Article Text

Abstract
Background Oesophageal cancer is one of the most deadly forms of cancer worldwide. Long non-coding RNAs (lncRNAs) are often found to have important regulatory roles.
Objective To assess the lncRNA expression profile of oesophageal squamous cell carcinoma (OSCC) and identify prognosis-related lncRNAs.
Method LncRNA expression profiles were studied by microarray in paired tumour and normal tissues from 119 patients with OSCC and validated by qRT-PCR. The 119 patients were divided randomly into training (n=60) and test (n=59) groups. A prognostic signature was developed from the training group using a random Forest supervised classification algorithm and a nearest shrunken centroid algorithm, then validated in a test group and further, in an independent cohort (n=60). The independence of the signature in survival prediction was evaluated by multivariable Cox regression analysis.
Results LncRNAs showed significantly altered expression in OSCC tissues. From the training group, we identified a three-lncRNA signature (including the lncRNAs ENST00000435885.1, XLOC_013014 and ENST00000547963.1) which classified the patients into two groups with significantly different overall survival (median survival 19.2 months vs >60 months, p<0.0001). The signature was applied to the test group (median survival 21.5 months vs >60 months, p=0.0030) and independent cohort (median survival 25.8 months vs >48 months, p=0.0187) and showed similar prognostic values in both. Multivariable Cox regression analysis showed that the signature was an independent prognostic factor for patients with OSCC. Stratified analysis suggested that the signature was prognostic within clinical stages.
Conclusions Our results suggest that the three-lncRNA signature is a new biomarker for the prognosis of patients with OSCC, enabling more accurate prediction of survival.
- Oesophageal Cancer
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
-
What is already known about this subject?
-
Long non-coding RNAs (lncRNAs) have important regulatory roles in cancer formation and development.
-
Some lncRNAs have been found to be associated with the survival of patients of various cancers.
-
The tumour node metastasis staging system which relies on anatomical and pathological features has limitations in the prognosis of patients with oesophageal squamous cell carcinoma (OSCC).
-
In many cancers, miRNA and mRNA prognostic signatures, which robustly predict the survival of patients, have been identified, but whether the lncRNA signature might also predict survival of patients with cancer remains unknown.
-
What are the new findings?
-
LncRNA expression profile in OSCC tissues is profoundly different from that in normal oesophageal epithelial tissues.
-
A three-lncRNA signature was identified which can reliably predict the survival of patients with OSCC.
-
Like mRNAs and miRNAs, the lncRNA signature could be used as a biomarker for the prognosis of patients with cancer.
-
How might it impact on clinical practice in the foreseeable future?
-
The lncRNA signature might help to predict the survival of patients with OSCC more accurately in clinical practice than previously possible.
Introduction
Oesophageal cancer ranks as the world's sixth most deadly cancer.1 It has two major histological types: adenocarcinoma and squamous cell carcinoma (OSCC). In China, over 90% of the cases of oesophageal cancer are OSCC, which is the fourth most prevalent cancer of the country.2 OSCC is a highly aggressive malignancy with poor prognosis. Better understanding of the genetic and molecular disorders of the disease is the key to early diagnosis, appropriate treatment and improved prognosis of patients with OSCC.
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides not translated into proteins.3 ,4 In recent years, lncRNAs have attracted increasing scientific interest and are believed to be implicated in diverse biological processes,5 by promoting or repressing transcription,6 or by acting as modulators of mRNA translation.7 LncRNAs affect the transcription of numerous genes located throughout the genome,6 the regulatory mechanisms being diverse and complex. Some lncRNAs regulate the transcription of nearby genes in cis, while others act in trans. Some lncRNAs regulate transcription through epigenetic pathways, while others interact directly with RNA polymerases or transcription factors.8 The well-known lncRNA HOTAIR is overexpressed in breast cancer where it induces genome-wide retargeting of polycomb repressive complex 2 (PRC2).9 This results in altered histone H3K27 methylation and gene expression, which further promotes cancer invasiveness and metastasis.9 A large number of human lncRNAs have been identified, but their characteristics and functions remain largely unknown.10
An increasing number of studies have suggested deregulation of lncRNAs in cancers,9 ,11 ,12 and reports on lncRNA expression profiles in specific cancers are beginning to be published. Studies on lncRNA expression profiles in five pairs of liver cancer and normal tissues,13 six pairs of renal clear cell carcinoma and corresponding normal tissues,14 and one glioblastoma tissue with one normal brain tissue from an age-matched donor15 found large numbers of lncRNAs significantly deregulated in cancer tissues. A clear understanding of the alterations in lncRNA expression occurring in cancers will require larger-scale studies than those yet reported and as far as we know, our study is the first to employ more than 100 sample pairs. Microarray assay is a popular and reliable method of profiling lncRNA expression. Compared with RNA sequencing, microarray has the advantages of low cost, ‘lower technical variation and better detection sensitivity for low-abundance transcripts’ and the ability to quantify antisense single-exon lncRNAs.16
For most solid cancers, including OSCC, clinical stage of the cancer is still the main predictor of survival for patients who have received surgery, but it does not provide an accurate prediction. Cancers are heterogeneous at the molecular and genetic levels,17 ,18 and patients of the same stage and who have received similar treatment, may nonetheless have quite different clinical outcomes. A number of studies have shown that messenger RNAs (mRNAs) and microRNAs (miRNAs) can be powerful predictors of survival in patients with cancer, particularly those mRNA or miRNA signatures consisting of multiple markers.19 ,20 However, up to now, whether an lncRNA signature might have similar prognostic power to that of mRNA and miRNA signatures for patients with cancer is not known.
