approaches, varying sampling sites (faeces or mucosa along the intestine), heterogeneity in clinical phenotypes, and variable statistical power, the overall consensus is that the diversity of dominant bacterial species is reduced in CD, notably among members of the Firmicutes phylum.
Largescale metagenomic sequencing (as in MetaHit and the Human Microbiome Project), which analyses whole genomic DNA directly extracted from human intestinal communities, offers a new dimension for the characterisation of these communities from a functional point of view, and represents an enormous reservoir for the discovery of novel signatures that could be used as biomarkers and predictors for different disease phenotypes or stages.16–18 Beyond functional metagenomics, metaproteomic studies will reveal the true expression of metabolic and cellular functions that govern physiology, become disrupted in disease, and can have a profound impact on molecular diagnosis. Therefore, while there are unmet needs for diagnosis, treatment and patient monitoring in CD, especially while no molecular maker has advanced to clinical use in CD,19 and considering that the intestinal microbiota is recognised as an essential contributor to disease initiation and perpetuation, we here demonstrate the feasibility of discovering and validating a range of CD-associated bacterial proteins by using a proteomic approach from discovery to validation. With protein profiling providing assays closer to activated functions, such metaproteome-wide association studies have the potential to become an important tool in modern medicine, and could answer major yet unmet clinical needs.
Methods
Subjects and samples
We conducted a cross-sectional study including six patients with CD (four women and two men, aged 26 through 41 years) and six healthy controls (HC) matched for age, sex and tobacco use (table 1). Patients were followed and selected in the gastroenterology unit of the Saint-Antoine Hospital (Paris). We made a conscious selection of different phenotypes to avoid an unnaturally uniform patient population for this pilot study. Exclusion criteria, however, were active disease with a Harvey–Bradshaw score >5, and any use of antibiotics within the preceding 2 months. The control group comprised healthy volunteers with neither symptoms nor a family history of gastrointestinal disease, and with no use of medication. All participants gave informed consent to the protocol that was approved by the ethics committee of the hospital.
Gender and age of matched participants and clinical characteristics of Crohn's disease patients at the time of stool collection
Every participant was asked to provide a fresh stool sample collected at home in a Stomacher 400 plastic bag (Seward Medical), which was left open in a one-litre hermetic plastic box containing a catalyst (Anaerocult, Merck, Darmstadt, Germany) to generate anaerobic conditions. This faecal material was maintained in a coolbox and transferred within 2 h into an anaerobic chamber (90% N2, 5% H2 and 5% CO2) for processing. We had verified in preliminary assays, that measurements at a single time point gave a reliable picture of individual metaproteomes, which showed little variation over time (see online supplementary figure S1).
Preparation of bacterial fractions and diversity profiling
Given the high complexity of faecal samples that contain bacterial, dietary and host proteins, we first extracted bacterial communities, to focus on the collected bacterial proteomes. Bacterial fractions were extracted in duplicate, at low temperature and in an anaerobic atmosphere (see online supplementary method 1, supplementary figure S2), from freshly collected stool specimens. The final bacterial pellets, as well as 150 mg stool aliquots, were kept at −80°C until further analyses. Diversity profiling was performed by 16S rRNA gene pyrosequencing (see online supplementary method 2).
Discovery of CD-associated gut microbial proteins using 2D-DIGE/LC-MSMS
We used two-dimensional differential gel electrophoresis (2D-DIGE), coupled with tandem mass spectrometry (MS/MS) and searches against metagenomic databases (MetaHit) as a non-targeted comprehensive approach to discovering CD-associated proteins. Briefly, microbial fractions were extracted in duplicate for the 12 participants leading to 24 samples, which were analysed in a dye-swap design comprising 12 gels in total (see online supplementary table S1 and supplementary method 3). For differential expression analysis, we applied two complementary methods, both established and commonly used in microarray gene expression analysis: a hierarchical analysis of variance (ANOVA) (false discovery rate (FDR) <10%) and an empirical Bayes moderated single-group t test per gene (FDR <10%). They represent different approaches to the challenge of comparing small sets of samples for thousands of variables (see online supplementary method 4).21 ,22 The complementary candidate lists were combined to yield a set of protein spots identified by at least one of the methods as showing significant differences between CD and HC.
For protein identification, nine gels (see online supplementary table S1) were poststained with SYPRO Ruby (BioRad), and spots of interest were robotically excised under computer-assisted visual control. In-gel trypsin digestion and LC-MS/MS analyses are detailed in online supplementary method 5. Finally, we used the X!TandemPipeline to identify and group the differentially expressed proteins (see online supplementary method 6).
Validation of CD-associated candidate proteins using selected reaction monitoring (SRM)-based targeted proteomics
A targeted LC-SRM assay was developed to validate a subset of 13 candidate proteins discovered in the original 2D-DIGE non-targeted comprehensive survey. The subset of proteins was defined by choosing the ones containing at least two specific peptides for a protein or a group of proteins with identical function in phylogenetically close bacterial strains, and that at the same time, had already been identified in previous label-free shotgun experiments run on equivalent samples, preferentially without prefractionation. Details on sample preparation, the SRM-assay development, the list of transitions, chromatographic and acquisition conditions, data processing and statistical analysis of the quantitative datasets using MSstats,23 are given in online supplementary method 7. The 284 optimised transitions measured for the 13/46 targeted proteins/peptides are detailed in online supplementary table S2.
