Article Text

Abstract
At least 250 million people worldwide are chronically infected with HBV, a small hepatotropic DNA virus that replicates through reverse transcription. Chronic infection greatly increases the risk for terminal liver disease. Current therapies rarely achieve a cure due to the refractory nature of an intracellular viral replication intermediate termed covalently closed circular (ccc) DNA. Upon infection, cccDNA is generated as a plasmid-like episome in the host cell nucleus from the protein-linked relaxed circular (RC) DNA genome in incoming virions. Its fundamental role is that as template for all viral RNAs, and in consequence new virions. Biosynthesis of RC-DNA by reverse transcription of the viral pregenomic RNA is now understood in considerable detail, yet conversion of RC-DNA to cccDNA is still obscure, foremostly due to the lack of feasible, cccDNA-dependent assay systems. Conceptual and recent experimental data link cccDNA formation to cellular DNA repair, which is increasingly appreciated as a critical interface between cells and viruses. Together with new in vitro HBV infection systems, based on the identification of the bile acid transporter sodium taurocholate cotransporting polypeptide as an HBV entry receptor, this offers novel opportunities to decipher, and eventually interfere with, formation of the HBV persistence reservoir. After a brief overview of the role of cccDNA in the HBV infectious cycle, this review aims to summarise current knowledge on cccDNA molecular biology, to highlight the experimental restrictions that have hitherto hampered faster progress and to discuss cccDNA as target for new, potentially curative therapies of chronic hepatitis B.
- HEPATITIS B
- DNA DAMAGE
- CHRONIC VIRAL HEPATITIS
- MOLECULAR MECHANISMS
Statistics from Altmetric.com
Introduction
At least 250 million people worldwide are chronically infected with HBV1 and at a greatly increased risk to develop liver fibrosis, cirrhosis and hepatocellular carcinoma, causing an estimated 650 000 deaths per year.2 While an efficient prophylactic vaccine is available,3 current treatments for chronic hepatitis B are limited to type 1 interferons and five approved nucleos(t)ide analogues (NAs), which target the viral polymerase, P protein, a multifunctional reverse transcriptase (see below). Due to severe side effects, only a fraction of patients are eligible for interferon therapy, and <10% of them show a sustained virological response, measured as loss of hepatitis B surface antigen (HBsAg; see below).4 NAs are much better tolerated, and the most potent drugs, entecavir and tenofovir, can reduce viraemia by 5–6 logs, often below detection limit, and with low rates of viral resistance development.5 However, HBsAg clearance is very rare (0–5%) even after prolonged treatment,4 and the frequent viral rebound upon therapy withdrawal indicates a need for lifelong treatment.6 Reactivation can even occur, upon immunosuppression, in patients who resolved an acute HBV infection decades ago,7 indicating that the virus can be immunologically controlled but is not eliminated.
The virological key to this persistence is an intracellular HBV replication intermediate, called covalently closed circular (ccc) DNA, which resides in the nucleus of infected cells as an episomal (ie, non-integrated) plasmid-like molecule that gives rise to progeny virus. A cure of chronic hepatitis B will therefore require elimination of cccDNA. However, despite >30 years of research, little is known about the molecular mechanisms of cccDNA formation and degradation, foremostly due to the lack of suitable experimental systems. Recent discoveries are about to change this situation, particularly the identification of a liver-resident bile acid transporter, sodium taurocholate cotransporting polypeptide (NTCP; also known as SLC10A1), as an entry receptor for HBV and hepatitis delta virus (HDV), which usurps HBV’s envelope to enter cells8 ,9 (box 1). Various aspects of this finding have recently been reviewed.10 ,11
A second key for HBV persistence is a flawed immune response, typically including the functional exhaustion and depletion of cytotoxic T cells, a lack of adequate CD4+ T cell help, and failure to mount neutralising antibodies. While immune restoration will likely be indispensable even if other ways are found to reduce cccDNA,12 for more information readers are referred to pertinent reviews.13–16 The focus here will be on a brief history on cccDNA research and its experimental difficulties, and on recent developments and how they may translate into new, curative treatments for chronic hepatitis B.
HBV infection and replication: a short overview
HBV is the prototypic member of the hepadnaviridae, a family of small enveloped hepatotropic DNA viruses sharing a similar genome organisation and replication strategy. The mammalian animal viruses (orthohepadnaviridae) include, for example, woodchuck hepatitis virus (WHV) and ground squirrel hepatitis virus and, more recently discovered, HBVs of woolly monkeys17 and bats.18 Bird viruses (avihepadnaviridae) include, among others, those of Pekin ducks (duck HBV (DHBV)) and heron HBVs. However, no hepadnavirus has been found in established experimental animals such as mice, rats or chicken.
As shown in figure 1A, HB virions,19 ,20 or ‘Dane particles’21, comprise an outer envelope of the lipid-embedded small (S), middle (M) and large (L) surface proteins (HBsAg in serology) and an inner nucleocapsid (core particle; hepatitis B core antigen in serology) whose icosahedral shell is formed by 120 dimers of the core protein.22–25 Its interior harbours the viral genome as a partially double-stranded, circular but not covalently closed ‘relaxed circular’ (RC) DNA in which the 5′ end of the (-)-strand is covalently linked to the viral P protein26; formation of this unusual structure by protein-primed reverse transcription is outlined below.

Basic molecular features of HBV. (A) Virion structure. Infectious virions consist of an outer envelope with the lipid-embedded surface proteins small (S), middle (M) and large (L), and an icosahedral inner nucleocapsid harbouring the viral genome as a relaxed-circular DNA that is covalently linked to the terminal protein (TP) domain of P protein. (B) Genome organisation. Outer lines denote viral transcripts, arrowheads transcription starts; ε symbolises the RNA encapsidation signal on pregenomic (pg)RNA. The two DNA strands are shown as present in relaxed circular (RC)-DNA to highlight the relative positions of RC-DNA-typical features; however, the viral RNAs encoding the open reading frames (ORFs) depicted in the centre are actually transcribed from covalently closed circular DNA. Note the highly compact organisation with overlapping ORFs and regulatory elements (green arrows, promoters; Enh I/Enh II, transcriptional enhancers; direct repeat (DR)1, DR2, direct repeats; wiggly red line, RNA primer on (+)-DNA). (C) Domain structures of HBV structural proteins. Numbers represent amino acid positions, based on genotype D, subtype ayw. The entry inhibitor Myrcludex B is derived from the N terminal part of the PreS1 domain. The middle surface protein M consists of PreS2 plus S. GAG, glycosaminoglycans; NTCP, sodium taurocholate cotransporting polypeptide; P, POL/RT, DNA polymerase/reverse transcriptase; RH, RNase H; L, large surface protein comprising PreS1, PreS2 and S domain; myr, myristoyl.