This study reports the first examination of lncRNA expression profiles in paired tumour and normal tissues in a large cohort of more than 100 patients with OSCC. We identified a three-lncRNA signature with the ability to predict the overall survival of patients with OSCC and validated its prognostic value in an independent cohort of 60 patients.
Patients and methods
Patients and samples
We retrospectively collected paired cancer and adjacent normal tissues from 119 patients with OSCC with follow-up information (minimum of 5 years) and examined the lncRNA expression profile of the tissues by microarray analysis. All patients had surgically proven primary OSCC and received oesophagectomy (R0 resection) at the Cancer Institute and Hospital of the Chinese Academy of Medical Sciences (CAMS) between December 2005 and December 2007. Samples were obtained with informed consent. To validate the prognostic signature, we enrolled an independent cohort of 60 patients with OSCC who underwent surgery at the Cancer Institute and Hospital, CAMS between January 2008 and December 2008 and examined the lncRNA expression level of their paired tumour and normal tissues using the same microarray assay as used for the original 119 patients. Details of the patient enrolment procedure are given in online supplementary methods and figure S1; clinical and pathological information of the patients is shown in online supplementary table S1. The study was approved by the medical ethics committee of the Cancer Institute and Hospital, CAMS.
RNA extraction, amplification, labelling and array hybridisation
Total RNA was first extracted from the tumour and normal tissues (see online supplementary methods) and used to produce labelled cDNA (see online supplementary methods). Array hybridisation using the labelled cDNA was performed in a CapitalBio BioMixerTM II hybridisation station (see online supplementary methods).
All the experimental procedures were done blinded to the clinical and pathological information and to the survival information of the patients.
Microarray processing and statistical analysis
LncRNA expression profiling was performed using the Agilent human lncRNA+mRNA array V.2.0 platform. After a filtering procedure, 8900 human lncRNAs (annotated by GENCODE(V13) database, lincRNAs from Cabili et al,21 and the University of California Santa Cruz database) were selected for the following analysis (see online supplementary methods). First, quantile normalisation of the microarray data (containing the 8900 lncRNAs and all mRNAs in the microarray) of all 119 paired tumour–normal samples was carried out. Then, the data was log 2-scale transformed. Missing values were imputed using the random Forest unsupervised classification algorithm (see online supplementary methods). The data of the 60 sample pairs in the independent cohort were processed independently in the same way.
Hierarchical clustering of the lncRNA profiles was performed using cluster 3.0.22 The normalised expression values of the lncRNAs were centred on the median before performing unsupervised hierarchical clustering. Clustering was done with complete linkage and centred Pearson correlation.
On the whole, lncRNAs have lower expression level than mRNAs. The average expression level of lncRNAs (after quantile normalisation and log 2 transformation) for the 119 paired tumour-normal samples was 5.93, while that of mRNAs was 10.19. In this study, we were only concerned with the lncRNAs with high and median expression values. LncRNAs with average expression value lower than five in both tumour and normal tissues of the 119 patients were deleted. Further, lncRNAs with invariable expression level (coefficient of variance <0.03) in 119 paired tissues were also filtered out. Finally, 4874 lncRNAs were left for further analysis.
For prognostic signature analysis, the 119 patients were first assigned into groups with good (47 patients) or poor prognosis (72 patients) according to an expected survival time of >5 or <5 years. They were then randomly divided into a training set (n=60) and a test set (n=59) using the random_shuffle function from C++ standard template library.
The 909 lncRNAs differentially expressed between tumour and normal tissues with absolute fold change >2 (false discovery rate adjusted p value of Student's t test <0.10 for all) in the 60 patients of the training set were selected from the 4874 lncRNAs (figure 1A,B). To reduce the influence of heterogeneity among different patients, the expression level of tumour minus normal was used for the following analysis.