Other general statistical analyses are detailed in online supplementary method 8.
Results
Pyrosequencing and quality of the microbial extracts
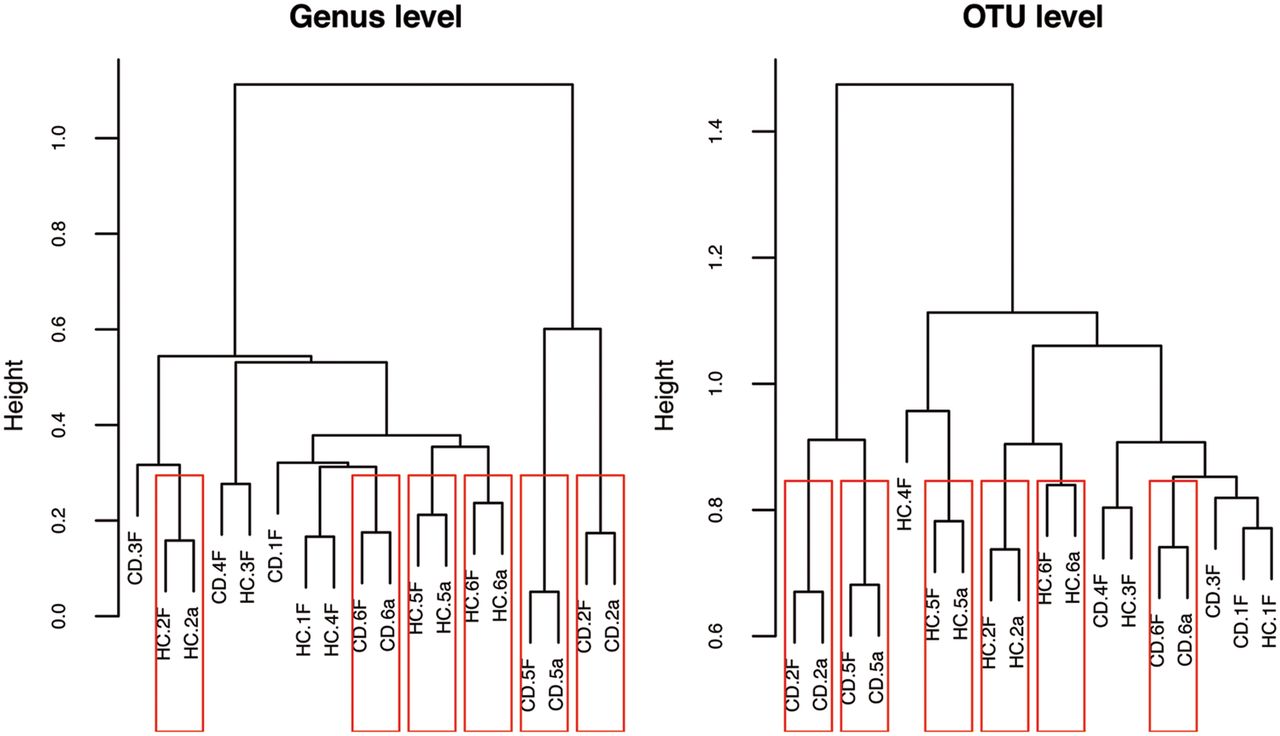
As illustrated by the dendrogram produced by hierarchical clustering of the 16S rRNA pyrosequencing data at the genus and operational taxonomic unit (OTU) levels (figure 1), microbial extracts were closely related to the corresponding stool total 16S rRNA. This illustrates the ability of our extraction method to preserve the microbial diversity observed in the raw sample material. On the other hand, samples did not cluster by clinical diagnosis, CD, or HC, based on their 16S rRNA gene profile alone. This highlights the need for a complementary proteomics viewpoint.

Structure of all crude samples and half the corresponding extracted microbial pellets profiled by 16S rRNA gene pyrosequencing. Hierarchical clustering of the 16S rRNA gene pyrosequencing dataset (at genus and OTU level) showed a high similarity between population structure of crude samples and those of the corresponding microbial extracts, but did not allow distinguishing between clinical status, CD or HC. HC.1 to HC.6 and CD.1 to CD.6 denote HC and CD patients, respectively; suffix letters, F and a, denote native faeces and bacterial cell extracts, respectively. CD, Crohn's disease; HC, healthy controls.
