Article Text

Abstract
Objective Commensal bacteria and innate immunity play a major role in the development of colorectal cancer (CRC). We propose that selected commensals polarise colon macrophages to produce endogenous mutagens that initiate chromosomal instability (CIN), lead to expression of progenitor and tumour stem cell markers, and drive CRC through a bystander effect.
Design Primary murine colon epithelial cells were repetitively exposed to Enterococcus faecalis-infected macrophages, or purified trans-4-hydroxy-2-nonenal (4-HNE)—an endogenous mutagen and spindle poison produced by macrophages. CIN, gene expression, growth as allografts in immunodeficient mice were examined for clones and expression of markers confirmed using interleukin (IL) 10 knockout mice colonised by E. faecalis.
Results Primary colon epithelial cells exposed to polarised macrophages or 4-hydroxy-2-nonenal developed CIN and were transformed after 10 weekly treatments. In immunodeficient mice, 8 of 25 transformed clones grew as poorly differentiated carcinomas with 3 tumours invading skin and/or muscle. All tumours stained for cytokeratins confirming their epithelial cell origin. Gene expression profiling of clones showed alterations in 3 to 7 cancer driver genes per clone. Clones also strongly expressed stem/progenitor cell markers Ly6A and Ly6E. Although not differentially expressed in clones, murine allografts positively stained for the tumour stem cell marker doublecortin-like kinase 1. Doublecortin-like kinase 1 and Ly6A/E were expressed by epithelial cells in colon biopsies for areas of inflamed and dysplastic tissue from E. faecalis-colonised IL-10 knockout mice.
Conclusions These results validate a novel mechanism for CRC that involves endogenous CIN and cellular transformation arising through a microbiome-driven bystander effect.
- Colon Carcinogenesis
- Colonic Bacteria
- Genetic Instability
- Macrophages
- Stem Cells
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Commensal bacteria and innate immunity play a major role in the aetiology of colorectal cancer (CRC). Mechanisms by which commensals drive genomic damage through innate immunity leading to cellular transformation and CRC, however, are lacking.
-
Enterococcus faecalis, a human intestinal commensal, can polarise macrophages to produce a bystander effect that causes double-stranded DNA breaks, tetraploidy and chromosomal instability (CIN) in target cells and induces inflammation and CRC in interleukin (IL) 10 knockout mice.
-
CRC, like other solid tumours, consists of tumour stem cells that are responsible for sustaining tumour growth. We investigated whether E. faecalis-polarised macrophages could transform primary colon epithelial cells to cancer-initiating cells.
What are the new findings?
-
Exposure of primary colon epithelial cells to commensal-polarised macrophages or 4-hydroxy-2-nonenal, an endogenous mutagen, induces heritable mutagenesis and CIN—features characteristic of CRC. These cells, when transplanted as allografts, grow into poorly differentiated carcinomas.
-
Transformed clones show altered expression of three to seven cancer driver genes per clone with increased expression of stem/progenitor cell markers Ly6A and Ly6E. Allografts are strongly positive for the tumour stem cell marker doublecortin-like kinase 1 (Dclk1).
-
Dclk1 and Ly6A/E are expressed by epithelial cells in colon biopsies from inflamed and dysplastic tissue in E. faecalis-colonised IL-10 knockout mice.
How might it impact on clinical practice in the foreseeable future?
-
Understanding mechanisms by which commensals initiate and promote CRC will permit new preventive strategies for decreasing the incidence of this common human cancer.
Introduction
Sporadic, colitis-associated and inherited forms of colorectal cancer (CRC) arise from somatic mutations and/or epigenetic alterations in tumour suppressors and proto-oncogenes.1 ,2 The origin of mutations and epigenetic changes that lead to these cancers, however, remains ill-defined. The initiation of chromosomal instability (CIN) and epithelial cell transformation by the colonic microbiome represents an endogenous mechanism for driving colon carcinogenesis.3 Several human commensals produce toxins that damage epithelial cell DNA, increase intestinal barrier permeability, and/or activate T cell responses to promote colorectal carcinogenesis.4–6 Enterococcus faecalis is a human commensal that can drive endogenous mutagenesis leading to CRC in interleukin (IL)-10 deficient mice.7–9 This commensal polarises macrophages to produce diffusible clastogens (or chromosome-breaking factors) that break double-stranded DNA, disrupt mitotic spindles and generate CIN through a bystander effect (BSE).8–10
Commensal-triggered BSE mechanistically links key events in colorectal carcinogenesis to the microbiome. This theory proposes that polarisation of colon macrophages by commensals initiates CIN and transforms colonic epithelial cells through BSE. Colon macrophages are normally quiescent and help maintain immunological tolerance to commensals.11 These cells, however, are also part of the host defence against invading pathogens and can be polarised to M1 or M2 phenotypes. M1 polarised macrophages secrete proinflammatory cytokines, superoxide and nitrogen radicals in response to infection.12 In contrast, M2 (or alternatively polarised) macrophages express anti-inflammatory phenotypes that participate in parasite clearance, tissue remodelling and, for tumours, cancer progression.