Identification of the long non-coding RNA (lncRNA) signature in the training set. (A) After microarray processing, the microarray data was described by an 60×8900 matrix with a ‘good’ or ‘poor’ label column. (B) After two filtering procedures, 909 lncRNAs remained for further analysis. (C) Selection process for the nine lncRNAs with highest classification power for patient survival. A random Forest supervised classification algorithm was used to narrow down the number of lncRNAs by several iterative steps, in which one-third of the least important lncRNAs were discarded at each step according to their importance score. (D) Development of prognostic classifier for all combinations (N=29−1=511) of the nine lncRNAs using the nearest shrunken centroid algorithm. Vg and Vp are the mean expression profiles of the lncRNA combination (g1 g3 g4 g6) for good-prognostic samples and poor-prognostic samples, respectively. Vi is the expression profile of sample i. The Euclid distances d(Vi,Vg) and d(Vi,Vp) are used to classify sample i into a low- or high-risk group. (E) The procedure for identifying the final signature. The accuracies of all 511 signatures were calculated and the nine highest accuracies for k=1, 2, …, 9 are shown in the plot. The signature containing three lncRNAs was selected as the final signature.
Using random Forest supervised classification algorithm, nine lncRNAs mostly related to the prognostic classification were selected among the 909 lncRNAs (figure 1C) according to the permutation important score by the software Random Jungle (see online supplementary methods).23
There were 29−1=511 combinations of the nine lncRNAs and we developed a signature for each combination from the training set using the nearest shrunken centroid algorithm. For each combination, two centroids (‘good’ and ‘poor’) were created using the mean gene expression profile of the lncRNAs based on the patients with good prognosis and those with poor prognosis, respectively. Then, the Euclid distances between all samples and the two centroids were calculated. If dig<dip (dig is the Euclid distance between sample i and the centroid ‘good’, dip is that between sample i and the centroid ‘bad’), sample i was predicted as ‘good’ (low-risk group); otherwise predicted as ‘poor’ (high-risk group) (figure 1D).
After the construction of all 511 signatures, we compared their classification accuracies in the training set. Because the sample size was not balanced between the ‘good’ and ‘poor’ groups, the classification accuracy was defined as the average of classification accuracy of the group with good prognosis and that of the group with poor prognosis. First, for signatures constructed by specific number of lncRNAs (k=1, 2, …, 9), the one with the highest classification accuracy was selected for each k (figure 1E). One of these selected signatures was then defined as the final signature, considering a balance between classification accuracy and the number of lncRNAs.
Quantitative RT-PCR
Quantitative RT-PCR (qRT-PCR) was performed to validate the microarray results. The reverse transcription reactions were carried out with reverse transcriptase (SuperScript III, Invitrogen) and quantitative PCR reactions were then performed on ABI 7900 (see online supplementary methods and supplementary table S2).
Results
LncRNA expression profiles display significant differences between OSCC tissues and adjacent normal tissues
We first compared the lncRNA expression profiles of OSCC tissues and adjacent normal tissues using unsupervised hierarchical clustering in 119 patients. In total, 6389 lncRNAs with a coefficient of variance >0.10 were selected from the 8900 lncRNAs for clustering analysis. Hierarchical clustering of these 6389 lncRNAs based on centred Pearson correlation clearly separated OSCC tissues from normal tissues (figure 2). Only 12 samples (six tumour samples and six normal samples) were misclassified by the clustering analysis. Among all the lncRNAs, 799 showed at least a twofold change in the OSCC tissues compared with the normal tissues (355 being upregulated and 444 downregulated).