Discovery of protein signatures of CD-associated gut microbiota by 2D-DIGE
The electrophoretic profile was well conserved across individual samples, and the internal standard (see online supplementary figure S3 and magnified regions thereof in online supplementary figure S4), making it possible to accurately compare spot volumes across the entire experiment. After image alignment and spot co-detection, 2007 protein spots were validated and simultaneously quantified in all 36 images derived from measuring three image channels for each of the 12 gels. There were no pronounced systematic differences between biological replicates (samples from a clinical group), and most protein spots (93%) were unchanged between patients and controls allowing reliable normalisation of the data (see online supplementary figure S5). A cluster tree based on the pairwise distances between 2D-DIGE profiles is shown in figure 2. Microbial fractions prepared in duplicate from the same stool specimen always clustered together, reflecting good technical reproducibility, and pairs of duplicates tended to cluster by clinical status, indicating a clinically relevant strong signal. A list of 141 candidate spots (7% of total, 53 increased and 88 decreased in CD patients) was obtained by hierarchical ANOVA or empirical Bayes moderated single-group t test, and all visible selected spots were repeatedly excised from nine SYPRO Ruby poststained gels for LC-MS/MS-based identification (see online supplementary table S1). Eighty-nine spots were found to contain bacterial proteins from a single functional category, and which could be attributed to a defined bacterial subpopulation, with most proteins being from Bacteroides/Parabacteroides species, or Prevotella species, or members of the order Clostridiales (see online supplementary table S3). For robust reporting, however, only a subset of 59 spots were retained (30 increased and 29 decreased in CD patients), which could be identified independently in several gels containing different patient-control pairs (see online supplementary figure S6 for the sequential spot selection process, and see online supplementary table S3 for lists of proteins and peptides). Human proteins were identified in five additional spots with differential signal. Results are summarised in the heat map of figure 3, showing the normalised volumes of those 59 bacterial and 5 human protein spots.

Cluster tree based on the pairwise distances between 2D-DIGE profiles. Similarities between patterns (normalised volumes of 2007 spots) were assessed by unsupervised hierarchical clustering. HC.1 to HC.6 and CD.1 to CD.6 denote HC and CD patients, respectively; g01–12 denote gel numbers. Microbial fractions prepared in duplicate from the same stool specimen always clustered together, reflecting good technical reproducibility of our method, and pairs of duplicates tended to cluster by clinical status, CD or HC, indicating a clear clinically relevant signal in the proteomics data. CD, Crohn's disease; HC, healthy controls; 2D-DIGE, two-dimensional difference gel electrophoresis.

Cluster heat map constructed from the normalised volumes of spots with significant different intensities between HC and CD patients, and that could be robustly identified. Spot numbers and meaningful names for the associated functions are in the right margin. Similarities between patterns are visualised by unsupervised hierarchical clustering. HC.1 to HC.6 and CD.1 to CD.6 denote HC and CD patients, respectively; g01–12 denote gel numbers. Since all spot variables were centred at the mean (the mean has been subtracted to each value), the new mean for each spot variable is now at 0, making half the values negative as indicated in the colour key. Blue and red tones therefore signify under-represented and over-represented, respectively. Proteins highlighted in yellow in the right margin are those that were chosen for SRM validation. As different forms of the same protein (typically TonB-dependent receptors of Bacteroides) may occur in different spots, the number of highlighted spots exceeds 13. Proteins annoted ‘surface’, ‘TonB’, ‘OMP’ and ‘lipoprotein’ in the right margin, may be grouped into ‘cell envelope proteins’ in the text when several categories are concerned, including proteins of unknown function that have specific features known to be characteristic of cell envelope localisation. CD, Crohn's disease; HC, healthy controls; SRM, selected reaction monitoring.
Of the 30 bacterial protein spots which were increased in CD patients, 25 were from the phylum Bacteroidetes, essentially Bacteroides species, three were from the phylum Firmicutes, order Clostridiales, and two were from the phylum Proteobacteria (see the lower half of figure 3). Human proteins IgA immunoglobulins and carboxypeptidase A1 were identified in two additional spots that we found to be over-represented in CD patients (lower half of figure 3). Proteins that were identified in these spots are reported in table 2, where they are organised according to their lineage and function. Of the 29 bacterial protein spots which were decreased in CD patients, 18 were from the phylum Firmicutes, invariably Clostridiales whenever order or lower phylogenic affiliation could be determined, three were from Prevotella species, and three others from undefined Bacteroidales members, one was from Escherichia coli, and four from unknown bacteria (see the upper half of figure 3). Another interesting result was the presence of fragments of human GP2 (pancreatic glycoprotein 2 of zymogen granule membranes) in three under-represented protein spots (upper half of figure 3). Proteins that were identified in these spots are listed in table 3 with their lineage and function.