Colon macrophages ordinarily resist polarisation by commensals, but in the absence of IL-10 can be activated by E. faecalis to generate BSE. Using the Il10−/− model of CRC, we showed that colon macrophages were polarised to a M1 phenotype by E. faecalis.10 In addition, when colon macrophages were depleted using rectally administered liposomal clodronate, inflammation and CRC were prevented, confirming the essential contribution of these cells to BSE and commensal-driven carcinogenesis.
One mediator for BSE that is produced by E. faecalis-infected macrophages is trans-4-hydroxy-2-nonenal (4-HNE),13 a mutagenic breakdown product of ω-6 polyunsaturated fatty acids.14 This reactive aldehyde is generated in a cyclo-oxygenase (COX)-2 dependent manner and can readily diffuse into neighbouring cells to damage DNA and disrupt mitotic spindles.13 ,15 These findings link COX-2, a key target for drugs that prevent CRC,16 ,17 with BSE.
The polarisation of colon macrophages by intestinal commensals to generate BSE is a novel mechanism for endogenous CIN and cellular transformation in the adenoma-to-carcinoma sequence.1 ,2 We previously showed that a single non-cytotoxic dose of 4-HNE disrupted mitotic spindles and induced tetraploidy in primary colon epithelial cells; one dose, however, failed to produce heritable CIN.13 In this study, we investigated whether E. faecalis-infected macrophages or purified 4-HNE could lead to CIN and cellular transformation in primary colon epithelial cells. Surprisingly, we found that it only took 10 weekly doses of 4-HNE, or exposure to E. faecalis-infected macrophages, to produce CIN and transform cells that could grow as poorly differentiated and invasive carcinomas when injected into immunodeficient mice. Gene expression profiling identified networks involving inflammation, cell cycle regulation, proliferation, cancer cell growth and driver genes for cancer.1 ,2 Finally, Ly6e, a member of the Ly6 gene family of haematopoietic stem/progenitor cell markers,18 and doublecortin-like kinase 1 (Dclk1), a tumour stem cell marker,19 were upregulated in the colonic epithelium of E. faecalis-colonised Il10−/− mice. These findings demonstrate that polarised macrophages, or purified 4-HNE, potently induces CIN and cellular transformation in primary colon epithelial cells and validates BSE as a mechanism for endogenous carcinogenesis.
Materials and methods
Cell lines, bacteria, 4-HNE
Young adult mouse colonic (YAMC) epithelial cells (Ludwig Institute for Cancer Research), murine macrophage RAW264.7 cells, and HCT116 human colon cancer cells (American Type Culture Collection) were grown as previously described.9 YAMC cells proliferate at 33°C in the presence of interferon-γ but not at 37°C in the absence of this cytokine (see below). E. faecalis OG1RF was grown as previously described.9 4-HNE was purified from infected macrophages as previously described.13
Treatment of YAMC cells
RAW264.7 cells were infected with OG1RF at a multiplicity of infection of 1000 as previously described.8 YAMC cells were co-cultured with E. faecalis-infected macrophages, or uninfected macrophages as control, for 72 h in a dual-chamber co-culture system, and recovered for 96 h, as previously described.8 YAMC cells were dosed with purified 4-HNE at 1 µM for 1 h, which resulted in <5% cytotoxicity as previously reported.13 Cells were allowed to recover for 1 week and 16 treatment cycles were performed.
Mutant fraction assay
YAMC cells harbour the H-2Kb class I gene promoter fused to the SV40 tsA58 early region.20 As a consequence, cells die by senescence within 10 days when grown at 37°C. Cells acquiring mutations in tsA58, however, grow at this temperature. Mutant fractions were assayed following every other treatment and compared with controls as previously described with slight modification.20 In brief, 1×105 cells were seeded with complete RPMI 1640 medium in the absence of interferon-γ at 33°C overnight to allow adherence. Cells were incubated at 37°C for 10 days. Randomly selected colonies were expanded. Remaining colonies were fixed, stained and used to calculate mutant fractions. Data were expressed as means with the SD. Student t test was used for comparison between experimental groups and controls. Analysis of variance was used for multiple comparisons. p Values <0.05 were considered statistically significant.
Fluorescent-activated cell sorting
Ploidy assays were performed as previously described.9 Mitotic cells were stained with phosphorylated histone 3 antibody (Cell Signaling Technology) and DNA contents were determined by propidium iodide staining. Ly6A/E expression by cells was assessed using fluorescein isothiocyanate (FITC)-conjugated rat antimouse Ly6A/E antibody (BD BioSciences).
Fluorescence in situ hybridisation
Two-color fluorescence-labelled probes were used to detect mouse chromosomes 11 and 18 according to manufacturer's instructions (Applied Spectral Imaging). Metaphase plates were prepared as previously described.9 Images were collected by fluorescence microscopy (Nikon).
In vitro transformation
Anchorage-independent growth was determined by spheroid formation in soft agar using CytoSelect 96-well Cell Transformation Assay, Cell Recovery Compatible Kit according to manufacturer's instructions (Cell Biolabs). Randomly selected single clones were recovered from soft agar using a matrix solubilisation solution and then grown in RPMI 1640 without interferon-γ at 37°C and expanded in RPMI medium with 10% fetal bovine serum prior to allografting.
