Article Text

Abstract
Objective Although direct-acting antiviral agents (DAAs) have markedly improved the outcome of treatment in chronic HCV infection, there continues to be an unmet medical need for improved therapies in difficult-to-treat patients as well as liver graft infection. Viral entry is a promising target for antiviral therapy.
Design Aiming to explore the role of entry inhibitors for future clinical development, we investigated the antiviral efficacy and toxicity of entry inhibitors in combination with DAAs or other host-targeting agents (HTAs). Screening a large series of combinations of entry inhibitors with DAAs or other HTAs, we uncovered novel combinations of antivirals for prevention and treatment of HCV infection.
Results Combinations of DAAs or HTAs and entry inhibitors including CD81-, scavenger receptor class B type I (SR-BI)- or claudin-1 (CLDN1)-specific antibodies or small-molecule inhibitors erlotinib and dasatinib were characterised by a marked and synergistic inhibition of HCV infection over a broad range of concentrations with undetectable toxicity in experimental designs for prevention and treatment both in cell culture models and in human liver-chimeric uPA/SCID mice.
Conclusions Our results provide a rationale for the development of antiviral strategies combining entry inhibitors with DAAs or HTAs by taking advantage of synergy. The uncovered combinations provide perspectives for efficient strategies to prevent liver graft infection and novel interferon-free regimens.
- HCV
- HEPATITIS C
- LIVER
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Direct-acting antivirals (DAAs) increase the response to IFN-based antiviral therapy against HCV genotype 1 but also lead to selection of drug-resistant HCV variants.
-
Given their important side effects and drug–drug interactions, DAAs against HCV are not approved for patients undergoing liver transplantation (LT), HCV/HIV coinfected patients or paediatric patients.
-
Although early clinical trials have demonstrated impressive outcomes for combinations of DAAs in IFN-free regimens for treatment-naive patients, there will be a need for novel antivirals addressing resistance, treatment of patients with comorbidity, co-medication or immunosuppression and patients undergoing LT.
-
HCV entry into target cells is a promising target for preventive and therapeutic antiviral strategies.
What are the new findings?
-
Given their complementary mechanism of action, entry inhibitors inhibit viral infection in a synergistic manner in combination with DAAs both in cell culture models and in human liver-chimeric uPA/SCID mice.
-
Synergy between entry inhibitors and DAAs or other host-targeting agents holds promise for a variety of possibilities of combination therapies for prevention of HCV liver graft infection and might be also useful for treatment of hepatitis C.
How might it impact on clinical practice in the foreseeable future?
-
Novel combinations based on synergy may widen the therapeutic arsenal against HCV infection for prevention of liver graft infection, treatment of difficult-to-treat-patients and provide alternatives for patients with contraindications to particular compounds of standard of care or future IFN-free regimens.
Introduction
The current standard of care (SOC) in chronic genotype 1 HCV infection consists of pegylated IFN-α, ribavirin (RBV) and a protease inhibitor—telaprevir or boceprevir.1–3 Furthermore, direct-acting antivirals (DAAs) sofosbuvir and simeprevir have been licensed very recently. Although expected to revolutionise HCV treatment by offering cure in the very large majority of treated patients,4 DAAs appear to have distinct limitations in certain difficult-to-treat patient subgroups, such as patients with advanced liver disease, transplant, HIV/HCV-coinfected and immune-compromised patients.2 Hurdles in these groups include resistance and side effects.2 ,4 Furthermore, not all genotypes respond similar to DAA combinations and high costs limit access to therapy in low-income, middle-income and high-income countries.2 ,4
Among the most difficult-to-treat patient groups are individuals with HCV-induced end-stage liver disease undergoing liver transplantation (LT).1 ,2 Due to viral evasion from host immune responses and absence of preventive antiviral strategies, graft reinfection is universal. Moreover, IFN-based therapies have limited efficacy and tolerability in LT recipients.5 The use of telaprevir and boceprevir is limited by drug–drug interactions if combined with immunosuppressive agents.6 ,7 Furthermore, emergence of resistant strains will certainly be higher in these patients because of lower efficacy of SOC and greater need for dose adjustments.5 These challenges define a need for additional combinations of therapeutics, ideally targeting complementary steps of the viral life cycle, with improved efficacy and safety.