List of proteins that were discovered to be over-represented in CD, using the without a priori 2D-DIGE approach
List of proteins that were discovered to be under-represented in CD, using the without a priori 2D-DIGE approach
Validation of protein signatures of CD-associated gut microbiota by SRM
A subset of 13 proteins (highlighted in yellow on figure 3) found to be differentially abundant between CD and HC on the basis of 2D-DIGE were selected to be validated using a targeted LC-SRM assay. Totally, 46 peptides were chosen and 284 transitions were finely optimised using heavy isotope-labelled synthetic peptides spiked into a sample pool in order to allow the precise relative quantification of the 13 candidate proteins in the sample cohort without further sample fractionation other than a stacking gel (see online supplementary table S2). Thus, all 13 proteins could be unambiguously detected with 2–6 specific peptides in single injections of the total bacterial protein extracts. Results are summarised in figure 4 representing the fold change value (differential expression) and the adjusted p value for each targeted protein from the triplicate analyses of the six CD versus six HC individual samples. Details on individual peptide quantification are given in online supplementary table S4. The differential expression of all candidates detected in the discovery experiment was validated and fold changes spanning 14–28 were detected with a very high confidence.23
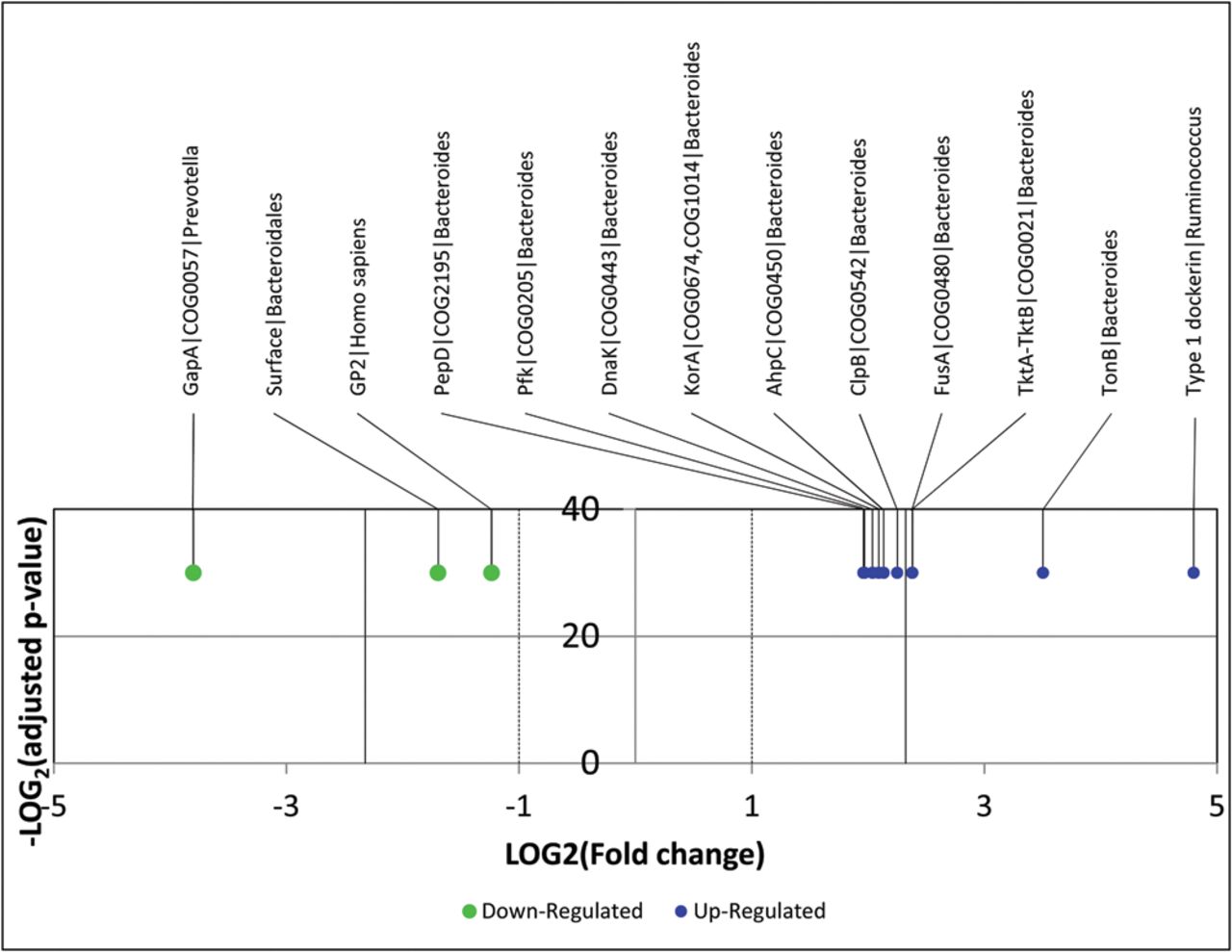
Volcano plot representing results of the LC-SRM assays on the 13 targeted proteins. The logarithmic fold changes (CD vs HC) are plotted against negative logarithmic adjusted p values calculated with the R package MSstats and performed from triplicate injections.23 All targeted proteins were found to be either upregulated or downregulated in CD patients compared with controls, and the results validated all candidates identified in the discovery experiments. CD, Crohn's disease; HC, healthy controls; SRM, selected reaction monitoring.
Clearly, we could validate in CD patients a significant elevation of Bacteroides proteins that participate in the protection against oxidative stress (AhpC), in protein synthesis, folding and repair (FusA, DnaK and ClpB), in energy saving, and the maintenance of a high carbon flux within both glycolysis and pentose phosphate pathways (PPi-dependent PfK and TktA-TktB), in the biosynthesis of precursors through the reductive branch of the tricarboxylic acid cycle (KorA), in nutrient acquisition and sensing of the environment (TonB), and in adhesion and colonisation (PepD), while some of these proteins (DnaK, AhpC and TonB-dependent receptors) are recognised for their strong immunogenic properties. We also confirmed elevation of type 1 dockerin from members of the family Ruminococcaceae, in CD patients. Other confirmed proteins included the glycolytic enzyme GapA of Prevotella, a cell surface protein of undefined Bacteroidales members, and the human protein GP2, which were all depleted in CD patients (figure 4).