Allografts
Animal studies were approved by the Institutional Animal Care and Use Committees at the University of Oklahoma Health Sciences Center and Oklahoma City Veterans Affairs Medical Center. For allografts, 1×106 cells for clones derived from treatments with polarised macrophages or 4-HNE were either directly subcutaneously injected into the flanks of 6-week-old female NOD/scid mice (Jackson Laboratory) or injected after mixing with matrigel (BD Biosciences). Untreated YAMC and HCT116 colon cancer cells served as negative and positive controls, respectively. Tumour masses were resected when flank masses reached 10% of body weight or at 20 weeks postengraftment and fixed in 10% formalin.
Staining
Immunohistochemical and immunofluorescent staining of allografts and colon biopsies were performed as previously described.21 Cytokeratins, Ly6A/E and Dclk1 were stained using mouse anti-pancytokeratin monoclonal antibody (Novus Biologicals), rat anti-Ly6A/E (BD BioSciences) and rabbit polyclonal antibody to Dclk1 (Abcam). Rabbit anti-nitric oxide synthase 2 (Enzo Life Sciences), anti-arginase 1 (Sigma), and anti-MSH2 (Santa Cruz Biotechnology) polyclonal antibodies were used for Western blots.
Gene expression
Total RNA was extracted from clones and YAMC cells using AllPrep DNA/RNA/Protein Mini Kit (Qiagen). Gene expression microarrays were performed using Mouse WG-6 v2.0 Expression BeadChip according to manufacturer's instructions (Illumina). Differentially expressed genes were screened using a 5% false discovery rate. Gene expression was compared for each clone and compared with averages for controls. Genes with the greatest degree of differential expression were further analysed by averaging all transformed clones and comparing results with control averages using p<0.001. Response networks were analysed by Ingenuity Pathways Analysis software (Qiagen).
Results
E. faecalis-infected macrophages and 4-HNE cause mutations
E. faecalis-infected murine macrophages (RAW264.7) strongly expressed inducible nitric oxide synthase 2 and did not express arginase 1 (figure 1A). In addition, our previous study showed substantial TNFα production in supernatants of E. faecalis-infected macrophages,21 indicating M1 polarisation of RAW264.7 cells by E. faecalis. To evaluate the mutagenic potential of E. faecalis-polarised macrophages, we initially exposed YAMC cells to infected macrophages using tsA58 as a target for mutagenesis. Mutant fractions increased significantly after only four treatments compared with cells co-cultured with uninfected macrophages (figure 1B, p<0.01). The highest mutant fractions occurred after 10 treatments (56.8±10.6 per 1×105 cells). To determine the role of 4-HNE—a diffusible endogenous mutagen, clastogen and spindle poison produced by polarised macrophages13 ,15—we exposed YAMC cells to 1 µM 4-HNE for 1 h once a week. Compared with shams, 4-HNE-treated cells showed significantly increased mutant fractions after six doses with peak fractions occurring after 12 treatments (311.3±18.3 per 1×105 cells, p=0.02; figure 1C). Of note, the fractions steadily decreased after 12 treatments for E. faecalis-infected macrophages and 14 treatments with 4-HNE (figure 1B,C), possibly due to accumulating mutations leading to cell death and/or senescence. These data show that, up to a point, repetitive in vitro exposure of primary epithelial cells to E. faecalis-polarised macrophages or 4-HNE causes cumulative genotoxicity.

Enterococcus faecalis-infected macrophages and 4-hydroxy-2-nonenal (4-HNE) are mutagenic to primary colon epithelial cells. (A) Western blotting shows expression of nitric oxide synthase 2 (Nos2), but not arginase 1, in RAW264.7 cells after infection with E. faecalis, indicating M1 polarisation. (B) Mutant fractions for tsA58 significantly increase following weekly exposure of YAMC cells to E. faecalis-infected macrophages (closed squares) compared with uninfected macrophages (open squares). (C) Mutant fractions for tsA58 also increase following eight weekly treatments with 1 µM 4-HNE (closed circles) compared with untreated controls (open circles).
E. faecalis-infected macrophages and 4-HNE induce aneuploidy and CIN
Aneuploidy and CIN are common features of CRC.22 We previously showed that a single exposure of YAMC cells to E. faecalis-infected macrophages was unable to produce aneuploidy or tetraploidy,9 possibly due to robust cellular defence and repair mechanisms. To determine whether repeated exposures could produce CIN, we analysed YAMC cells exposed to E. faecalis-polarised macrophages or 4-HNE by fluorescent-activated cell sorting. Compared with shams, we found significantly increased percentages of aneuploid cells after repetitive treatment using either modality (figure 2A, p<0.001 and p<0.01, respectively).

Enterococcus faecalis-infected macrophages and 4-hydroxy-2-nonenal (4-HNE) induce aneuploidy and chromosomal instability (CIN) in primary colon cancer cells. (A) After 8 weekly treatments, the rate of aneuploidy significantly increases in YAMC cells co-cultured with E. faecalis-infected macrophages (red squares) compared with untreated control (blue circles). The proportion of aneuploid cells increases after only two treatments with 1 µM 4-HNE (green triangles). (B) Representative histograms of mitotic cells by fluorescent-activated cell sorting show increased numbers of aneuploid cells (R1 and R3 windows) in YAMC clones isolated after 10 treatments with E. faecalis-infected macrophages (37M10-3, middle) and 8 treatments with 4-HNE (H8-4, right) compared with sham-treated cells (left). (C) and (D), fluorescence in situ hybridisation analysis shows aberrant karyotypes with chromosomal translocations (arrows). Red, chromosome 11; green, chromosome 18.