A promising antiviral target complementary to targets of SOC and DAAs is viral cell entry. HCV entry is the first step of virus–host cell interactions and is required for dissemination and maintenance of infection.8 Viral entry plays an important role in the pathogenesis of HCV infection, especially during HCV reinfection of the graft after LT.9 ,10 Targets for entry inhibitors include CD81, scavenger receptor class B type I (SR-BI), tight junction proteins CLDN1 and occludin, epidermal growth factor receptor (EGFR)8 ,11 and Nieman-Pick C1-Like 1.12 Entry inhibitors exhibit a broad pan-genotypic activity.10 ,12–21 By acting through a complementary mechanism of action (MOA), entry inhibitors may synergistically act with SOC. To explore the future application of entry inhibitors for combination therapy in IFN-free regimens, we investigated the antiviral efficacy of entry inhibitors in combination with DAAs or other host-targeting agents (HTAs) in state-of-the-art cell culture models and in human liver-chimeric uPA/SCID mice.
Material and methods
Cell lines
Huh7.5.1 cells and primary human hepatocyte (PHH) culture have been described.13
Antibodies and inhibitors
CLDN1- (OM-7D3-B3),22 SR-BI- (NK-8H5-E3)14 and CD81-specific (QV-6A8-F2C4)16 monoclonal antibodies (mAbs) have been described. Erlotinib and dasatinib were from LC Laboratories, and IFN-α2a and IFN-α2b were from Roche and Merck. Alisporivir, telaprevir, boceprevir, danoprevir, simeprevir, daclatasvir, mericitabine and sofosbuvir were synthesised by Acme Bioscience.
Analysis of antiviral activity of compounds and combinations on HCV infection
Each compound was tested individually or in combination with a second compound using the cell culture-derived HCV (HCVcc)-Huh7.5.1 model.14 ,23 Production of HCVcc has been described.13 For prophylactic/prevention combination experiments, Huh7.5.1 cells were preincubated with IFN-α, DAAs or HTAs for 1 h at 37°C before incubation for 4 h at 37°C with HCVcc and both compounds. For therapeutic/treatment combination experiments, Huh7.5.1 cells were infected with HCVcc during 5 days to establish chronic persistent HCV infection before adding compounds for additional 5 days.23 Viral infection was analysed by assessing luciferase activity.10 ,13 ,22
Analysis of synergy
Synergy was assessed by two independent methods: the combination index (CI)24 and/or the Prichard and Shipman method.25 A CI <0.9, 0.9–1.1 and >1.1 indicates synergy, an additive effect and antagonism, respectively.24 For the Prichard and Shipman method, a surface >20% above the zero plane indicates synergy and a surface <20% below the zero plane indicates antagonism.25 Validity of the assay and methods was confirmed by non-synergistic or antagonistic combinations.
HCV cell culture persistence assay
HCV RNA (Jc1 or Luc-Jc1) was electroporated into Huh7.5.1 cells to establish persistent HCV infection. Then, 1% DMSO was used to differentiate the cells and maintain them in culture without passage for up to 20 days.26 Medium containing compounds at the indicated concentrations was replenished every 4–5 days until the end of the experiment. Luciferase activity or RT-PCR was used to monitor viral infection and viral load.27
In vivo experimentation
Human liver-chimeric uPA/SCID mice were transplanted with PHH at 3 weeks of age by intrasplenic injection of 106 cells suspended in PBS as described previously.28 Successful engraftment was determined by measuring the human albumin (HA) concentration in the serum of transplanted mice by specific ELISA (Bethyl, Catalogue No. E80-129). Mice with HA levels >1 mg/mL were used for IV inoculation with HCV Jc1-containing infectious mouse serum (6×103 IU). Eight weeks later, the mice were allocated to different treatment groups. Mice received telaprevir (300 mg/kg) or vehicle (carboxymethylcellulose 0.5%, tween-80 0.2%) per os twice a day and were intraperitoneally injected with 500 µg of control or anti-SR-BI mAb (NK8-H5-E3) twice a week for 2 weeks. Blood was collected by retro-orbital puncture every 5–10 days under isoflurane anaesthesia for the determination of serum HCV RNA level and HA concentration. Experiments were performed in the Inserm Unit 1110 animal facility according to local laws and ethical committee approval (AL/02/19/08/12 and AL/01/18/08/12).