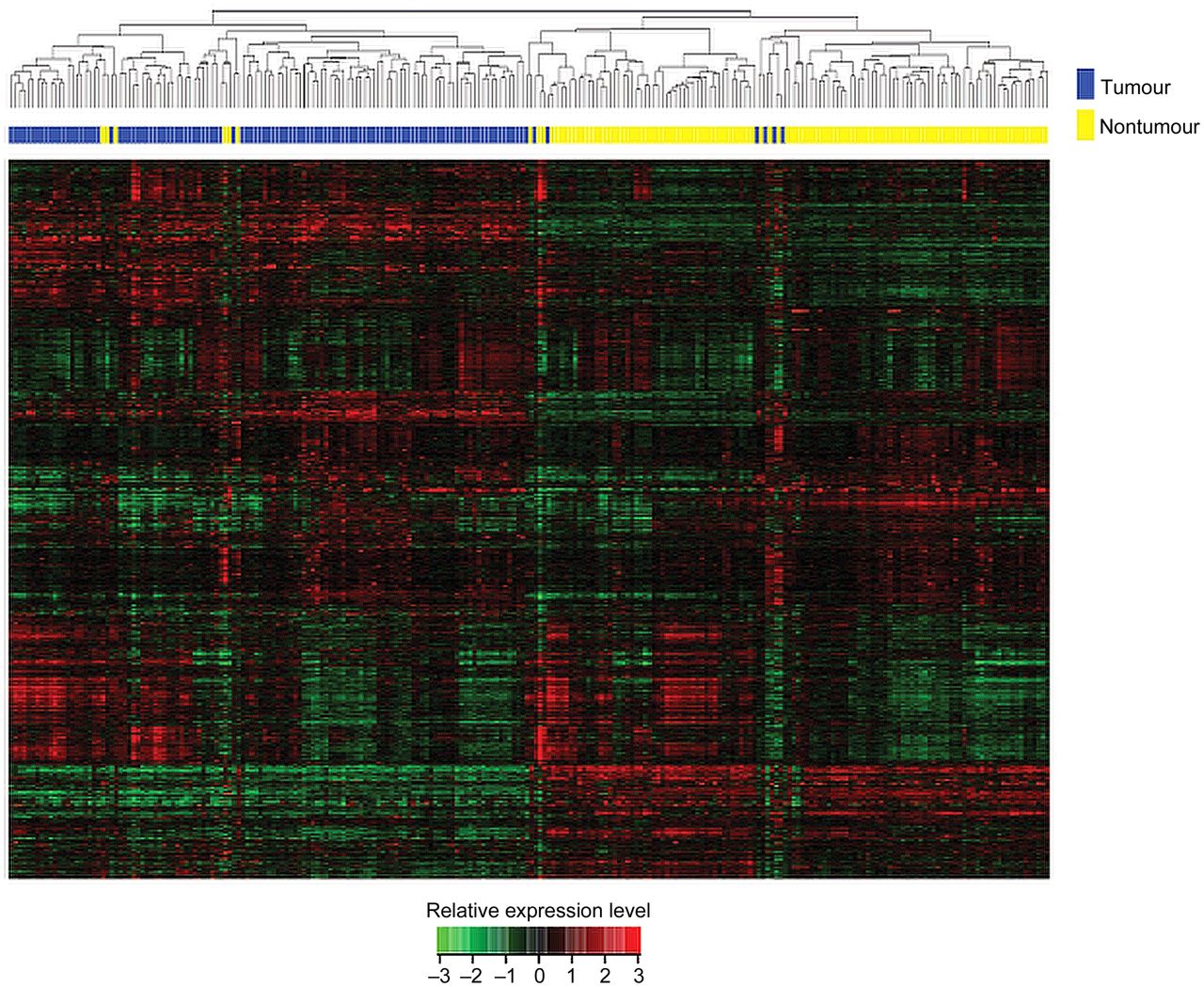
Unsupervised hierarchical clustering of the 119 pairs of tissues. The normalised expression data of the 6389 lncRNAs with coefficient of variance >0.10 was used for clustering analysis. Hierarchical clustering clearly separated tumour (blue bar) and normal (yellow bar) samples. Only six tumour samples and six normal samples were misclassified.
Derivation of a three-lncRNA prognostic signature from the training set
We next explored the association between lncRNA expression and the overall survival of patients with OSCC. A three-lncRNA signature including ENST00000435885.1, XLOC_013014 (annotated by Cabili et al21) and ENST00000547963.1) was selected from the training set considering a balance between accuracy and the number of lncRNAs (figure 1E). The expression level of the three lncRNAs measured by microarray was verified by qRT-PCR (see online supplementary results and supplementary figure S2). In this signature, the ‘good’ and ‘poor’ centroids were (−2.11, −1.35, 3.38) and (−0.57, −2.50, 2.38), which represented the average expression level of the three lncRNAs for the patients with good and poor prognosis, respectively. The signature was defined as follows:

 where
where  denoted the expression level of ENST00000435885.1, XLOC_013014, ENST00000547963.1 for sample i, respectively. A patient was classified as ‘low risk’ if dig<dip according to the patient's three-lncRNA expression value and as ‘high risk’ if not.
denoted the expression level of ENST00000435885.1, XLOC_013014, ENST00000547963.1 for sample i, respectively. A patient was classified as ‘low risk’ if dig<dip according to the patient's three-lncRNA expression value and as ‘high risk’ if not.
A three-lncRNA signature predicts survival of patients with OSCC
With the three-lncRNA signature, patients of the training group were divided into a high-risk group (n=33) or a low-risk group (n=27). Patients with the high-risk signature had significantly shorter overall survival than those with the low-risk signature (median survival 19.2 months vs >60 months, p<0.0001) (figure 3A,D). There was no significant difference in clinical and pathological characteristics between high- and low-risk group patients (table 1).
Clinical and pathological characteristics of patients with OSCC with high- or low-risk lncRNA signature in the three datasets

The three-lncRNA signature predicts overall survival of patients with OSCC. Heat maps (A–C) of the relative expression level (tumour minus normal) after z-score transformation for each lncRNA, and Kaplan–Meier survival curves (D–F) of patients classified into high- and low-risk groups using the three-lncRNA signature. p Values were calculated by log-rank test. (A, D) Training set, 60 patients. (B, E) Test set, 59 patients. (C, F) Independent cohort, 60 patients. OSCC, oesophageal squamous cell carcinoma.
The three-lncRNA signature was then tested for its prognostic value in the test group of 59 patients. The same model and criteria as those derived from the training group classified 25 and 34 patients of the test group into the high-risk and low-risk groups, respectively. As in the training group, the overall survival time of the high-risk group patients was significantly shorter than that of low-risk group patients (median survival 21.5 months vs >60 months, p=0.0030) (figure 3B,E). The two groups of patients differed significantly in N stage (p=0.0290), tumour node metastasis (TNM) stage (p=0.0378) and arrhythmia (p=0.0055), but not in other clinical and pathological factors (table 1).