To determine whether these treatments generated heritable CIN, we analysed cells for ploidy. Clones were generated by expanding single cells at 37°C after 6–10 treatments with either E. faecalis-infected macrophages or purified 4-HNE. Eighteen of 22 clones (82%) displayed an aneuploid karyotype (table 1 and figure 2B). In addition, fluorescence in situ hybridisation showed numerous chromosome translocations, indicating CIN (figure 2C,D). To determine whether MSH2, a mismatch repair gene commonly implicated in CRCs with microsatellite instability (MIN), was altered in clones, we performed Western blotting for this protein. Increased MSH2 expression was noted compared with controls (see online supplementary figure S1), suggesting that this form of genomic instability was unlikely in these clones.
Genomic instability and tumour growth for YAMC clones and control cell lines
Cellular transformation
Anchorage-independent growth is a hallmark of cellular transformation. To investigate whether repeated exposure of YAMC cells to E. faecalis-infected macrophages or 4-HNE led to this phenotype, we tested cells for growth in soft agar after every two treatments. Untreated cells failed to grow in soft agar while treated cells showed anchorage-independent growth (figure 3A).

Enterococcus faecalis-activated macrophages and 4-hydroxy-2-nonenal (4-HNE) cause cellular transformation. (A) Anchorage-independent growth (upper graph) significantly increases for YAMC cells grown in soft agar after two treatments with 1 μM 4-HNE (pink) or E. faecalis-infected macrophages (blue) compared with sham-treated controls (green). HCT116 cells are a positive control (red). Growth of multicellular spheroids for YAMC cells is evident after 10 treatments with 4-HNE (middle lower) or E. faecalis-infected macrophages (left lower), and as a control following no treatment for HCT116 cells (right lower). (B) Allografts grow in flanks of NOD/scid mouse injected with clone M15 (right) compared with no growth for untreated YAMC cells (left); excised tumours for clone M15 (below). (C) H&E staining of clone M15 allograft shows poorly differentiated carcinoma (20×). (D) Staining for cytokeratins (brown) confirms epithelial origin (20×). (E) Staining for Dclk1 shows abundant expression of the tumour stem cell antigen (20×). (F) PCR for SV40 large T antigen gene in allografts further confirms YAMC as the cells of origin.
To further assess the oncogenic and transformation inducing abilities of E. faecalis-polarised macrophages or 4-HNE, we engrafted 25 clones into the flanks of NOD/scid mice. Injection of HCT116 human colon cancer cells (as controls) resulted in large tumours (see online supplementary figure S2). No tumour growth was noted for YAMC cells and no clone, except M17 (see online supplementary figure S3A–D), formed tumours when injected directly into mice. However, when clones were premixed with matrigel, 10 of 25 clones developed flank masses (table 1, figure 3B). Of note, all were derived from clones exposed to at least 10 treatment cycles. Eight of 10 masses were poorly differentiated carcinomas (figure 3C) with 3 tumours invading skin and/or muscle (see online supplementary figure S3E,F). One mass was lymphoid and may represent a spontaneously formed neoplasm known to develop in NOD/scid mice.23 Immunohistochemical staining using a pan-keratin reagent confirmed 8 of 10 flank masses as epithelial in origin (figure 3D). Staining of each carcinoma was also positive for Dclk1 (figure 3E). Finally, these tumours were verified as being derived from YAMC cells by amplifying the gene for SV40 large T antigen (figure 3F). For clones H3 and 37M10-3, weakly positive PCRs likely represented a small number of transformed YAMC cells that had persisted within larger flank masses. In aggregate, these findings indicated that exposure of a primary colon epithelial cell line to commensal-polarised macrophages or to 4-HNE resulted in clones that grew as poorly differentiated invasive carcinomas expressing the tumour stem cell marker Dclk1.