The most remarkable features of the genome (figure 1B) are its tiny size (∼3 kb) and extremely compact organisation, with each nucleotide (nt) having coding function in one or even two (overlapping) open reading frames (ORFs); by necessity, all regulatory elements for gene expression and numerous cis-elements for replication overlap with coding information. Notably, avihepadnaviruses lack a functional X protein, which in the mammalian viruses appears essential to establish infection (see below); furthermore, they produce L and S but no M surface protein. The domain structures of HBV’s structural proteins are outlined in figure 1C.
A key feature shared by all hepadnaviruses is their narrow host range, which for HBV essentially comprises humans and the Great Apes. This restriction has greatly hampered functional studies and established the importance of the animal HBVs as more tractable surrogates. WHV in woodchuck is a relevant preclinical model for new HBV antivirals,27 and DHBV has been indispensable in establishing fundamental aspects of hepadnavirus infection and replication.28
Complemented by studies employing transfection with human HBV expression vectors of human hepatoma cell lines, for example, HepG2 and Huh7, that support replication (but are not susceptible to infection), this has resulted in a generalised scheme for the HBV infectious cycle.29 Apart from a few recent HBV-specific findings (NTCP as receptor, likely function of HBV X protein (HBx)) indicated in figure 2, the basics are likely to apply to all hepadnaviruses.

Simplified scheme of the HBV infectious cycle. (A) Attachment and entry. (B) Release of the nucleocapsid into the cytoplasm. (C) Nucleocapsid-mediated nuclear transport and release of P-linked relaxed circular (RC)-DNA at the nuclear pore into the nucleus. (D) RC-DNA to covalently closed circular (ccc)DNA conversion. (E) Transcription of viral RNAs. (F) RNA nuclear export. (G) Translation. (H) Prevention of cccDNA transcriptional silencing by HBV X protein (HBx). (I) Co-packaging of P and pregenomic (pg)RNA into newly forming nucleocapsids. (J) First-strand DNA synthesis (inhibited by nucleos(t)ide analogue (NAs)), pgRNA degradation and second strand DNA synthesis leading to new RC-DNA. (K) Envelopment of mature RC-DNA containing nucleocapsids. (L) Secretion of new virions assisted by components of the multivesicular body (MVB) machinery. (M) Secretion of hepatitis B e antigen (HBeAg) and excess subviral particles (SVP) constituting the bulk of hepatitis B surface antigen (HBsAg). GAG, glycosaminoglycans; L, large; M, middle; NTCP, sodium taurocholate cotransporting polypeptide; S, small.
In brief, HB virions are concentrated on susceptible cells via interaction of an exposed region in the S domain that overlaps with the immunodominant ‘a-determinant,’ bearing most of the HBV neutralising epitopes, and cell surface glycosaminoglycans.30 ,31 Then, a high-affinity interaction between the myristoylated N terminal PreS1 region of the L protein with NTCP, and likely further host factors,32 triggers uptake (likely by endocytosis) of the virion. Notably, the PreS1 domain is also essential for nucleocapsid envelopment, which is accounted for by its dual topology (figure 1A), with one part facing the virion interior and the other the virion surface.33 For the interaction with NTCP, about 50 aa from the PreS1 N terminus and N terminal fatty acylation are sufficient (figure 1C), the basis for entry inhibition by PreS1-derived lipopeptides (‘Myrcludex B’11). Following virion uptake, the RC-DNA containing nucleocapsids are released into the host cell cytoplasm. The process is poorly understood but likely to yield to new in vitro infection systems based on NTCP-transfected hepatoma cells. Exposure of nuclear localisation signals in the Arg-rich C terminal domains (CTDs)34 of the core protein (figure 1C), possibly regulated by CTD phosphorylation and/or completion of the incomplete (+)-strand in RC-DNA, enables transport of the nucleocapsid to the nuclear pore where the capsid shell disintegrates,35 releasing the viral polymerase-bound RC-DNA into the nucleoplasm; there conversion into cccDNA and formation of a nucleosome-bound minichromosome, likely associated with HBx and core protein, occur.36 As on cellular DNA, this provides numerous options for dynamic epigenetic control of cccDNA transcriptional activity, as highly schematically outlined in figure 3. These include DNA modifications such as methylation, repressive and activating post-translational modifications (PTMs) of the histones such as acetylation, methylation, phosphorylation and others,37 nucleosome spacing and likely more recently discovered mechanisms acting via non-coding RNAs38 or replacement of normal histones by specific variants.39 Notably, in the absence of HBx, cccDNA appears to be rapidly silenced, whereas HBx promotes a transcriptionally active state40 that correlates with the presence of some of the known activating histone PTMs.41 ,42 However, the mechanism of HBx-mediated de-silencing is unclear, and the full repertoire of potentially activating versus repressing regulation is largely unexplored, as is the question of whether