To validate the prognostic value of the three-lncRNA signature, we used the lncRNA expression values and survival data of an independent cohort of 60 patients. The patients of the independent cohort were classified as high-risk (37 patients) or low-risk (23 patients) according to their three-lncRNA signature (median survival 25.8 months vs >48 months, p=0.0187) (figure 3C,F). The two groups of patients did not differ significantly in clinical and pathological characteristics (table 1).
Survival prediction by the three-lncRNA signature is independent of clinical and pathological factors
To assess whether the survival prediction ability of the three-lncRNA signature is independent of other clinical or pathological factors of the patients with OSCC, multivariable Cox regression analysis was performed using a stepwise variable selection method. Selected covariables included age, sex, tobacco use, alcohol use, tumour location, tumour grade, T stage, N stage, TNM stage, postoperative complications, adjuvant therapy and the lncRNA signature. Because adjuvant therapy information was missing for some of the patients, we used the multiple imputation method of Markov chain Monte Carlo to impute the missing value of adjuvant therapy in the Cox regression analysis (see details in online supplementary methods and supplementary table S3).24–,26 The results from the training set showed that the high-risk three-lncRNA signature (HR=8.486, 95% CI 3.550 to 20.284, p<0.0001), older age (HR=2.366, 95% CI 1.191 to 4.701, p=0.0140) and postoperative anastomotic leak (HR=5.805, 95% CI 1.605 to 21.000, p=0.0073) was significantly correlated with poor overall survival of the patients with OSCC (table 2). Combined test and independent datasets showed that the three-lncRNA signature (HR=2.203, 95% CI 1.330 to 3.649, p=0.0022), adjuvant therapy (HR=2.328, 95% CI 1.299 to 4.172, p=0.0045) and age (HR=1.674, 95% CI 1.033 to 2.713, p=0.0365) were independent prognostic factors for patients with OSCC (table 2). The results of the multivariable Cox regression analysis thus indicated that the predictive ability of the three-lncRNA signature is independent of other clinical and pathological factors for the survival of patients with OSCC.
Univariable and multivariable Cox regression analysis of the lncRNA signature and survival in the training set (n=60) and in the combined test and independent cohort (n=119)
The three-lncRNA signature has prognostic value within clinical stages
We next carried out a stratified analysis in TNM stage II and III patients to evaluate whether the three-lncRNA signature could predict survival of patients within the same clinical stage. Log-rank test of stage II patients in both the training group (p<0.0001, figure 4A) and the combination of test and independent cohort (p=0.0257, figure 4B) showed that the signature could classify stage II patients with OSCC into high- and low-risk groups. For patients with stage III OSCC, the three-lncRNA signature showed similar prognostic value in the training (p=0.0104, figure 4C) and the combined test and independent (p=0.0105, figure 4D) datasets. Because of limited sample size (n=10), the stratified analysis was not performed for stage I patients.

Survival prediction in stage II and III patients. Kaplan–Meier survival curves of stage II and III patients with OSCC classified into high- and low-risk groups based on the three-lncRNA signature. (A) Stage II patients, training set (n=22). (B) Stage II patients, combined test set and independent cohort (n=55). (C) Stage III patients, training set (n=36). (D) Stage III patients, combined test set and independent cohort (n=56). OSCC, oesophageal squamous cell carcinoma.