Correlating functional shifts with the structure of the bacterial community
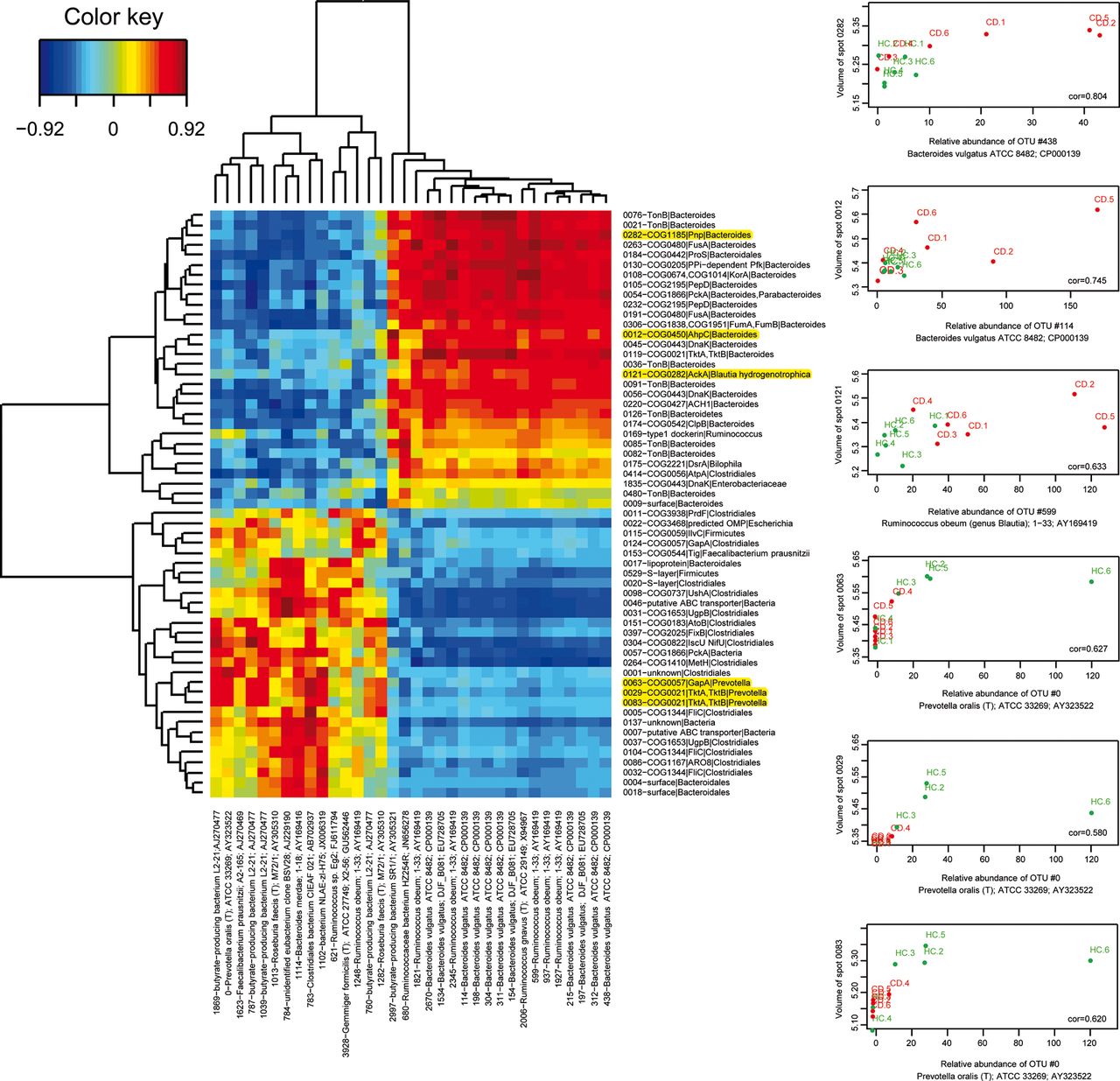
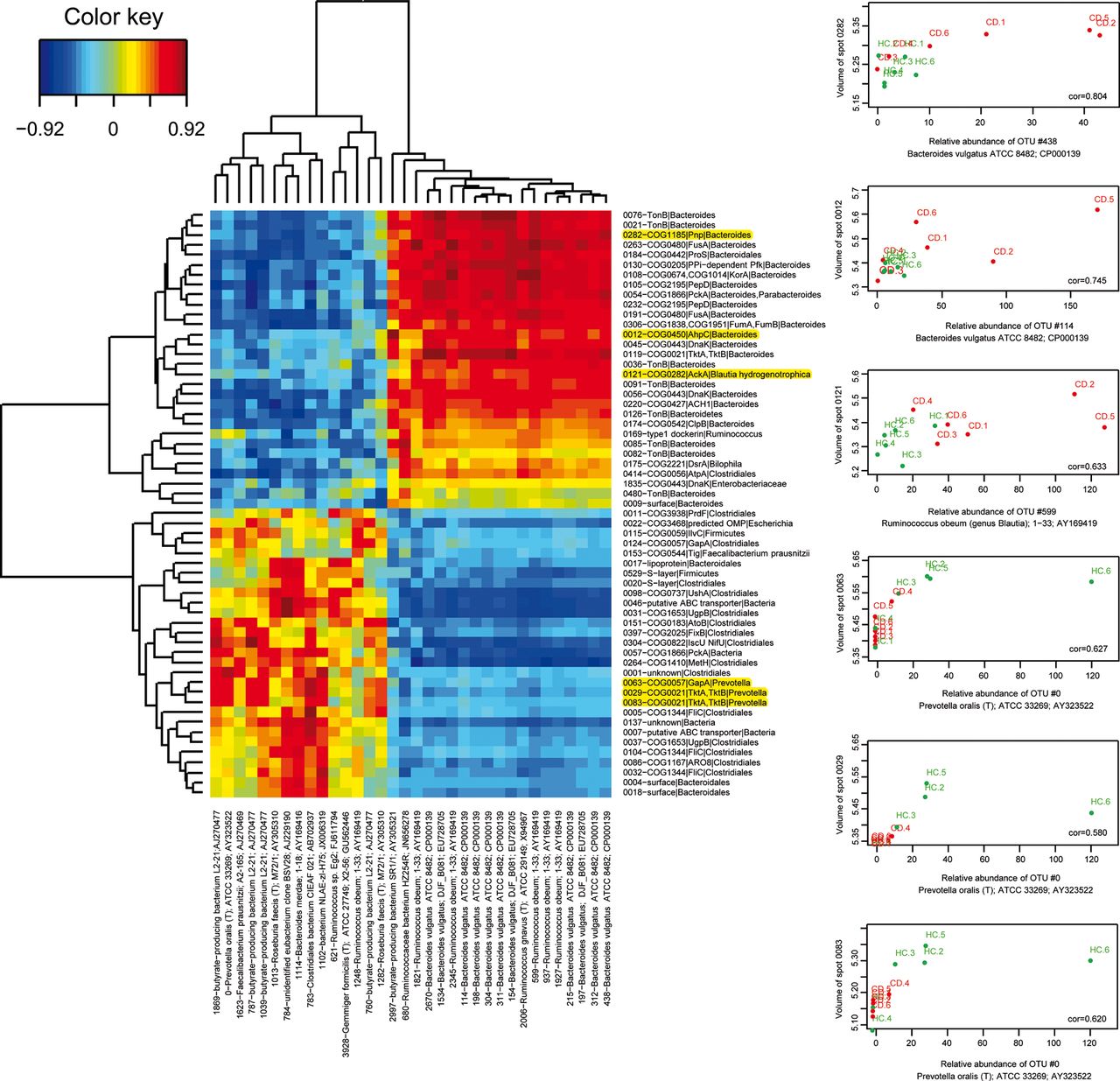
We then investigated the question whether the imbalance in bacterial protein abundance that we observed in CD corresponded to changes in gene expression in a stable bacterial community, or whether they reflected a remodelling of the population structure, or whether both events could have occurred. OTU richness estimated by the bias-corrected Chao 1 richness estimator, was significantly lower (p=0.015) in the CD group (840 OTUs, SD 259) compared with the HC group (1371 OTUs, SD 286), but the α diversity Simpson index did not significantly differ between the two groups. This means that a lower number of species was present, which was more evenly distributed in the CD group. Specific traits of CD microbiota are illustrated by figure 5. Thirty-four OTUs varied or tended to vary in abundance (see online supplementary table S5). Those that were increased in CD were related to Bacteroides vulgatus and Ruminococcus obeum (genus Blautia) and interestingly included one OTU similar to the potentially anti-inflammatory butyrate-producing bacterium SR1/1, while those that were decreased in CD were related to the butyrate-producing bacterium L2-21, to Roseburia faecis (T) M72/1, Faecalibacterium prausnitzii A2-165, or other clostridial members, and also included one OTU most similar to Prevotella oralis. The heat map of figure 6 shows a positive correlation between the abundance of most of the varying protein groups and the abundance of the related varying OTUs. There were, however, a number of interesting exceptions suggesting additional effects at work, for instance a subset of TonB proteins attributed to Bacteroides members that were increased in CD patients independently of a modulation of the corresponding bacterial populations (see the green-yellow bands on the right middle part of figure 6).