chromatinisation can already be initiated on RC-DNA. Next, cccDNA exerts its key role as template for host RNA polymerase II-mediated transcription of subgenomic RNAs (for the surface proteins and HBx) and two greater-than-genome-length RNAs, that is, the pregenomic (pg) RNA and the precore RNA that gives rise to the precore protein precursor of the secretory hepatitis B e antigen (HBeAg). All RNAs are 5′ capped and 3′ polyadenylated, like cellular mRNAs. Efficient cccDNA transcription is regulated by liver-specific transcription factors43 yet also depends on HBx,40 which likely counteracts transcriptional silencing of the cccDNA (see below). Following nuclear export, the RNAs are translated. The envelope proteins are directed to the endoplasmic reticulum from where they enter the secretory pathway to yield a huge excess of empty envelopes over complete virions. These subviral particles constitute the bulk of HBsAg and are the basis of the hepatitis B vaccine. Translation of the bicistronic pgRNA yields core protein plus P protein, for DHBV likely by a shunting mechanism that does not seem to apply to the human virus.44
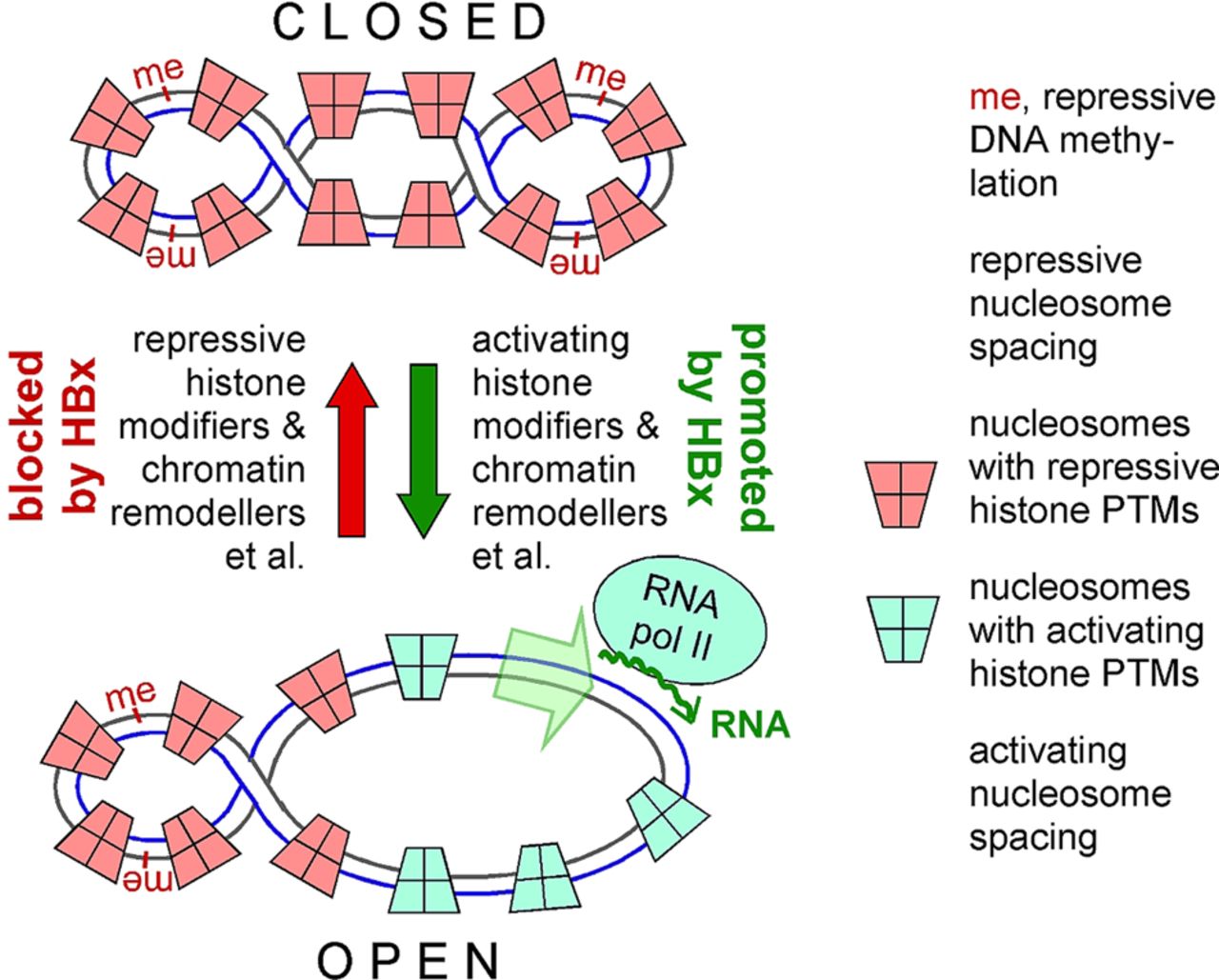
Epigenetics of covalently closed circular (ccc)DNA. Nuclear cccDNA is found as a mini-chromosome, associated with around 15–16 nucleosomes (consisting of the core histones H2A, H2B, H3, H4 plus the linker histone H1) and non-histone proteins, probably including the viral core protein and HBV X protein (HBx). The resulting chromatin architecture, together with potential DNA modifications such as methylation (‘me’), strongly affects accessibility of regulatory elements on the DNA to transcription factors and RNA polymerase and thus can control transcriptional activity. Important factors are nucleosome spacing, itself modifiable by chromatin remodellers, and various post-translational histone modifications (PTMs), which regulate the extent of DNA compaction. On cellular DNA, and perhaps on HBV cccDNA, additional regulation (denoted as ‘et al’ in the figure) can occur via variant histones or non-coding RNAs. In the absence of HBx, cccDNA appears to be rapidly silenced (closed state); HBx promotes de-silencing (open state), either by stimulating activating modifications, or by blocking repressive modifications (or both); potential contributions by other epigenetic controls remain to be explored.
Establishment of the unusual features of RC-DNA
The next steps establish the unusual structure of RC-DNA in progeny nucleocapsids and virions. P protein binding to the 5′ proximal ε stem-loop on the pgRNA triggers copackaging of RNA and P into newly forming nucleocapsids and protein-primed initiation of reverse transcription.29 ,45 In the priming reaction, a Tyr-residue in the unique terminal protein domain of P protein (figure 1C) substitutes for the 3′ terminal OH group of the nucleic primer (eg, a host tRNA) employed by conventional reverse transcriptases as acceptor for the first nt of the (-)-strand DNA; the result is a covalent tyrosyl-5′-DNA-phosphodiester linkage that is maintained to the very end of RC-DNA formation. The first 3–4 nt of the growing (-)-strand DNA are templated by a bulged region within ε (figure 4, top), followed by transfer to a matching acceptor motif in the 3′ proximal direct repeat 1* (DR1*). Extension from there to the 5′ end of the pgRNA template (figure 4, (-)-DNA completion) yields a unit length (-)-strand DNA copy of the pgRNA carrying a small (∼10 nt) terminal redundancy (‘r’). As DNA synthesis progresses, most of the pgRNA is degraded by P protein’s RNase H activity (a potential new therapeutic target46). The non-degraded 5′ residues of the pgRNA then act as primer for (+)-strand DNA synthesis. In a minority of nucleocapsids, direct (‘in situ’) extension of the RNA primer yields a double-stranded linear (dsL) DNA molecule (figure 4, in situ primed (+)-DNA synthesis). Its linear structure cannot give rise to new pgRNA; however, unless degraded, dsL-DNA can be circularised by the non-homologous end-joining (NHEJ) DNA repair pathway into cccDNA-like molecules. As NHEJ is error-prone,47 ,48 many of these molecules are functionally defective. Alternatively, dsL is the dominant substrate for integration into the host chromosome,49 frequently seen in HBV-associated hepatocellular carcinoma.50 Different from retroviruses, integration is neither virus-catalysed (for lack of an integrase gene) nor is it obligatory for hepadnavirus replication; rather, the functional analogue to retroviral provirus integration is cccDNA formation. Replication proper requires transfer of the RNA primer (which carries the DR1 sequence) to the sequence-identical DR2, and extension from there to the 5′ end of the (-)-strand DNA (figure 4, properly primed (+)-DNA synthesis). As ‘r’ at the other end has the identical sequence, exchange of the two ends allows (+)-strand DNA synthesis to proceed, eventually yielding RC-DNA with a slightly overlength (-)-strand carrying covalently linked P protein at its 5′ end, and an incomplete (+)-strand whose 5′ end is constituted by the RNA primer. These structural features are key elements to be considered for RC-DNA to cccDNA conversion.