Toxicity assays
Huh7.5.1 cells and PHH were incubated with compounds for 48 h and/or 5 days.22 ,23 Cytotoxic effects were analysed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay13 ,22 or PrestoBlue assay (Invitrogen) with flavopiridol or anti-Fas antibody as positive controls.22 The 50% cytotoxic concentrations (CC50) of entry inhibitors were calculated by regression analysis.
Statistical analysis
Statistical analysis and CI estimations have been run under Bayesian paradigm. Results are given as mean and (95% credible interval). Data were analysed by IC (50/75/90). Group comparisons were based on the mean difference. Normality was assessed with a Shapiro–Wilk test. When required, data transformation was used to reach normality. Each data set was analysed using hierarchical (mixed) model with fixed group effects and random treatment effect as described.29 The whole data set was analysed using a two-stage hierarchical model, with the fixed group effects and two random effects that were treatment and IC (50/75/90), in order to take account of both levels of repeated measurements. Dummy variables, representing the IC studied (50/75/90), had also been considered as fixed effects to test differences between CI in each case. For all of these models, uninformative priors for coefficients were used: Gaussian distributions with mean 0 and precision 0.001, gamma distribution with parameters 0.1 and 0.1 for the model precision. Hyperpriors for random effects were also uninformative: normal with mean 0 and precision 0.001, and a uniform distribution (0.100) for dispersion parameters. Assumption of homogeneous dispersions in random effects was respected. Computations were run with R 3.00 and WinBUGS 1.4. For each analysis, a single MCMC chain with 5000 iterations as burn-in and 100 000 iterations was used to generate the posterior distribution. Convergence was checked and present in every case. Unless otherwise stated, results are shown as means±SEM from three independent experiments performed in triplicate. For the Prichard and Shipman method, one representative experiment performed in triplicate is shown.
Results
Synergy of entry inhibitors and DAAs uncovers novel combinations for IFN-free regimens
A major effort of current drug development is to develop IFN-free treatments based on the combination of DAAs with or without RBV.1 Addressing these concepts, we studied the combined antiviral effect of entry inhibitors with clinically licensed protease inhibitors telaprevir,30 ,31 boceprevir,32 ,33 simeprevir34 and danoprevir—a protease inhibitor in late-stage clinical development35 using the HCVcc cell culture model. The antiviral effect of each molecule was tested alone or in combination to determine the CI. Combination of telaprevir or boceprevir with a sub-IC50 concentration of all entry inhibitors tested—which exerts only minimal inhibition on HCV infection—resulted in synergy with CIs of 0.48–0.71 at IC90 (figure 1A and online supplementary table S1). Calculation of 95% credible intervals indicates that the differences between CI values did not occur by chance (figure 1A and online supplementary table S1). Synergy was also observed at IC50 and IC75 (figure 1A and online supplementary table S1). Inversely, combination of telaprevir and boceprevir was additive (CI of 0.94, 95% credible interval (0.84 to 1.04); figure 1A) confirming the validity of the assay. Combination of simeprevir or danoprevir with entry inhibitors resulted in synergy at all inhibitory concentrations (CIs of 0.06 to 0.65 at IC90; figure 1A and online supplementary table S1), demonstrating the relevance of adding an entry inhibitor to improve antiviral efficacy in preventing HCV infection. Highly effective combinations included inter-alia combinations of telaprevir with erlotinib (figure 2A), and boceprevir or simeprevir with anti-CLDN1 mAb (figure 2B, C).