Box plots of the relative abundances of faecal bacterial populations found by 454 pyrosequencing. Differences between Crohn's patients (□) and HCs (□) at the different phylogenetic levels were considered *significant for p≤0.05, and (*)tendencies were reported up to p≤0.10 (‘glm’ with the ‘quasibinomial family’). Specific traits of CD microbiota were significantly increased abundances of members in the lineage Betaproteobacteria-Burkhoderiales-Alcaligenaceae, a tendency towards increased abundances of Bacteroidaceae-Bacteroides and Blautia, significantly lower numbers of Roseburia, and a tendency towards lower numbers of Alphaproteobacteria, Prevotellaceae-Prevotella and Oscillospira. On the other hand, inter-individual variability was higher in CD patients, which is in agreement with heterogeneity of CD. CD, Crohn's disease; HCs, healthy controls.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Heat map of the correlation matrix between abundance of the varying protein groups and the abundance of the related varying OTUs (left panel). Red and hotter orange tones indicate a positive correlation between abundance of a protein group and abundance of the corresponding OTUs; green and yellow tones indicate absence of correlation as for example, TonB proteins and other uncharacterised surface proteins of Bacteroides in spots 0082, 0480 and 0009, and DnaK of Enterobacteriaceae in spot 1835 (right middle part of the image). Activities that were found to be increased and decreased in CD patients fell in the upper and lower halves of the image, respectively. OTUs that were found to be increased and decreased in abundance in CD patients fell into the right and left halves of the image, respectively. Examples of positive correlations are detailed on right panel and highlighted in yellow on panel A. CD, Crohn's disease.