Establishment of the unusual structural features of relaxed circular (RC)-DNA via protein priming. Protein priming at 5′ ε generates a short ε bulge-templated DNA oligo that is phosphodiester-linked to the terminal protein (TP) domain in P protein and transferred to direct repeat 1* (DR1*) for extension. During (-)-DNA completion, pregenomic (pg)RNA is degraded except for the capped 5′ end, yielding single-stranded (ss) DNA. The undegraded RNA oligo serves as (+)-DNA primer. Direct (in situ) extension yields double-stranded linear (dsL) DNA. Properly primed (+)-DNA synthesis proceeds by transfer of the RNA primer to DR2; after reaching the 5′ end of the (-)-DNA template, the small terminal redundancy r enables exchange against the sequence-identical 3′ r sequence such that (+)-strand synthesis can go on to generate RC-DNA. See text for details.
The infectious cycle is completed by envelopment of the mature RC-DNA containing nucleocapsids (though not immature RNA or ssDNA containing nucleocapsids51) by the surface proteins; different from subviral particles, virion secretion involves host factors of the multivesicular body pathway.52 Insufficient supply of surface proteins promotes, instead, recycling of the new RC-DNA to the nucleus, amplifying cccDNA copy number.53 For lack of suitable experimental systems for HBV, most present knowledge on this process is derived from the duck—DHBV model.
Scope and limitations of current experimental systems to study cccDNA
Productive hepadnavirus infection depends on the formation of cccDNA. Hence, controllable cell culture infection systems would provide a means to investigate cccDNA formation, yet owing to HBV’s narrow host-range until recently only a few and complex in vitro infection systems were available.54 In brief, one suitable system are primary hepatocytes from a matching host, that is, humans for HBV; major drawbacks are availability and highly variable quality of these cells. An alternative are primary hepatocytes from tree shrews (Tupaia belangeri),55 ,56 yet appropriately keeping these animals is demanding and costly. Surprisingly, hepatocytes from most monkeys appear refractory to HBV infection although these animals are all much more closely related to humans than tupaias. A recent explanation is the higher local similarity of tupaia NTCP to the human protein in two regions that are critical for the interaction with HBV PreS1.57 Searching these sequences in NTCP from other monkeys may reveal access to new infection models. A general drawback of primary hepatocytes is their rapid loss of susceptibility that correlates with a loss of NTCP expression.8 ,9 Longer-term HBV susceptibility can be achieved by xenotransplanting human or tupaia hepatocytes into immunodeficient mice58 ,59 but is technically very elaborate. A breakthrough was the discovery of the human liver-derived HepaRG cell line,60 ,61 which becomes susceptible to HBV (and HDV) upon a lengthy differentiation procedure61 that induces, inter alia, NTCP expression,8 ,9 yet without detectable viral spread.
That expression of human NTCP is sufficient to render the widely used HepG2 and Huh7 cell lines susceptible to HBV and HDV infection8 ,9 marks therefore a major further advance, even though high infection rates currently require a huge (up to 104-fold) excess of virus particles over cells.9
In contrast, non-infection-based systems, such as transfection of HBV plasmids into hepatoma cells, or hydrodynamic transfection of mice with plasmid-borne HBV genomes62 suffer all from very low or absent cccDNA formation. Rather than cccDNA, the transfected plasmid serves as the dominant transcription template (figure 2). Similarly, viral transcription in HBV-transgenic mice appears restricted to the integrated HBV sequences despite long-term viral replication at substantial levels. Potential explanations for the failure of mouse hepatocytes to generate cccDNA include the lack, or incompatibility with HBV, of murine host factors required for RC-DNA to cccDNA conversion, or the presence of restriction factors that, in a broad sense, could include factors or conditions that affect nuclear transport of properly matured nucleocapsids, for example, by premature cytoplasmic release of the RC-DNA. Also, levels of cccDNA could be low owing to rapid degradation. Hence, more than expressing human NTCP is required to generate a mouse model of HBV infection.
Different from HBV, DHBV generates easily detectable levels of cccDNA in all systems tested, including avian63 and human hepatoma cell lines,64 whereas unambiguous detection of HBV cccDNA remains challenging (see below). Moreover, primary duck hepatocytes for DHBV infection53 are readily available, as are ducks for in vivo experiments; the few chimp studies with human HBV have been instrumental in elucidating HBV immunobiology13 but are now largely banned.
Importance of DHBV for understanding cccDNA biology
Owing to its supercoiled structure, cccDNA exerts a higher electrophoretic mobility than equally sized linear or RC forms, as is well known from plasmid DNA preparations. Hence, Southern blotting provides for a unique distinction of cccDNA from the other forms of the hepadnaviral genome (figure 5A). Using this assay, cccDNA was first observed in infected livers of ducks,65 ,66 groundsquirrels67 and humans68 as a viral DNA that was not associated with viral particles, exclusively nuclear, and in chronically infected duck hepatocytes was present in up to 50 copies per cell. Anecdotally, an early chimp study concluded, mistakenly, that cccDNA was present in Dane particles69 and that these represented the true infectious virions. However, only the DHBV model provided the means to experimentally evaluate cccDNA function. A non-comprehensive list of landmark results includes the dependence of cccDNA formation on viral reverse transcription rather than on semiconservative DNA-to-DNA replication70; generation of a pool of cccDNA, that is, more than one copy per nucleus, via intracellular amplification53; regulation of the copy number via the levels of large envelope protein63; and the development of methods to determine cccDNA copy numbers in single cells.71 This work, corroborated by studies in the woodchuck model, established the crucial role of cccDNA in the replication cycle as the hepadnaviral persistence reservoir.

Direct versus indirect detection of covalently closed circular (ccc)DNA. (A) Southern blotting. Vectors for wild-type (+ENV) and envelope-deficient (-ENV) duck HBV (DHBV) were transfected into the avian hepatoma cell line LMH. Viral DNAs from cytoplasmic nucleocapsids (cytopl) and in nuclear DNA were detected using a 32P-labeled DHBV DNA probe; nuclear DNAs were treated with Dpn I to digest bacterially derived transfected plasmid, and with Plasmid-Safe nuclease that degrades linear but not circular DNA; relaxed circular (RC)-DNA with a nearly completed (+)-strand is not degraded. Note (i) the characteristically distinct mobility of cccDNA from all other viral DNA forms and (ii) the drastically enhanced cccDNA levels generated from the envelope-deficient DHBV. (B) ‘cccDNA-specific PCR’. Primers (purple and green arrows) are chosen to span the region containing the gap in the (+)-strand and the nick in the (-)-strand of RC-DNA. cccDNA provides a continuous template for exponential amplification. On RC-DNA, linear extension of the individual primers generates shorter but overlapping products that can anneal to each other and subsequently form an identical amplicon as that from cccDNA.