Combination of direct-acting antivirals and entry inhibitors results in a synergistic activity in preventing HCV infection. Huh7.5.1 cells were preincubated for 1 h with serial concentrations of (A) telaprevir, boceprevir, simeprevir or danoprevir, (B) daclatasvir or (C) mericitabine or sofosbuvir and 0.01 µg/mL of receptor-specific (anti-CD81, anti-SR-BI or anti-CLDN1) mAbs or 0.1 µM protein kinase inhibitors (erlotinib or dasatinib) before incubation with HCVcc Luc-Jc1 in the presence of both compounds. HCVcc infection was analysed by luciferase activity as described.22 The CIs at IC50, IC75 and IC90 are indicated in online supplementary table S1. Means±95% credible intervals from three independent experiments performed in triplicate are shown.

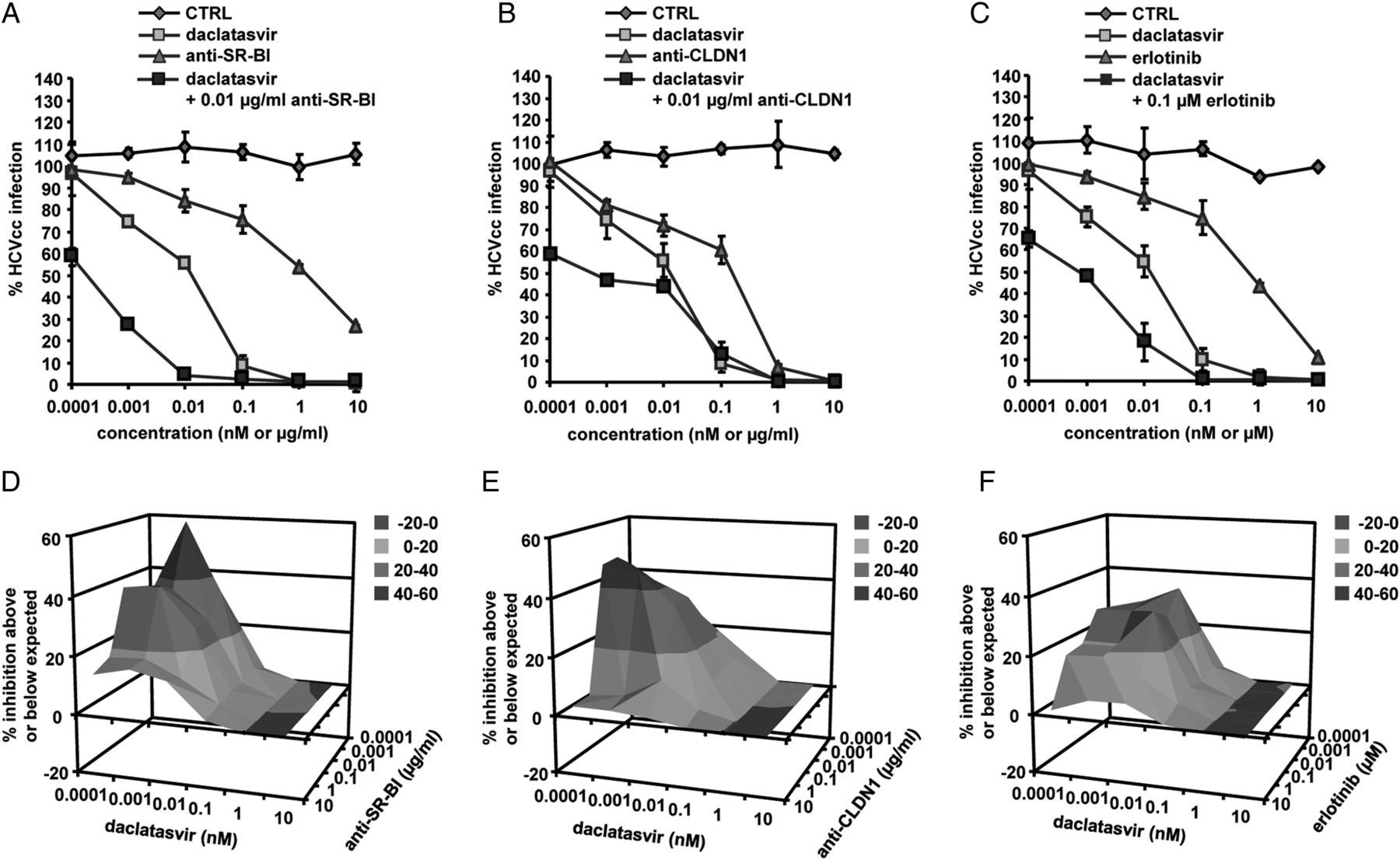
A number of NS5A and polymerase inhibitors have reached early-stage to late-stage clinical development including clinical licensing of the first compounds. As the first NS5A inhibitor, daclatasvir36 has shown potent antiviral activity against HCV genotype 1 in mono- and combination therapy.37 ,38 Marked synergy at all inhibitory concentrations was observed for combination of daclatasvir with entry inhibitors (CIs of 0.27–0.89 at IC90; figure 1B, online supplementary table S1C). Effective combinations included inter-alia combinations of daclatasvir and anti-SR-BI, anti-CLDN1 mAbs or erlotinib decreasing its IC50 up to 60-fold (figure 3A–C).