Survival prediction power: comparison of TNM stage and the three-lncRNA signature
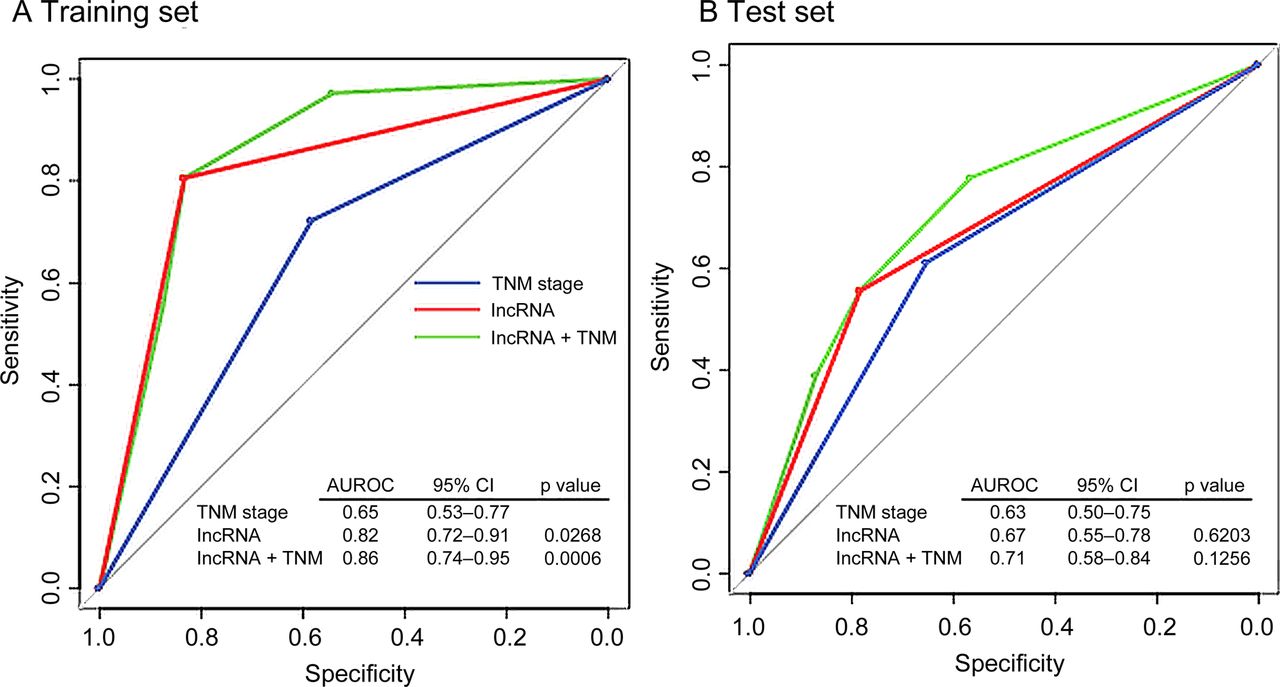
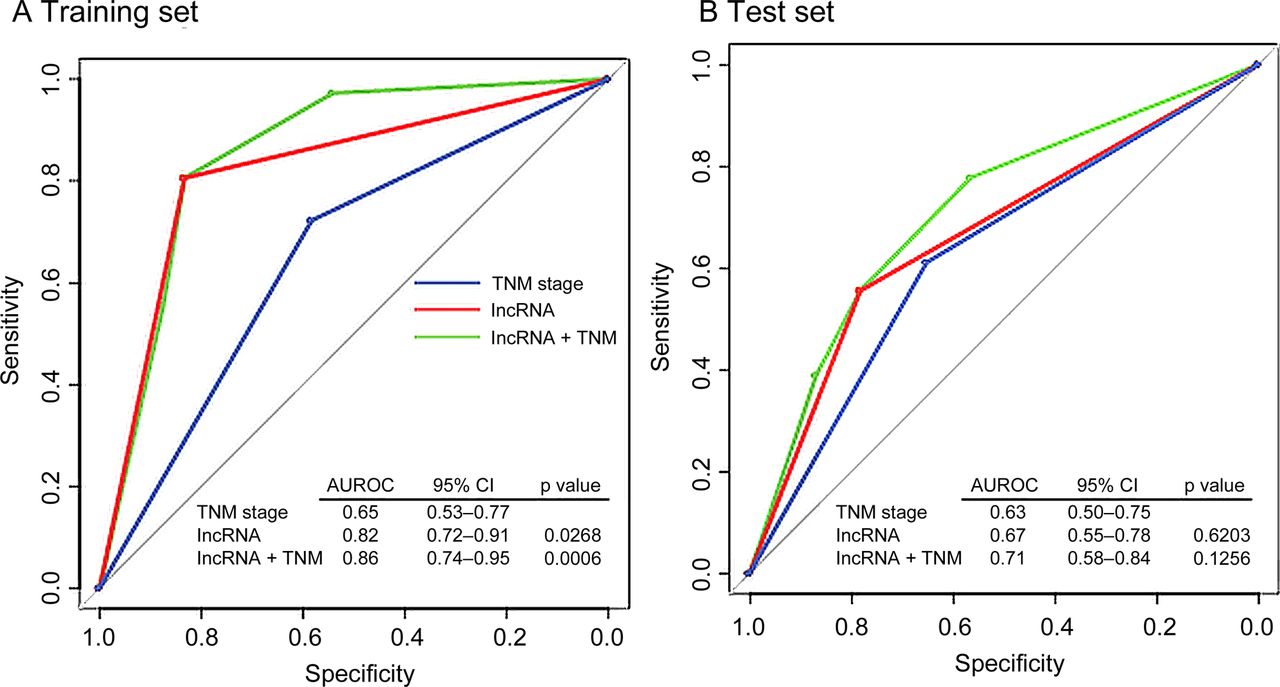
To compare the sensitivity and specificity in survival prediction between TNM stage and the three-lncRNA signature, we performed receiver operating characteristic (ROC) analysis (see online supplementary methods).20 We also constructed a prognostic model combining the two factors and compared the predictive ability. In the training set, predictive ability of both three-lncRNA signature and the combined model were significantly better than TNM stage alone (p=0.0268, p=0.0006, respectively, figure 5A). In the test set, no significantly different predictive ability between the TNM stage and the signature was found. The combined model had a higher area under the ROC curve than the TNM stage (0.71 vs 0.63, figure 5B); however, the difference was not significant (p=0.1256), probably owing to limited sample size. ROC analysis was not performed for the independent cohort because the follow-up period of these patients was <5 years.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of sensitivity and specificity for survival prediction by the three-lncRNA signature, TNM stage and combination of the two factors. The three receiver operating characteristics (ROC) curves in the training set (A) and test set (B). p Values show the area under the ROC (AUROC) of TNM stage versus the AUROC of the three-lncRNA signature, or the combination of signature and TNM. TNM, tumour node metastasis.