Gene expression in transformed clones
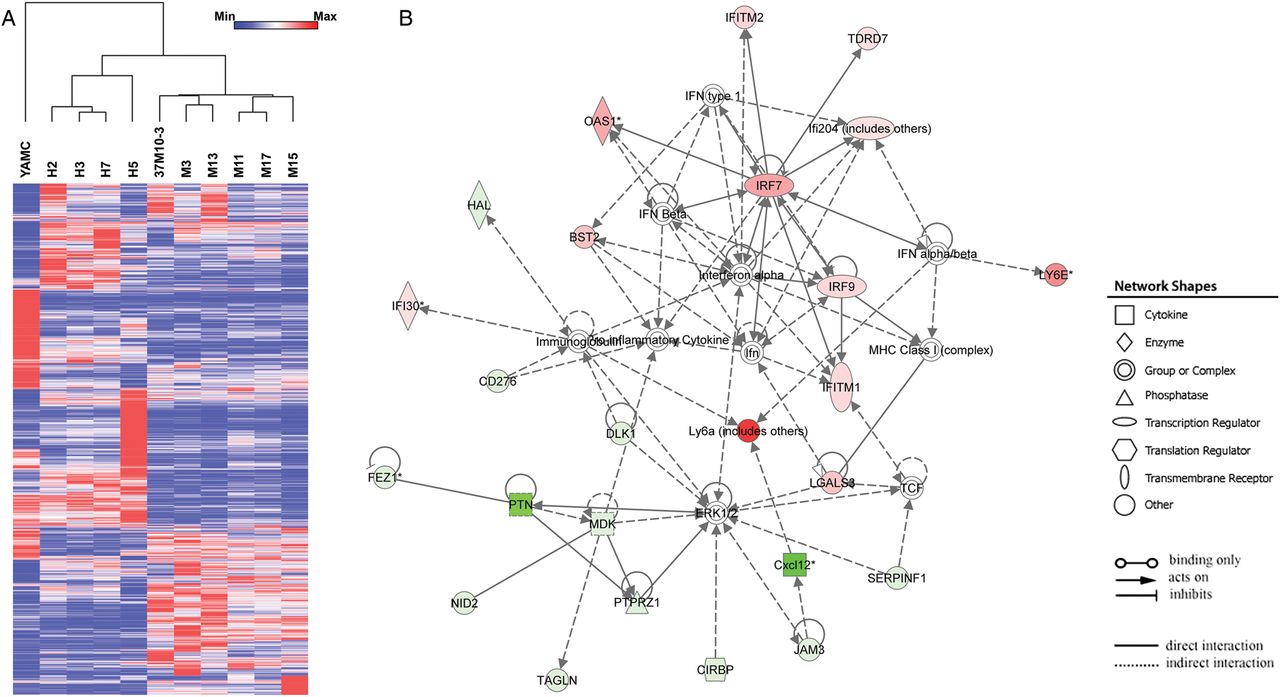
To explore gene expression associated with cellular transformation, whole-genome profiling was performed on 10 transformed clones. Expression data were normalised and comparisons made with untreated YAMC cells. Hierarchical clustering was performed for 11 000 of the most variable probes and correlations established using two controls (r=0.9), four clones from 4-HNE treatments (r=0.4–0.7) and six clones from polarised macrophage treatments (r=0.3–0.5). We filtered these probes down to 2391 that had at least a twofold change compared with controls and identified 1974 differentially expressed genes (figure 4A). Of these, 567 genes were unique for 4-HNE generated clones, 688 for macrophage induced clones and 719 that were shared by all 10 transformed clones. Compared with controls, each transformed clone contained three to seven cancer driver genes (table 2).2 Finally, Dclk1 was not differentially expressed in any clone.
Driver genes for cancer in transformed clones

Gene expression in transformed YAMC clones. (A) Heat map shows a total of 2391 probes representing 1974 genes differentially expressed in 10 transformed clones compared with untreated YAMC cells. Dendrogram indicates strong correlations among four clones isolated from 4-HNE-treated YAMC cells and six clones from YAMC cells repetitively exposed to Enterococcus faecalis-polarised macrophages. (B) The most highly significant network consists of 25 differentially expressed genes. Green, decreased expression; red, increased expression.
To identify response networks, we averaged gene expression for these same 10 transformed clones and compared the results with untreated YAMC cells. There were 151 significantly differentially expressed genes of which 62 were upregulated and 89 downregulated (see online supplementary table S1). Ingenuity pathway analysis identified eight response networks (see online supplementary table S2). The first ranked network contained 25 genes involved in infectious diseases and cell morphology (figure 4B). Other networks included cell cycle regulation, cell growth and proliferation, and cancer development (see online supplementary table S2). Finally, several gene regulators were identified including Mapk1, Ifna2, Trim24, IfnG and Trp53.