Finally, we investigated synergy between entry inhibitors and the polymerase inhibitor sofosbuvir,39 which recently obtained FDA approval and has the potential to become the cornerstone for an efficacious, all-oral combination regimen for many HCV-infected patients.40 ,41 Combination of sofosbuvir with entry inhibitors resulted in synergy at all inhibitory concentrations (CIs of 0.41–0.61 at IC90; figure 1C, figure 4A, B, D, E and online supplementary table S1). Potent combinations included inter-alia combinations of sofosbuvir and anti-SR-BI or anti-CLDN1 mAbs, decreasing its IC50 up to 18-fold, and combinations of sofosbuvir with erlotinib or dasatinib decreasing its IC50 up to 210-fold (figure 4A,B,D,E). These data demonstrate the potential of combining sofosbuvir with entry inhibitors to improve its antiviral activity. Similar results were obtained when combining mericitabine35 with entry inhibitors (CIs of 0.18–0.68 at IC90; figure 1C and online supplementary table S1).

Entry inhibitors enhance the antiviral activity of sofosbuvir in a synergistic manner. Combination of sofosbuvir and (A) anti-SR-BI mAb, (B) anti-CLDN1 mAb, (D) dasatinib or (E) erlotinib was performed as described in figure 1. (C, F) Synergy was confirmed using the Prichard and Shipman method.25
To further confirm synergy over a broad range of concentrations of two compounds, we performed combinations of each DAA with a defined entry inhibitor, testing a full checkerboard of compound dose–response curves.25 In particular, combinations of low doses of two compounds resulted in an antiviral effect well above the expected value (figures 2D–F, 3D–F and 4C,F). These results demonstrate that adding an entry inhibitor markedly increases the antiviral activity of different DAAs currently evaluated in IFN-free regimens, thereby defining novel antiviral combinations for further preclinical and clinical development in IFN-free regimens.