More recent studies included the generation of avian hepatoma cell lines such as DStet5,72 which carry a stably integrated envelope-deficient DHBV (DHBVenv−) under a tetracycline (Tet)-responsive promoter. Withdrawal of Tet induces transcription from the integrate and subsequent replication, including cccDNA formation. Adding back Tet shuts off integrate transcription such that cccDNA takes over the template function.73 As the competing pathway of virion secretion is blocked by the envelope deficiency, cccDNA levels can reach several hundred copies per cell. Unexpectedly, DHBV maintains the ability to generate high levels of cccDNA even in human hepatoma cells,64 ,74 allowing to search for the human host factors involved without the technical issues plaguing HBV cccDNA detection. Notably, for HBV the boosting effect of envelope deficiency on cccDNA accumulation is much less pronounced than for DHBV.64 ,75
The challenge of unambiguous human HBV cccDNA detection
Single-cell analysis of liver biopsies from a chronically infected duck showed a mean of around 10 cccDNA copies per cell, with a broad distribution between individual cells and temporal fluctuations during the course of infection.71 Copy numbers in human chronic hepatitis B appear markedly lower (mean 0.1–1.0 copies per cell) and similar numbers apply to human liver chimeric mice,76 limiting the use of Southern blotting as an unequivocal means to detect true cccDNA. As a rule of thumb, Southern blotting using 32P-labeled probes can detect a few picograms of virus DNA per lane; 2 pg correspond to roughly 106 molecules. Assuming about 106 cells per well of a six-well plate, and transfected or infected cells representing 10% of the total cell count, this translates into a detection limit of around 10 cccDNA copies per infected/transfected cell. The need for a more sensitive cccDNA detection technique was addressed early on77 by a PCR technique referred to as ‘over-gap’ or, misleadingly, as ‘cccDNA-specific’ PCR (figure 5B). Its key principle is the use of primers spanning a region that is contiguously double-stranded on cccDNA but carries the nick in the (-)-strand and the gap in the (+)-strand of RC-DNA, the most likely ‘contaminant’ (besides plasmid DNA in transfections or integrated HBV DNA in stable cell lines or transgenic animals). However, the entire sequence information of cccDNA is also present on RC-DNA (figure 5B), which can be present at 1000 copies or more per cell.78 Though exponential amplification will initially occur only on cccDNA, linear extension of the primers on the (-)-strand and (+)-strand DNA generates overlapping shorter products that can anneal to each other and then be amplified exponentially to an identical product as that from cccDNA. Hence, over-gap PCR provides relative, not absolute discrimination between cccDNA and RC-DNA. The originally achieved discriminatory power of around thousand fold77 can be increased by reducing the levels of RC-DNA in the template preparation, for example, pretreatment with Plasmid-Safe DNase,79 which degrades DNA with free ends; notably though, RC-DNA with a nearly complete (+)-strand is not a substrate (reference64 and figure 5A). Hence, without proper controls seemingly cccDNA-specific amplicons can be generated in the complete absence of cccDNA.
Rolling circle amplification is a potential alternative as it should depend on a circular template.80 However, our own data using pure DHBV RC-DNA suggest that this dependence is, again, not absolute. A non-PCR alternative is the Invader technology, which uses only one strand as template on which appropriately designed oligonucleotides assemble a triple-stranded structure that is a substrate for a structure-specific endonuclease; hence, it can discriminate whether the sequence of the HBV (+)-strand has a free 5′ end as in RC-DNA, or is part of a contiguous sequence as in cccDNA.81 At any rate, to maximise cccDNA selectivity of all these assays, a potential contamination with RC-DNA should be kept minimal. A simple means is to start from nuclear rather than whole cell DNA,64 possibly combined with an exonuclease treatment that also degrades RC-DNA but does not harm cccDNA; a single nick in one strand would suffice to relax supercoiling, falsely suggesting lower levels of cccDNA than might actually be present.
To derive cccDNA copy numbers per cell, it is equally important to quantify cellular DNA. Depending on the extraction procedure, this may be done by determining the copy number of a cellular gene such as β-globin76 or β-actin.82 If smaller DNAs were enriched over larger DNAs, as in the Hirt procedure,83 mitochondrial DNA may be used for an approximation,84 ,85 although the variable number of mitochondrial genomes per cell needs to be considered.86
Most importantly, researchers should be aware of the potential pitfalls of the specific method they use, and any indirect assay should be calibrated using a reference standard of RC-DNA and cccDNA whose molecular nature and concentrations have independently been confirmed by Southern blotting. A joint effort towards establishing a standardised high-sensitivity protocol would be highly desirable.
Readers, on the other hand, should be aware that claims of a further reduction in the already very low levels of HBV cccDNA are not always warranted by the data.
Longevity of cccDNA
The frequent rebound of viral replication upon withdrawal of NA therapy or immunosuppression, despite virtual loss of replicative intermediates, indicates that cccDNA can persist for decades,7 yet reliable numbers on the kinetics of cccDNA loss are difficult to derive from human patients. Studies in chronically WHV-infected woodchucks and DHBV-infected ducks87 ,88 consistently found half-lives between 33 and 57 days, and similar values were derived in primary hepatocytes.89 During the clearance phase of acute infection in chimps, cccDNA half-life was reduced to ∼3 days,90 although traces of cccDNA persisted for years. Together these data demonstrate the longevity of the cccDNA pool in chronic infection; however, the steady-state level of this pool is determined by so many parameters affecting production of new versus loss of existing cccDNA molecules that deconvolution is extremely demanding.90 ,91 Hence, at present, we do not know how long an individual cccDNA molecule lives, although this is a crucial issue for new therapeutic approaches aiming to directly target cccDNA (see below).