Stem/progenitor and tumour stem cell markers
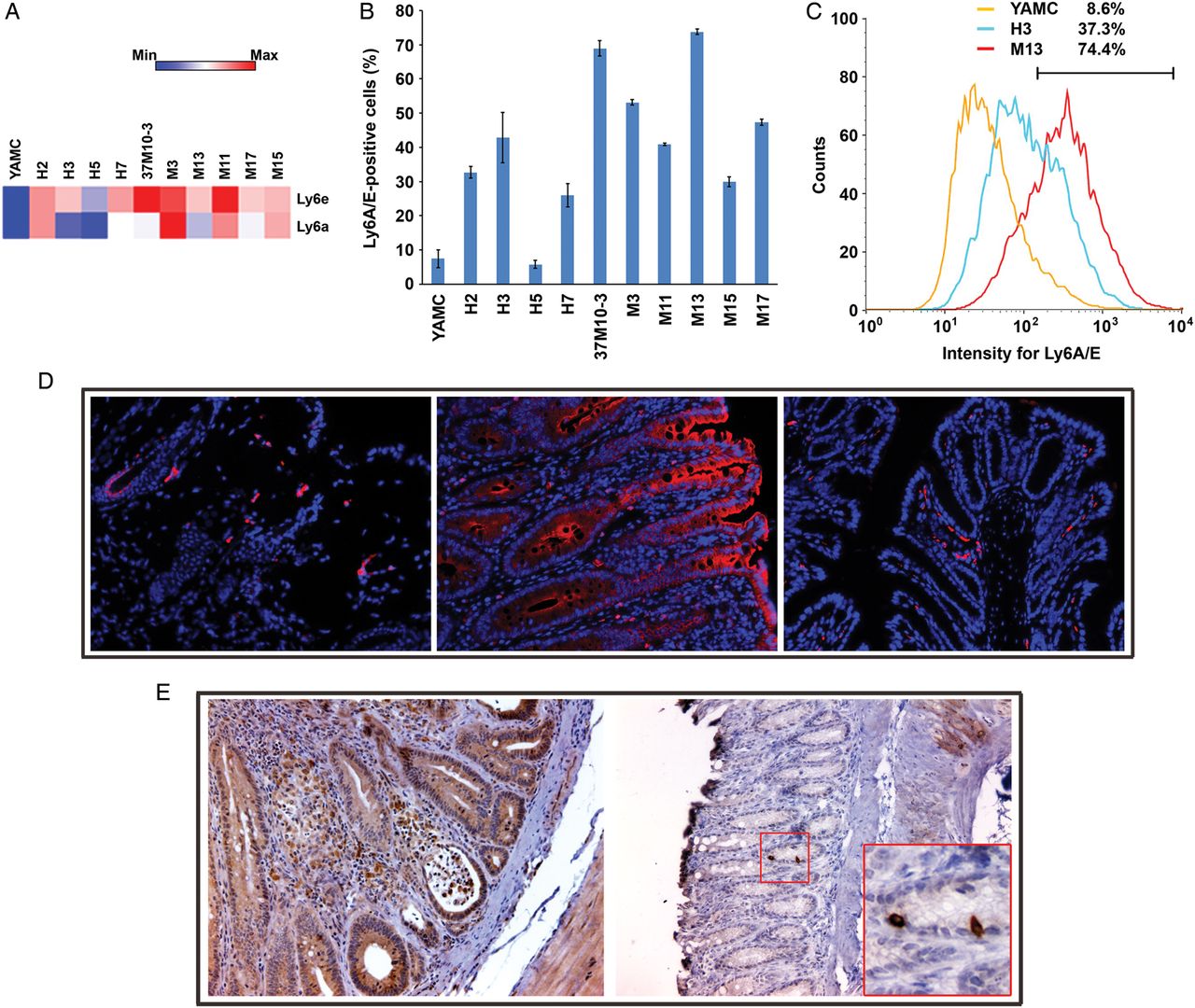
Ly6 genes code for haematopoietic stem/progenitor cell markers that are expressed in diverse cancers.18 Ly6e showed strong upregulation in nearly all transformed clones (average fold-change (±SD), 8.39±2.40 and 7.96±1.97 for each probe). In addition, Ly6a expression was also increased (average fold-change of 16.8±8.33 for one probe) (figure 5A). To confirm these data, we assayed transformed clones for Ly6A/E by fluorescent-activated cell sorting and compared results with untreated YAMC cells. We found that the percentage of positive cells was significantly increased for these surface markers in all clones except H5 (42.1±18.7 vs 7.4±2.6, p<0.001 by analysis of variance) (figure 5B,C). In addition, Ly6A/E-positive cells were noted in allografts (figure 5D, left and see online supplementary figure S4A). Compared with sham-colonised Il10−/− mice, Ly6A/E was strongly expressed by colonic epithelial cells from E. faecalis-colonised mice (figure 5D, middle and right; see online supplementary figure S4B,C). Although Dclk1 was non-differentially expressed in transformed clones, staining for the tumour stem cell marker was positive in numerous epithelial and stromal cells of E. faecalis-colonised Il10−/− mice (figure 5E). These findings confirm induction of stem/progenitor and tumour stem cell markers in murine allografts and epithelial and stromal cells from biopsies of a BSE-rich tissue microenvironment.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transformed clones show increased stem/progenitor cell marker expression. (A) Genes for haematopoietic stem/progenitor cell markers Ly6e and Ly6a are significantly upregulated in transformed clones compared with untreated YAMC cells. (B) The percentage of Ly6A/E-positive cells is increased in 9 out of 10 transformed clones compared with untreated YAMC controls. (C) A representative histogram shows increased Ly6A/E expression in transformed clones H3 and M13 compared with untreated YAMC cells. (D) Immunofluorescent staining shows Ly6A/E-positive cells (red) in the M11 allograft tumour (20×, left) with nuclei counterstained using 4′,6-diamidino-2-phenylindole (DAPI) (blue); inflamed and neoplastic epithelial cells (middle) strongly stain for Ly6A/E in colon from Enterococcus faecalis-colonised Il10−/− mice (20×); no Ly6A/E expression in normal colon epithelia from sham-colonised Il10−/− mice (right) although a few positively stained immune cells are visible in the lamina propria (20×). (E) Strong staining (brown) for Dclk1 is seen in epithelial and lamina propria cells from inflamed and dysplastic colons of E. faecalis-colonised Il10−/− mice (20×; left); minimal staining is seen in colon biopsies from sham-colonised mice (20×; right); inset (60×) shows rare Dclk1+ colon crypt cells, most likely representing tuft cells.