Discussion
The present work is a clear demonstration that environmental proteomics of gut microbes can provide molecular signatures of IBDs, becoming a powerful complementary tool for their study and, ultimately, their diagnosis and treatment. So far, long lists of candidate biomarker proteins, notably in oncology, remarkably never progressed from research discovery to clinical application because de novo development of multiple ELISA would have been prohibitive in time and money. SRM, by contrast, offers a valuable alternative to antibody-based validations, as it allows the accurate and robust multiplexed quantification of a range of proteins in complex samples without the need for antibodies and ELISA testing. This technology has, for instance, recently been used successfully for the verification of diagnostic and prognostic cancer biomarkers.24 ,25 In the present study, we first developed and validated a workflow for the discovery of protein signals specific to CD-associated gut microbial communities without any a priori assumption of the metabolic and/or cellular functions that can accompany CD. Then we developed a SRM assay to confirm a set of candidates, as de novo development of ELISA assays would not have been feasible in a reasonable timeframe and poorly adapted to multiple verifications.
Simple unsupervised hierarchical clustering of samples based on their 2D-DIGE profiles showed that five pairs of replicates out of six within each group, CD and HC, already clustered together, indicating a clear signal, whereas, hierarchical clustering based on 16S rRNA gene pyrosequencing (at the genus and OTU levels) did not allow a distinction between clinical status. This illustrates well that the metaproteomic approach is a powerful tool for highlighting functional imbalances even in the absence of clear major shifts in the dominant bacterial groups or species. The 2D-DIGE strategy further solved the specific question of detecting differences between groups, a problem that faces even greater challenges in label-free shotgun metaproteomics.26–31 A set of proteins from members of the Bacteroidetes phylum, largely Bacteroides species, were over-represented in CD microbiota. By contrast, under-represented proteins were mostly from Clostridiales (Firmicutes phylum), and more rarely from Prevotella and undefined Bacteroidales members. Functions that we found to be increased in the Bacteroides/Bacteroidales population from CD microbiota included proteins corresponding, in general, to functions related to strategic adaptation for survival in challenging environments. For instance, DnaKs and other chaperones, such as ClpB homologues, have a defensive role against oxidative, nitrosative, nutritional, osmotic and pH stresses that are likely to occur in the gut of CD patients.32–35 AhpCs represent another important defence mechanism to cope with reactive nitrogen intermediates and reactive oxygen species.36 ,37 An over-representation of proteins involved in the binding and import of nutrients (TonB-dependent receptors and other cell envelope proteins) could be related to an increased need for carbon substrates and/or for micronutrients to fuel increased central metabolism in Bacteroidales.38 ,39 Consistent with this, a set of key enzymes that maximise the energy yield from monosaccharide catabolism in Bacteroides (PPi-dependent Pfk, PEP-carboxykinase PckA and fumarate hydratase FumA-FumB) were also over-represented in CD-associated microbial proteomes.40 Finally, a higher abundance of PepD could contribute to increased surface colonisation by Bacteroides members observed in CD patients.41–44 Remarkably, the overexpression of 10 of these proteins was reliably confirmed by quantitative SRM measurements in unfractionated bacterial protein extracts. Therefore, we reach an attractive hypothesis, that a number of Bacteroidales members, essentially Bacteroides species, might be adapted or promoted under environmental conditions specific to the gut of CD patients, and that a set of overabundant bacterial proteins that can be quantified by SRM, could be regarded as bacterial signatures for CD. Moreover, all of them make sense in the context of our understanding of CD. Indeed, a number of these proteins have been proposed as major traits for allowing opportunistic pathogens, including Bacteroides fragilis, to colonise mucus layers, breach host barriers, and invade the mucosae. For instance, DnaK and AhpC, which are usually intracellular proteins, may also be found in the outer membrane where they bind human plasminogen and enhance its conversion into plasmin by host activators, a scenario which might promote colonisation and host invasion.45 DnaK proteins exhibit strong immunostimulatory properties both at the level of the innate and adaptative immune system, and can, moreover, promote the processing and presentation of other bacterial or food antigens by chaperoning them.46 AphC of several bacteria also demonstrate immunogenic properties as assessed by high titres of seric antibodies against purified/recombinant targets or whole proteome maps,47 and the β-barrel domains of TonB-dependent receptors are suspected to play an important role in the virulence of Gram-negative bacteria, exposing epitopes on the bacterial surface.48 Consistent with this and with a previous report,49 we found an over-representation of microbiota-associated secretory IgA in CD patients. These results encourage inspection of the immunome of the intestinal microbiota to capture those strongly IgA-coated bacteria as well as a set of derived antigenic peptides that could be used to map and distinguish specific antibody profiles in subgroups of IBD patients, and thus facilitate appropriate therapeutic options, evaluation of treatment efficacy, and long-term follow-up.