Conceptual evidence for an involvement of host DNA repair in RC-DNA to cccDNA conversion
RC-DNA from incoming virions is, in all likelihood, the direct precursor to cccDNA. Hence, the distinctive features of RC-DNA, that is, covalently bound P protein and RNA primer at the DNA 5′ ends and incompleteness of the (+)-strand and terminal redundancy of the (−)-strand, must be fixed, and the ends must be ligated (figure 6A). Conceivably, some of these steps might be performed by the viral P protein, foremostly filling the gap in the (+)-strand. However, inhibition of P protein by NAs did not, or only partly, inhibit initial cccDNA formation in infection models, suggesting that NA-insensitive host DNA polymerases can perform this function.92 ,93

Relaxed circular (RC)-DNA mimics damaged cellular DNA. (A) Structural peculiarities on RC-DNA. The (-)-strand contains the phosphotyrosyl bonded P protein, a nick, and a single-stranded flap (corresponding to r); the (+)-strand contains the gap and the RNA primer on its 5′ end. On cellular DNA, surplus structures are often removed by endonucleases (green lightning symbols), including from a distance to the lesion; phosphotyrosyl-linked adducts can be resolved by specific tyrosyl-DNA-phosphodiesterases (TDPs, red lightning symbol; see B). Nucleolytically induced gaps require fill-in by a DNA polymerase, possibly after end-polishing by polynucleotide kinase and/or phosphatase, and nick-sealing by DNA ligase. The same repair activities are predictably required during RC-DNA to covalently closed circular (ccc)DNA conversion. (B) Similarity of the linkage between P protein and RC-DNA to cellular trapped topoisomerase (TOP) cleavage complexes. 5′ linked TOP2 is either released by TDP2 (or perhaps in some instances by TDP1), or by nucleolytic repair. Principally the same holds for 3′ linked TOP1, except TDP1 appears to be the major TDP involved. Recent evidence strongly suggests that TDP2 is able to release P protein from RC-DNA; however, nucleolytic pathways may also occur. (C) Slowed-down RC-DNA to cccDNA conversion in HepG2 cells with reduced TDP2 levels. A vector for envelope-deficient duck HBV (DHBV) was transfected into naive (nai.) cells or cells in which TDP2 levels were reduced by 80–90% by stable expression of an anti-TDP2 shRNA (TDP2 kd). Kinetics of RC-DNA and cccDNA generation were compared by Southern blotting. The ratio of cccDNA:RC-DNA (indicated at the bottom; mean±SD, n=6) was significantly reduced in the TDP2 knock-down cells on day 2 and day 3 post transfection (adapted from Königer et al74).
Also, an ‘autocatalytic’ release of P protein from RC-DNA could be envisaged, enabled by the nature of this linkage as a tyrosyl-DNA-phosphodiester. Chemically identical bonds occur as covalent intermediates of topoisomerase activity (see below). Topoisomerases regulate DNA supercoiling by incising one (TOP1) or both DNA strands (TOP2), so as to reduce torsional stress during replication or transcription.94 In the process, TOP1 becomes linked through a tyrosyl-phosphodiester to the 3′ site of the DNA break, TOP2 to the 5′ site (figure 6B). The energy released by breaking the DNA backbone is stored in the newly formed tyrosyl-DNA-phosphodiester bond; hence, the reaction (a transesterification) is reversible such that the DNA is resealed and the enzyme is released. However, for the backreaction to occur the two DNA ends must be sterically in line, as demonstrated, for instance, by the dramatic increase in trapped covalent TOP-DNA adducts (‘TOP cleavage complexes’) caused by drugs that distort the DNA in the cleavage complex.94 ,95 Hence, for RC-DNA, P protein release and concomitant circularisation of the (-)-strand would require exact removal of the 3′ copy of the r redundancy in the (−)-strand, an activity hardly attributable to P protein. Likewise, P protein appears unsuitable to achieve removal of the RNA primer from the (+)-strand DNA and/or ligating the open ends in one or both strands. Hence, most, if not all, steps of RC-DNA to cccDNA conversion have to be performed by the host cell, which, indeed, contains all required activities as part of its DNA repair system.
Genome integrity is fundamental to all organisms to maintain viability yet under constant threat. While double-strand breaks (DSBs) are obviously detrimental, numerous other modifications including single-strand breaks, copying errors, cross-links, base modifications or covalent DNA adducts can impair replication and transcription and/or introduce lethal mutations. A single mammalian cell may experience >104 damage events per day.96 Hence, all cells are equipped with a sophisticated DNA repair machinery whose >250 components serve to detect DNA damage, signal to halt the cell cycle and allow time for repair, and fix the damage; in metazoans, failed repair will usually induce apoptosis (or else may result in cancerous cells). It is increasingly appreciated that viruses with a nuclear phase have to cope with this surveillance system either by usurping certain aspects for their own benefit or as a cellular defence to be overcome.97–100 Just two examples are papillomaviruses, which use the interconnection of DNA repair and cell cycle control to bring the host cell into an appropriate phase for viral genome replication and then block apoptosis101; or adenoviruses whose linear double-stranded DNA genomes are interpreted as DSBs by the cell, inducing repair into concatemers too large to be packaged. Using its early proteins, adenovirus counteracts this cellular response by inactivating key components of the DNA repair machinery,97 including the DSB-detecting MRN complex.102 ,103
Hence, it would not be surprising if the numerous unusual features of hepadnaviral RC-DNA (figure 6A) also call in a DNA damage response. However, the molecular characterisation of this process is just at its beginning.
Emerging experimental evidence for an involvement of host DNA repair in hepadnavirus cccDNA formation
The most obvious peculiarity of hepadnaviral RC-DNA is the large (∼90 kDa) covalently linked P protein. Its phosphodiester linkage to the 5′ end of (-)-strand DNA strikingly resembles the phosphotyrosyl bonds in trapped TOP cleavage complexes (figure 6B). Cellular repair of these protein adducts involves at least two distinct mechanisms. In the nucleolytic pathway (applicable to remove all kinds of chemical modifications), the lesion is excised together with a piece of neighbouring DNA by structure-specific nucleases94 ,104; the resulting gap is then filled-in and the ends are eventually ligated. One factor involved is the MRN complex, which contains itself a nuclease, MRE11, yet may also act in concert with associated nucleases.105–107 A second mechanism uses tyrosyl-DNA-phosphodiesterases (TDPs95), which specifically cut the phosphotyrosyl-linkage. TDP1 acts predominantly on 3′ linked TOP1 adducts, yet partly controversial results suggest it may also have 5′ activity, possibly dependent on its species origin. In 2009, TDP2 was discovered as the first TDP enzyme with a dominant 5′ substrate activity.108–110 Hence, either enzyme was a candidate for a cellular activity capable of releasing the 5′ phosphotyrosyl-bonded P protein from RC-DNA.
As we recently showed74 using recombinant TDP enzymes, human and chicken TDP2, yet only yeast TDP1, exerted high cleavage activity on a synthetic 5′ phosphotyrosyl model substrate. The same was observed on DHBV P protein to which short DNA oligonucleotides were linked via in vitro reconstitution of the protein-priming reaction,111 ,112 and on genuine DHBV and HBV RC-DNA from cell culture-derived nucleocapsids. These biochemical data firmly established TDP2, but not TDP1, as a cellular DNA repair factor having the appropriate enzymatic activity to cleave P protein from RC-DNA also during virus replication.
As P protein release from RC-DNA is mandatory for conversion into cccDNA, inactivating the responsible factor (if it is only one) should ablate cccDNA formation. We therefore generated HepG2 cells stably expressing TDP2-specific small hairpin (sh) RNAs, which enduringly reduced TDP2 expression by 80–90%. Next, these TDP2 knock-down cells and naive HepG2 cells were transfected with a vector for an envelope-deficient DHBV genome to initiate viral replication, and the kinetics of RC-DNA and cccDNA accumulation were compared by Southern blotting. As shown in figure 6C, reduced TDP2 levels correlated with significantly slowed-down RC-DNA to cccDNA conversion. Eventually, however, the same levels and ratios of both DNA forms were established in both cells.