Discussion
The majority of solid tumours, including CRCs, arise from the progressive accumulation of mutations in normal epithelial cells that lead to oncogenic transformation.1 ,2 Searches for the origin of mutations that initiate the adenoma-to-carcinoma sequence in CRC have usually focused on exogenous carcinogens or metabolically converted pro-mutagens in foodstuffs. Few studies, however, have investigated endogenous mutagens. This work shows that commensal-polarised macrophages,10 and a clastogen produced by them, 4-HNE,13 ,15 are potent initiators of CIN that lead to the transformation of primary colon epithelial cells through BSE.
BSE has been most thoroughly described following in vivo or in vitro irradiation.24 This phenomenon is defined by clastogens that diffuse into neighbouring cells to cause CIN. Sophisticated congenic sex-mismatch bone marrow transplant studies have confirmed that BSE occurs in vivo.25 In addition, BSE is observed in animal models where second primary tumours occur in organs remote from primary sites of carcinogenesis.26 ,27 We expanded our understanding of BSE by showing that polarisation of macrophages through infection can also generate this effect.10 ,13 Infection-induced BSE and radiation-induced BSE are similar in that both have been linked to COX-2.8 ,15 ,28 E. faecalis provides proof-of-principle that human commensals can polarise colonic macrophages to initiate CIN and cellular transformation. This is not meant to imply that other commensal bacteria cannot similarly promote BSE. Indeed, Escherichia coli, a Gram-negative bacillus, has been shown to stimulate macrophages to produce 4-HNE in vitro13 and, much like E. faecalis, a Gram-positive coccus, generate BSE and cellular transformation in colonised Il10−/− mice.7
Polarised macrophages are found following acute infection, during chronic inflammation and in association with tumours.12 ,29 M1 polarised macrophages help clear infections while M2 macrophages assist in parasite clearance and tissue remodelling. Of note, M2 macrophages can also promote the progression of extant cancers.12 Resting intestinal macrophages are neither M1 nor M2.11 Instead, under normal conditions, these cells are potently phagocytic but remain non-inflammatory. This distinctive phenotype is a result of the local intestinal microenvironment and helps maintain appropriate homoeostasis and tolerance to commensals. In the Il10−/− model, colon macrophages are polarised by colonisation with E. faecalis to a M1 phenotype. This phenotype appears essential to BSE.10 The interaction of polarised macrophages with other immune cells in colorectal carcinogenesis, including mast cells, dendritic cells, T cells or natural killer cells,5 ,30–35 is likely important to the regulation of BSE but not yet fully understood and merits additional investigation.
BSE is a plausible theory for sporadic and colitis-associated CRC. It directly addresses the origin of CIN, the most common genotype for these cancers, and links COX-2 reactivity to the initiation of the adenoma-to-carcinoma sequence. In this study, we found that repetitive exposure of primary colon epithelial cells to E. faecalis-polarised macrophages or 4-HNE induced aneuploidy and heritable CIN. These findings are consistent with the notion that long-term exposure of epithelial cells to clastogens initiates genomic instability in CRC. Only a minority of our transformed clones did not develop CIN (table 1). Of the many clones that were aneuploid, a majority failed to form tumours in NOD/scid mice. We speculate that this is a reflection of the known tumour-suppressive effects of aneuploidy.36
Previous studies have shown that CIN and MIN can be induced by specific carcinogens.37 For example, tissue culture cells surviving near-lethal doses of the exogenous mutagen 2-amino-1-methyl-6-phenylimidazo (4,5-b) pyridine acquire CIN.38 In contrast, cells resistant to treatment with N-methyl-N’-nitro-N-nitrosoguanidine have exhibited MIN. 4-HNE forms bulky adducts39 and in this study, as with 2-amino-1-methyl-6-phenylimidazo (4,5-b) pyridine, was primarily associated with CIN instead of MIN. These findings are consistent with predictions that bulky-adduct-forming mutagens preferentially cause CIN while methylating agents induce MIN.37 4-HNE is an α,β-unsaturated aldehyde that readily modifies proteins, forms DNA adducts and contributes to carcinogenesis by inhibiting DNA repair, inducing COX-2, and modulating mitogen-activated protein kinases (MAPK) and nuclear factor (NF)-κB signalling.14 In addition, 4-HNE causes double-stranded DNA breaks that may help initiate CIN.9 These breaks are difficult to repair, generate dicentric chromosomes through non-homologous end-joining repair, and result in anaphase bridging.40 The result can be lagging chromosomes, multipolar mitoses, and missegregation. When bridges fragment, chromosomes enter breakage-fusion-bridge cycles that produce rearrangements and aneuploidy.40 Finally, 4-HNE also disrupts mitotic spindles by activating stathmin, a key regulatory protein in microtubule kinetics, and thereby produces microtubule catastrophe.13 These overlapping mechanisms likely individually and jointly contribute to the initiation of CIN by 4-HNE.
Whole genome sequencing of human cancers has identified ∼90 mutations per tumour.2 Of the many genes affected, only 125 or so ‘driver’ genes seem important to tumour growth. Typically, two to eight driver mutations are required for malignant transformation.2 ,41 In our study, we found differential expression in three to seven driver genes for each transformed clone. In silico analyses detected altered expression in several gene regulators commonly found in cancer (eg, Trim24, Mapk1 and Trp53). We did not investigate, however, whether these changes were due to mutations, copy number alternations, epigenetic modification or disruption of regulatory pathways. Detailed characterisation of mutations and/or regulatory changes in these clones was beyond the scope of this investigation.