All three lncRNAs of the signature are essential for its prognostic value
To confirm that all of the three lncRNAs of the signature are required for its prognostic value, we constructed all possible ‘signatures’ containing from one to three lncRNAs (a total of seven signatures). The prognostic value of all signatures with fewer than three lncRNAs was evaluated by log-rank test in the training, test and independent datasets and compared with the original three lncRNA signature. The comparison showed that none of the signatures with fewer than three lncRNAs was consistently associated with patient survival in all three groups of patients (see online supplementary table S4). This indicates that all three lncRNAs are essential for the prognostic power of the signature.
Functional enrichment analysis of genes correlated with the signature lncRNAs
We next sought to explore the potential role of the lncRNAs of the prognostic signature in OSCC tumorigenesis and development. For this purpose, we examined the correlation between their expression values and those of the mRNAs in the original group of 119 patients and summarised the genes correlated with the three lncRNAs. The expression level of 292 protein coding genes was positively correlated (Pearson correlation coefficient >0.60) with that of at least one of the three signature lncRNAs. The 292 genes clustered most significantly in ectoderm development and epithelial cell differentiation in gene ontology (GO) biological process enrichment analysis27 ,28 (see online supplementary table S5). The same analysis of the 1572 genes negatively correlated with at least one of the three signature lncRNAs (Pearson correlation coefficient <−0.40) returned GO term cell cycle regulation and ubiquitin-protein ligase activity regulation (see online supplementary table S6). These results suggest that the lncRNAs of the signature may positively regulate genes which affect the development and differentiation of oesophageal epithelial cells and repress genes which affect cell cycle and ubiquitin-protein ligase activity.
Discussion
In this study, we examined the lncRNA profiles of OSCC tissues and paired adjacent normal tissues and identified a three-lncRNA signature which was closely related to the prognosis of patients with OSCC. The prognostic value of this signature was verified in the test set of 59 patients and in an independent cohort of 60 patients.
In recent years, an increasing number of lncRNAs have been identified and associations between lncRNAs and various diseases have been reported.29 The roles of lncRNAs in cancer development are increasingly being studied.9 ,30 ,31 However, the involvement of lncRNAs in OSCC has not been reported. Here, we present the first report on differential lncRNA expression in a cohort of 119 patients with OSCC. Through an analysis of tumour and normal tissues, we found that many lncRNAs were differently expressed in OSCC tissues compared with adjacent normal tissues, indicating that lncRNAs may have critical roles in OSCC tumorigenesis.
Our finding of a three-lncRNA signature in OSCC suggests that lncRNAs can be powerful predictors for survival of patients with cancer. The correlation of lncRNA expression levels with the prognosis of patients with cancer has recently been reported for several malignancies, such as hepatocellular carcinoma,13 breast cancer9 and colorectal cancer.30 In our study, the three-lncRNA signature identified in the training group showed similar prognostic value in both the test group and the independent cohort. Thus, we believe that the prognostic power of the signature has a solid basis in patients with OSCC. This is a pioneering study of the association between lncRNA expression and the survival of patients with cancer. Our findings are important because we show that lncRNA has a similar prognostic power to those of mRNA or miRNA for patients with cancer. Moreover, according to Du and colleagues in their recent report, the function of lncRNAs is more closely associated with their expression level compared with mRNAs as they do not encode proteins.16
For the statistical analysis of high-throughput biological data, the ‘curse-of-dimensionality’ problem (small sample size combined with a very large number of genes) is very common. In this work, we tried to reduce the effects of the ‘curse-of-dimensionality’ problem. At first, 909 lncRNAs differentially expressed between tumour and normal samples were filtered out and then subjected to random Forest supervised classification in order to further narrow down the number of lncRNAs associated with prognosis. The random sampling and ensemble strategies used in random Forest classification enable it to achieve accurate predictions while running efficiently on ‘curse-of-dimensionality’ datasets. In random Forest classification, the measures of gene importance are used to filter the original gene set iteratively, resulting in good performance in feature selection.
After the feature selection procedure, we constructed a classifier for each combination of the nine selected lncRNAs using the nearest shrunken centroid algorithm. In this study, we compared the performances of k-lncRNA signatures in the training set for all k=1,2,…,9 and the best accuracies for each k were listed. As shown in figure 1E, the accuracies were similar for k ≥ 3—between 81.3% and 84.7%. Although the signature with k=4 had the highest accuracy, we found that one lncRNA in the signature was redundant (see online supplementary results). Also the prognostic classification and performance of the four-lncRNA and three-lncRNA signatures were similar (see online supplementary results). Thus for the above reasons and the rule of Occam's razor, the signature with k=3 was selected as the final signature.