Conversely, many members of the order Clostridiales and some of the genus Prevotella appeared unable to meet the ecological challenge imposed in CD patients, as judged from the decrease in abundance of key proteins involved in diverse cellular and biochemical functions within this population. For instance, 25 different flagellin FliC proteins attributed to Clostridiales members were identified in three under-represented spots, while common enteric flagellins are proposed as major targets of the CD-associated aberrant immune response.50 The under-representation of trigger factor Tig attributed to Faecalibacterium prausnitzii further suggests that this numerically dominant and potentially anti-inflammatory subgroup,51 might fail to sustain efficient protein synthesis in CD patients. Finally, under-representation of key enzymes, notably GapA and TktA-TktB, attributed to Prevotella members could reflect the inability of this subpopulation in maintaining the flux of carbon within both glycolysis and pentose phosphate pathways. Interestingly, the CD-associated under-representation of GapA from Prevotella was confirmed by SRM. Consistent with this, we found that the relative abundances of members of the lineage Prevotellaceae-Prevotella tended to decrease in CD patients, and that one OTU assigned to Prevotella was under-represented in CD.
While sequencing of the 16S rRNA-encoding genes revealed many positive correlations between the abundances of the varying protein groups and the abundances of the related varying OTUs, it also highlighted a number of unexpected interesting deviations. In particular, some of the TonB-dependent receptors and other uncharacterised proteins localised in bacterial cell envelopes of Bacteroides species were increased in CD patients independently of a modulation of the corresponding bacterial populations. Therefore, findings in the present study clearly point to functional changes beyond what can be explained by a mere shift in populations, and extend earlier observations of possible dissociations between structural and functional changes in obese individuals29 ,30 or CD patients.31 The fact that completely different approaches independently identified Bacteroides membrane proteins as over-represented in CD,31 and moreover, that this was confirmed here by SRM, also gives strong support for regarding these proteins as possible relevant bacterial signatures for CD, and highlights the need for future work focusing on microbial cell envelopes in health and disease, inasmuch as this subcellular fraction constitutes the first line of interaction with the host.
Our study also provides the first and unexpected clue towards under-representation of intestinal microbiota-associated GP2 in CD patients. This was confirmed by SRM, and may favour adhesion of bacteria to the mucosa, and then promote inflammation. Indeed, it has been demonstrated recently that recombinant human GP2 binds Escherichia coli Type I fimbriae, a bacterial adhesin commonly expressed by members of the intestinal microbiota. A role in host defence has been proposed in which GP2 may serve as a physical barrier that prevents bacteria from binding to host cell receptors.52 Consistent with this, a higher bacterial biomass on the mucosa, and an adherent mucosal biofilm enriched with Bacteroides fragilis were shown to be prominent features in IBD patients.43 Finally, the question arises as to whether decreased GP2 binding to bacteria might be related to increased anti-GP2 titres reported in CD patients.53
In conclusion, our metaproteomics approach spanning discovery to confirmation demonstrates, for the first time, the feasibility of extracting bacterial protein signals relevant to CD, by interrogating myriads of intestinal bacteria, even from a small number of patients and HCs. We provide an initial list of CD-associated microbial proteins extracted from a typical group of patients, which could represent major common features in CD patients. The next step should be to validate the specificity and sensitivity of bacterial protein signals either in individual clinical trials with well-defined and homogenous CD populations, or in a comprehensive study with a larger heterogeneous patient cohort. Given that effects are harder to detect in smaller samples, one can expect that even more subtle differences could be detected in larger cohorts, and that inclusion and accurate quantification of additional predefined sets of proteins could be used to refine recognition of IBD entities in the very near future. The extraction and robust quantification of bacterial protein signals is also a way to identify disrupted protein networks that drive onset and perpetuation of CD and, therefore, candidate targets for IBD treatment based on gut-ecological intervention strategies.
Acknowledgments
We are grateful to Bertrand Nicolas for his help in the preparation of figures.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online methods
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
- Data supplement 5 - Online table 3
- Data supplement 6 - Online table 4
- Data supplement 7 - Online table 5
Footnotes
-
Contributors CJ, DPK, CC, HS, PS, LB, JD: conception and design of the study; CB, AG, SV, CC, SM, PL, FB, FL, WC, VL, NP: acquisition of data; CJ, DPK, CB, AG, SV, CC, SM, FB, WC, VL, NP: analysis and interpretation of data; CJ, DPK, CB, SV, CC: drafting of the manuscript; DPK, AG, CC, HS, PS, J-FG, SDE, AVD, JD, BV, PM, VM: critical revision of the manuscript for important intellectual content; CJ, DPK, SV, CC, OD, BS: statistical analysis; CB, AG: technical or material support; CJ, HS, PS, LB: study supervision.
-
Funding The Boku Chair of Bioinformatics acknowledges funding by the Vienna Science and Technology Fund (WWTF), Baxter AG, Austrian Research Centres Seibersdorf, and the Austrian Centre of Biopharmaceutical Technology. We acknowledge the ‘Fondation pour la Recherche Médicale’ for funding the triple quadrupole instrument for targeted proteomics experiments.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was conducted with the approval of the Ethics Committee of the St Antoine hospital, Paris.
-
Provenance and peer review Not commissioned; externally peer reviewed.