Primary epithelial cells from the bladder, cervix, colon, kidney, lung and breast of normal mice can acquire chromosomal aneuploidy and centrosomal instability upon prolonged in vitro passage (eg, 6–12 months).42 In contrast, the primary murine colon cells in our study were rapidly transformed upon exposure to polarised macrophages or 4-HNE. Techniques for the spontaneous transformation of cells commonly involve a 10-day refeeding protocol that creates nutrient starvation and oxidative/metabolic stresses that likely contribute to genotoxicity. The mechanism(s) for this phenomenon, however, remains to be defined. Our approach involved feeding YAMC cells on a normal schedule (ie, thrice a week) and consisted of only 10 treatments prior to identifying clones that grew as poorly differentiated carcinomas in immunodeficient mice. Unexposed YAMC cells were not transformed. Finally, it should be noted that epithelial tumours do not spontaneously develop in mice, including Il10−/− mice, unless carcinogens, inflammation, or changes in oncogenes and/or tumour suppressors are introduced. In contrast, colonisation of Il10−/− mice by E. faecalis triggers events that generate molecular signatures for BSE and within 6–9 months results in CRC.10 ,13
The expression of Dclk1 for clones that grew as carcinomas in immunodeficient mice was surprising since gene expression analyses failed to identify this gene. This lack of difference in mRNA may reflect regulation by post-translational processing and is an area of ongoing investigation. In addition to Dclk1 expression in allografts, we also found widespread expression in epithelial and stromal cells of dysplastic and cancerous tissues from E. faecalis-colonised Il10−/− mice. In the normal intestinal epithelium Dclk1 cells appear as fully differentiated epithelial cells and are synonymous with tuft cells.19 ,43 These quiescent cells have a long life span, a characteristic likely essential for acquiring multiple mutations as cells undergo transformation. Recent work using the ApcMin/+ model of intestinal tumorigenesis identified Dclk1-expressing cells as tumour stem cells.44 ,45 Deletion of Dclk1 cells resulted in the regression and elimination of intestinal tumours, suggesting that this marker was functionally required for tumour growth. Finally, Dclk1-positive stromal cells in colon biopsies from E. faecalis-colonised Il10−/− mice may represent cells undergoing the epithelial-mesenchymal transition.19 This process is defined by a conversion of epithelial cells to divergent phenotypes involved in wound healing, fibrosis and the metastatic spread of cancer. These observations are an area of ongoing investigation.
Another distinctive gene expression signature in transformed clones was Ly6A and Ly6E. These genes are part of a multigene family of glycosyl phosphatidylinositol-anchored cell surface proteins and considered stem/progenitor cell markers.18 In YAMC cells, members of the Ly6 superfamily are upregulated by interferon-γ, IL-22 and TNFα.46 Ly6A (or stem cell antigen-1) marks murine haematopoietic stem cells and is involved in cell-cell adhesion and signalling, stem cell self-renewal, and stress responses.18 It has no human homologue. Notably, overexpression of Ly6A occurs in several murine cancers including prostate and breast.47 ,48 Silencing Ly6a alters cell proliferation, migration and organisation.49 Increased expression of Ly6A and Ly6C has been previously reported in colon biopsies from mice with colitis.46 Ly6e, or locus E of the Ly6 multigene family, is an ortholog of LY6E in humans and was strongly expressed in nearly all transformed clones. Because LY6E is overexpressed in human pancreatic cancer stem cells and CRC,50 dysregulation of this gene may contribute to tumorigenesis. Finally, as in transformed YAMC clones, we observed marked upregulation of Ly6A/E in colon epithelial cells in Il10−/− mice that were colonised with E. faecalis.
In summary, repetitive exposure of primary colonic epithelial cells to commensal-polarised macrophages, or the endogenous clastogen 4-HNE, induced CIN, caused transformation via BSE, increased expression of tumour stem cell and stem/progenitor-like markers, and led to the formation of poorly differentiated and invasive tumours in immunodeficient mice. These findings are evidence for commensal-induced endogenous CIN and cellular transformation leading to CRC. Understanding mechanisms by which commensals initiate and promote CRC will permit new preventive strategies for decreasing the incidence of this common human cancer.
Acknowledgments
The authors thank Bart Frank at the Oklahoma Medical Research Foundation for gene expression analyses, Ravindranauth Sawh in the Department of Pathology at the University of Oklahoma Health Sciences Center, and Jim Henthorn in the Flow Cytometry Laboratory, and Histology and Immunohistochemistry Core in the Peggy and Charles Stephenson Cancer Center (NIH GM103639).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online tableS1
- Data supplement 3 - Online tableS2
Footnotes
-
Contributors MMH provided study supervision; XW and MMH are responsible for study design and concepts, and they analysed data, and wrote the manuscript; XW and YY performed the research.
-
Funding This study was supported by NIH CA127893 (MMH), Oklahoma Center for the Advancement of Science and Technology HR10-032 (XW), and Francis Duffy Endowment.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Microarray data has been deposited into Gene Expression Omnibus (Accession number GSE55233). No other unpublished data is available.