The current TNM staging system has critical limitations in predicting the survival of patients with OSCC. Thus molecular markers are needed to assist doctors in clinical practice. In the stratified analysis, the three-lncRNA signature showed prognostic value both in stage II and stage III patients. The three-lncRNA signature can classify patients of the same TNM stage into high- and low-risk groups with significantly different survival prospects, indicating that the signature can improve the accuracy of survival prediction. This finding might help doctors to select high-risk patients for adjuvant therapy in addition to traditional surgery, which can improve the outcome of OSCC.
In this study, we have analysed the prognostic value of the three-lncRNA signature. Whether this signature might be used to predict if adjuvant therapy would be of benefit for patients was not evaluated since accurate and complete information about adjuvant therapy after surgery was not available for some patients. Also, as the lncRNA signature was derived from patients who received R0 resection, whether it has prognostic value in suboptimal R1/R2 patients remains unknown. One limitation of our study is the generalisability of the three-lncRNA signature identified. Although this signature was generated and tested in the largest cohort of patients with OSCC by far and the patients enrolled were from different regions of China, datasets from other institutes and other countries are still necessary to verify its generalisability. Its validity should be further tested in prospective cohorts.
Most lncRNAs are not yet functionally annotated. However, we can infer the possible function of the lncRNAs in OSCC using the mRNA expression data of the same group of patients. Genes whose expression value positively correlated with the three lncRNAs were enriched for the GO biological process term ectoderm development and epithelial cell differentiation, and the negatively correlated genes clustered in cell cycle regulation and ubiquitin-protein ligase activity regulation GO terms. Thus it is a plausible inference that the three lncRNAs associated with survival of patients with OSCC may be involved in the development, differentiation and cell cycle regulation of oesophageal epithelia cells and their deregulation may lead to OSCC tumorigenesis and progress. Some of the ectoderm development and differentiation related genes correlated with the signature lncRNAs have already been reported to have tumour suppressive functions. For instance, ANXA1 gene encodes the Ca2+-dependent phospholipid-binding protein annexin I, which inhibits the cancer related NF-κB signal transduction pathway.32 Another gene clustered into the same GO term, PPL, is also a well-studied gene involved in tumour formation and development. Its protein product periplakin is a component of desmosomes involved in cell–cell junction.33 ,34
In conclusion, our study has shown that the lncRNA expression profile is altered in OSCC tissues compared with normal oesophageal tissues. The three-lncRNA signature we discovered robustly predicts the survival of patients with OSCC. Furthermore, this signature can predict the survival of patients with OSCC within same TNM stages. To our knowledge, it is the first lncRNA signature identified that predicts survival in patients with cancer. Further validation studies in prospective cohorts and in cohorts from different institutions are needed to test the prognostic power of the signature before it is applied clinically. Whether the signature is useful for the prediction of the benefit of adjuvant therapy after surgical resection for patients with OSCC requires study with a sufficient number of patients with clear postoperative adjuvant therapy information.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online figure
- Data supplement 4 - Online table 1
- Data supplement 5 - Online table 2
- Data supplement 6 - Online table 3
- Data supplement 7 - Online table 4
- Data supplement 8 - Online table 5
- Data supplement 9 - Online table 6
Footnotes
-
JL, ZC and LT contributed equally.
-
Contributors JH, RC, JL and ZC: contributed to the concept and design of the study. RC, JL and LT: contributed to interpretation of the data (statistical and computational analysis). JL, ZC and LT: contributed to the writing of the manuscript. JH, GS and ZC: contributed to the review and revision of the manuscript. JL, CZ, YZ, SW, FZ, JS and BZ: contributed to the RNA extraction and array hybridisation. CZ: contributed to the qRT-PCR. SS and XF: contributed to the pathological identification of the samples. MYH, YG, NS and ZL: contributed to the haematoxyloin and eosin staining of the samples. JD, RY, YY, XS and ML: contributed to the collection of clinical and pathological data of the patients. KS, NL, BQ, FT: contributed to the collection of samples. JH is the guarantor of the paper, who accepts full responsibility for the work and the conduct of the study. He has access to the data and controls the decision to publish.
-
Funding The study was supported by National High Technology Research and Development Program of China (2012AA02A502, 2012AA02A503, 2012AA02A207), International Science and Technology Corporation and Exchange Project (2010DFB30650), National Natural Science Foundation of China (81172336, 81101772, 81201856), Beijing Natural Science Foundation (7141011).
-
Competing interests None.
-
Ethics approval Medical ethics committee of the Cancer Institute and Hospital, Chinese Academy of Medical Science.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The microarray original data, processed data and the clinical and pathoogical data of our study have been submitted to the Gene Expression Omnibus with accession number GSE53625.