A straightforward explanation for delayed but not ablated cccDNA accumulation is that the residual TDP2 feeds less P protein-free RC-DNA per time into the conversion pathway (ie, P protein release becomes rate-limiting), slowing down cccDNA formation (see figure 7). However, the data are also compatible with more complex interpretations that account for the widespread redundancy in DNA repair.113 Given the fundamental importance of genome integrity, failure of one repair pathway is usually safeguarded by one or more others. For TOP cleavage complex repair, this is exemplified by the several nucleolytic pathways that can substitute for TDP-mediated repair.94 ,114

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Candidate therapeutic strategies to target HBV covalently closed circular (ccc)DNA. The circles represent P protein-linked and P protein-free relaxed circular (RC)-DNA and cccDNA in the host cell nucleus. (A) Prevention of cccDNA accumulation by blocking host factors involved in the multiple steps of RC-DNA to cccDNA conversion. (B) Prevention of nuclear import of RC-DNA by capsid-targeting drugs; cytoplasmic release of RC-DNA may, in addition, trigger DNA sensors like cyclic GMP-AMP synthase (cGAS) and activate STING to induce antiviral cytokines. (C) Silencing of cccDNA transcriptional activity by inducing the host cell’s epigenetic machinery, or by blocking the de-silencing activity of HBV X protein (HBx). (D) Degradation of existing cccDNA by immune-mediated mechanisms, perhaps via APOBEC enzymes, or (E) by direct targeting with designer nucleases as used in genome editing. See text for details.
Hence, such alternative pathways could contribute to P protein release from RC-DNA in the TDP2 knock-down cells and thus to ongoing, though slowed-down, cccDNA formation.
A distinction should be possible by complete abrogation of TDP2 expression. Genome-editing using designer nucleases now allows specific gene knockout in human cells115 but is complicated in HepG2 and Huh7 cells by their variable hyperploidy (MN and M. Leipoldt, Institute of Human Genetics, University of Freiburg; unpublished data); the strand-breaking editing procedures as such may further decrease genetic stability. Hence, results in one particular cell clone may not necessarily be generalisable, making confirmatory experiments in independently derived cell clones advisable. A specific problem during our own attempts to generate TDP2 knockout cells was formation of an N terminally truncated yet catalytically active TDP2; its expression was independent from the upstream editing site, designed to interrupt the ORF shortly after the supposed initiator codon.74 At any rate, generating well-characterised hepatoma cells with defined host factor knockouts will be a highly worthwhile investment for further-reaching studies into the interplay between HBV and the host DNA repair.
RC-DNA to cccDNA conversion as new therapeutic target?
The data described above suggest TDP2 and likewise any other host factors involved in RC-DNA to cccDNA conversion as potential new therapeutic targets whose inhibition should prevent formation of HBV's persistence reservoir (figure 7A). However, several issues need further clarification. The most important is whether the longevity of cccDNA relates to individual molecules or is achieved via turnover. Eventually, this will require monitoring the fate of single cccDNA molecules over time, a technically non-trivial task. Without cccDNA turnover, inhibiting RC-DNA to cccDNA conversion should work only during establishment of the initial infection, that is, prophylactically. Interestingly, treatment of human liver chimeric mice with the entry inhibitor Myrcludex B, starting after initiating a low-dose HBV infection, prevented not only virus spread yet also appeared to impair cccDNA amplification from RC-DNA in the already infected cells.76 While implying that de novo synthesis of cccDNA can occur, these results need to be confirmed in the clinical setting.
A second critical issue is that targeting a DNA repair factor could also impact cellular DNA damage repair, with an ambivalent role for the repair system's redundancies. If, for instance, P protein release from RC-DNA or any of the other conversion steps can be achieved through more than one mechanism, blocking just one of them will not prevent cccDNA formation. Conversely, redundancy may allow to identify a factor that is essential for the virus but has a back-up for cellular DNA repair. This will require a comprehensive knowledge on the host factors involved, making a case for high-throughput screening approaches, for example, via genetic inactivation through RNA interference or advanced knockout technologies and/or screening for small compound inhibitors. A recent study suggests indeed that small molecules can interfere with cccDNA formation, although the mechanism is unknown.116 Highly valuable for any such screening would be HBV reporter vectors that allow for non-invasive, sensitive and quantitative detection of cccDNA formation; for other viruses, including HCV, such reporter-bearing variants have been instrumental.117 However, manipulating the tiny, compactly organised HBV genome without disturbing its functionality has proven very difficult;118 ,119 hence, further research is highly warranted.
An alternative strategy would be to block the RC-DNA precursor from reaching the nucleus (figure 7B). As nuclear transport depends on the capsid, this may be achieved by capsid-targeting drugs,120 currently in development.54 Besides inducing assembly of empty capsids or irregular polymers, they may also destabilise existing nucleocapsids.121 ,122 RC-DNA released from cytoplasmically disassembled capsids may then be unable to reach the nucleus and, moreover, may become detectable by cytoplasmic DNA sensors such as cyclic GMP-AMP synthase , which can induce type 1 interferons via the stimulator of interferon genes STING.123 ,124 Shielding from this defence system may be a reason that HBV reverse transcription is restricted to the capsid interior, and that RC-DNA is escorted to the nucleus in intact capsid shells.35 Again, however, the existing cccDNA pool would only be affected if cccDNA turnover occurs.
Silencing cccDNA's transcriptional activity—likely a transient solution
Accumulating evidence suggests that transcriptional activity of cccDNA is subject to epigenetic control (figure 3); hence, manipulating the cell's epigenetic machinery125 may be exploited to functionally inactivate, rather than eliminate, cccDNA (figure 7C). Model studies suggest interferon-α treatment as one possibility.73 ,126 Alternatively, HBx may represent a target for direct-acting antivirals as it appears necessary to counteract the default silencing of cccDNA upon HBV entry.40 The mechanism is unknown and convoluted by a vast body of candidate HBx interactors (e.g. see reference127); their relevance in infection is often questionable because the test systems used were marginally, if at all, HBx (and cccDNA) dependent.128 One of the stronger candidates is DNA damage-binding protein DDB1129 that with its partner DDB2 recognises UV-induced DNA distortions and forms an E3 ubiquitin ligase complex that mediates degradation of multiple targets.130 Those relevant for the de-silencing activity of HBx are not known but may yield to the new HBx and cccDNA-dependent infection systems. Notably, though, blocking the interaction between HBx and a cellular partner would have to be sustained as long as cccDNA is present. Moreover, cells with transcriptionally inactive cccDNA appear more stable towards immune-mediated cccDNA clearance.131 Hence, more research is required to validate this concept.
Ridding the liver of cccDNA
Steady-state cccDNA levels are determined by the rates of formation versus loss. As long as the turnover kinetics are not settled, active elimination of existing cccDNA appears as the most straightforward approach. Two major current strategies are to mimic, in the chronic setting, the immune-mediated clearance of most of the cccDNA that occurs during self-limited acute HBV infection13 ,132 (figure 7D) and the employment of designer nucleases that have revolutionised genome editing (figure 7E).
Innate and adaptive immune responses are critical in clearing acute HBV infection. Restoring full activity of the insufficient immune responses typical for chronic HBV infection will thus remain highly relevant,12 likely on both the cellular level (to clear infected hepatocytes) and the humoral level (to prevent reinfection). Given the breadth of the field, only a few general and simplified considerations are discussed here (for more comprehensive accounts, see references14 ,15 ,133 ,134).
The two extreme scenarios for cccDNA clearance from hepatocytes are non-cytolytic elimination (‘curing’), or destruction of (nearly) all cells harbouring cccDNA by T cells (‘killing’) and replacement by non-infected cells.132 Frequent liver damage during chronic hepatitis B argues against curing as the only explanation; conversely, the fast recovery from acute infection would require that the entire liver be turned over within a few weeks—while maintaining functionality. Hence, likely both mechanisms exist, yet their relative contributions are still debated, owing to the multiple, difficult to assess parameters involved.90 ,91 ,135 Examples include the fate of cccDNA during cell division,136 specifically if and how cccDNA re-enters the reforming nucleus; the turnover time of cccDNA-free versus cccDNA bearing cells and, for the latter, the impact of cccDNA transcriptional activity; or the origin, fraction and proliferation characteristics of cells that are refractory to infection, or refractory to immune-mediated clearance. Irrespective of these difficulties, cytokines such as interferons and their downstream effectors appear to play an important role, although the exact mechanisms are not firmly established. While various steps of the replication cycle might be affected,132 a recent study137 suggested that very-high-dose interferon-α, or more potently activation of the lymphotoxin-β receptor, could directly target cccDNA integrity via APOBEC3A and 3B-mediated deamination of the (-)-strand and subsequent degradation. Though some aspects are controversial,138 ,139 the worthiness of activating innate responses is underlined by promising preclinical results with the Toll-like receptor 7 agonist GS-9620.140
Notably, in various settings of immune-mediated cccDNA decline, a fraction of the cccDNA pool appeared refractory to further reduction.88 ,84 ,131 ,137 ,141 This might reflect properties of the cccDNA harbouring cell, or cccDNA may per se exist in distinct forms that differ, for example, in methylation, chromatinisation or some unknown property (figure 3). Possibly, non-natural ways to induce cccDNA degradation might be able to also target this resilient reservoir.
Key role of HBV covalently closed circular (ccc)DNA in viral persistence and chronic hepatitis B
Chronic hepatitis B, caused by persistent infection with HBV, puts >250 million people at risk to develop terminal liver disease.
HBV persistence is mediated by an intranuclear, episomal form of the viral genome called cccDNA.
cccDNA is the template for viral RNAs and subsequent generation of progeny virions.
A few copies of cccDNA per liver can (re)initiate full-blown infection.
cccDNA is not targeted by current treatments—but a cure of chronic hepatitis B will require elimination of cccDNA.
Recent advances, including identification of a liver-specific HBV receptor and evidence for HBV's interaction with cellular DNA damage repair, promise to greatly expand the limited knowledge on cccDNA biology.
Major unresolved issues in HBV covalently closed circular (ccc)DNA biology
Does the longevity of cccDNA relate to individual molecules, or is there cccDNA turnover? If turnover occurs, by which route (intracellular? cell-to-cell? with an extracellular phase?) and with which kinetics?
How is cccDNA ‘cleared’ in acute self-limiting hepatitis B?
If cells can be immunologically cured from cccDNA, by which mechanism(s)?
Does cccDNA survive cell division—and how?
What restricts HBV cccDNA formation/accumulation in most human hepatoma cell lines and particularly in mouse hepatocytes?
Why is this restriction much less pronounced for duck HBV, even in human cells?
Which mechanism(s) prevent infinite cccDNA amplification even in duck HBV model systems? Could such mechanisms be harnessed to reduce cccDNA copy number?
Advances in genome editing using designer nucleases115 have prompted studies harnessing these new tools for targeting cccDNA, for example, by zinc-finger nucleases,142 ,143 transcription activator-like endonucleases144 or the RNA-guided clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system.85 However, numerous issues are unresolved, foremost efficient access of the nucleases to all cccDNA molecules. Unless cccDNA-bearing cells can specifically be targeted, the nucleases must be delivered to all hepatocytes. Off-target effects, including chromosomal integration of the linearised viral DNA, could adversely affect hepatocyte function, especially when long-term presence of the effector nucleases is necessary. Also, while NHEJ-mediated repair of the nuclease-induced DSBs is error-prone,47 a fraction of repair events will result in the reformation of intact cccDNA. Not the least, it is unclear how the excess RC-DNA in the same cells affects the targeting efficiency for cccDNA. Again, much more research is required and there is ample room for other strategies including therapeutic vaccination or anti-sense and RNA-interference-based approaches.54
Conclusions and perspectives
The importance of cccDNA as persistence reservoir of HBV is firmly established and so is the realisation that any strategy towards a cure of chronic hepatitis B will have to cope with this long-lived molecule. Current knowledge on cccDNA formation and degradation is still very limited, yet in particular the emerging cell culture infection systems, and the possible development of small animal infection models, promise to dramatically change this situation. Still, the current gap in knowledge is so large that many facets of cccDNA biology are open to new discoveries (box 2). It appears unlikely that a single magic bullet will turn up that causes cccDNA to completely disappear; however, the combination of new knowledge on cccDNA biochemistry, a molecular understanding of how the body's immune system deals with cccDNA during clearance of acute HBV infection, and new technologies for targeted DNA manipulation hold promise to achieve this goal. An indispensable premise is appropriate funding for basic HBV research. New opportunities might come from the renewed interest of pharmaceutical industry in HBV. It is hoped that fair and as far as possible open interactions between academia and industry will make the quest for a cure of chronic hepatitis B a similar success as in the case of chronic hepatitis C.145
Acknowledgments
I apologise to numerous colleagues whose original contributions could not or only partly be referenced for space limitations.
References
Footnotes
Correction notice This article has been corrected since it published Online First. The article is no longer Open Access.
Funding Work in the author's laboratory was supported by the Deutsche Forschungsgemeinschaft (DFG) via grant NA154/12-2 within the Collaborative Research Unit FOR1202 (persistence of hepatotropic viruses) and by the European Union via the FP7 Infect-ERA programme (project ID hepBccc).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.