Combination of HTAs results in a synergistic antiviral effect
It is still not clear whether DAA-based therapies will be effective and safe in difficult-to-treat populations including patients with comorbidity, complex co-medication, immunosuppression and patients undergoing LT. Combinations of HTAs are a promising alternative to DAAs for IFN-sparing regimens, allowing increasing the genetic barrier to resistance. The clinically most advanced HTA is cyclophilin A inhibitor alisporivir.42 Combination of alisporivir with entry inhibitors resulted in a marked synergy (CIs of 0.19–0.69 at IC75 and 0.06–0.50 at IC50; figure 5D and online supplementary table S2). Effective combinations included inter-alia alisporivir and erlotinib or anti-CLDN1 mAb (figure 5E). Indeed, very low concentrations of these entry inhibitors markedly increased the antiviral effect of alisporivir by decreasing its IC50 up to 3000-fold. Synergy of alisporivir and anti-CLDN1 mAb was confirmed using the Prichard and Shipman method (figure 5F). Taken together, these data demonstrate the promise of combining HTAs acting on complementary steps of the viral life cycle and open perspectives for HTA-based DAA or IFN-free regimens. These combinations may be of particular interest for/to patients who are resistant to SOC, or cannot tolerate SOC or combinations of DAAs. Furthermore, combinations of two entry inhibitors resulted in synergy on HCVcc infection at all inhibitory concentrations (CIs of 0.13–0.68 at IC90; figure 5A–C and online supplementary table S2) except for combination of anti-CLDN1 and anti-CD81 mAbs that resulted in an additive effect (CI of 0.95, 95% credible interval (0.85–1.06) at IC90; figure 5A).

Synergy of host-targeting agents on HCV infection. (A) Combination of receptor-specific (anti-CD81, anti-SR-BI or anti-CLDN1) mAbs and protein kinase inhibitors (erlotinib or dasatinib) was performed as described in figure 1 and the CIs at IC50, IC75 and IC90 are indicated in online supplementary table S2. Means±95% credible intervals from three independent experiments performed in triplicate are shown. (B) Combination of anti-CLDN1 mAb and dasatinib (left) or erlotinib (right). (C) Synergy was confirmed using the Prichard and Shipman method.25 (D) Combination of alisporivir and entry inhibitors was performed as described in figure 1 and the CIs at IC50, IC75 and IC90 are indicated in online supplementary table S2. Means±95% credible intervals from three independent experiments performed in triplicate are shown. (E) Combination of alisporivir with erlotinib (left) or anti-CLDN1 mAb (right). (F) Synergy was confirmed using the Prichard and Shipman method.25
Entry inhibitors potentiate the antiviral activity of IFN-α in a synergistic manner
Since IFN-α is the key component of current SOC, we also investigated whether HCV entry inhibitors potentiate the antiviral activity of IFN-α by combining an entry inhibitor with IFN-α2a or IFN-α2b. Combination of IFN-α2a or IFN-α2b with a sub-IC50 concentration of receptor-specific mAb resulted in a synergistic activity at IC90 in inhibiting HCVcc infection (CIs of 0.16–0.53) (see online supplementary figure S1 and table S3). Synergy combining IFN-α2a or IFN-α2b with erlotinib has been described elsewhere.23 Synergy was also observed at IC50 and IC75 for all combinations tested (see online supplementary figure S1 and table S3). Conversely, in line with previously reported data,43 combination of IFN-α2a or IFN-α2b with sorafenib, a different protein kinase inhibitor (PKI) that inhibits the antiviral effects of IFN, resulted in antagonism (CI of 1.23; 95% credible interval (1.13–1.34) and 1.27, 95% credible interval (1.16–1.38); online supplementary figure S1A,B and table S3), demonstrating that the observed synergies are specific for the combinations and not related to technical issues of the model. Calculation of 95% credible intervals indicates that the differences between CI values did not occur by chance (see online supplementary table S3). These data demonstrate that entry inhibitors enhance the antiviral activity of IFN-α.
Synergy in experimental design for treatment of HCV infection
Finally, to explore whether synergy between DAAs or HTAs and entry inhibitors also occurs in experimental approach mimicking treatment of chronic infection, we used a well-established design of persistent infection.23 Similar to the prevention design shown before, combining DAAs or alisporivir with entry inhibitors also resulted in high synergy when compounds were added postinfection in chronically infected cells (figure 6) as well as in chronically infected DMSO-differentiated hepatoma cells2,6 (figure 7), which are more physiologically relevant than normal hepatoma cells. These data demonstrate the potential of combining DAAs or HTAs with entry inhibitors to improve their antiviral activity for the treatment of chronic hepatitis C.
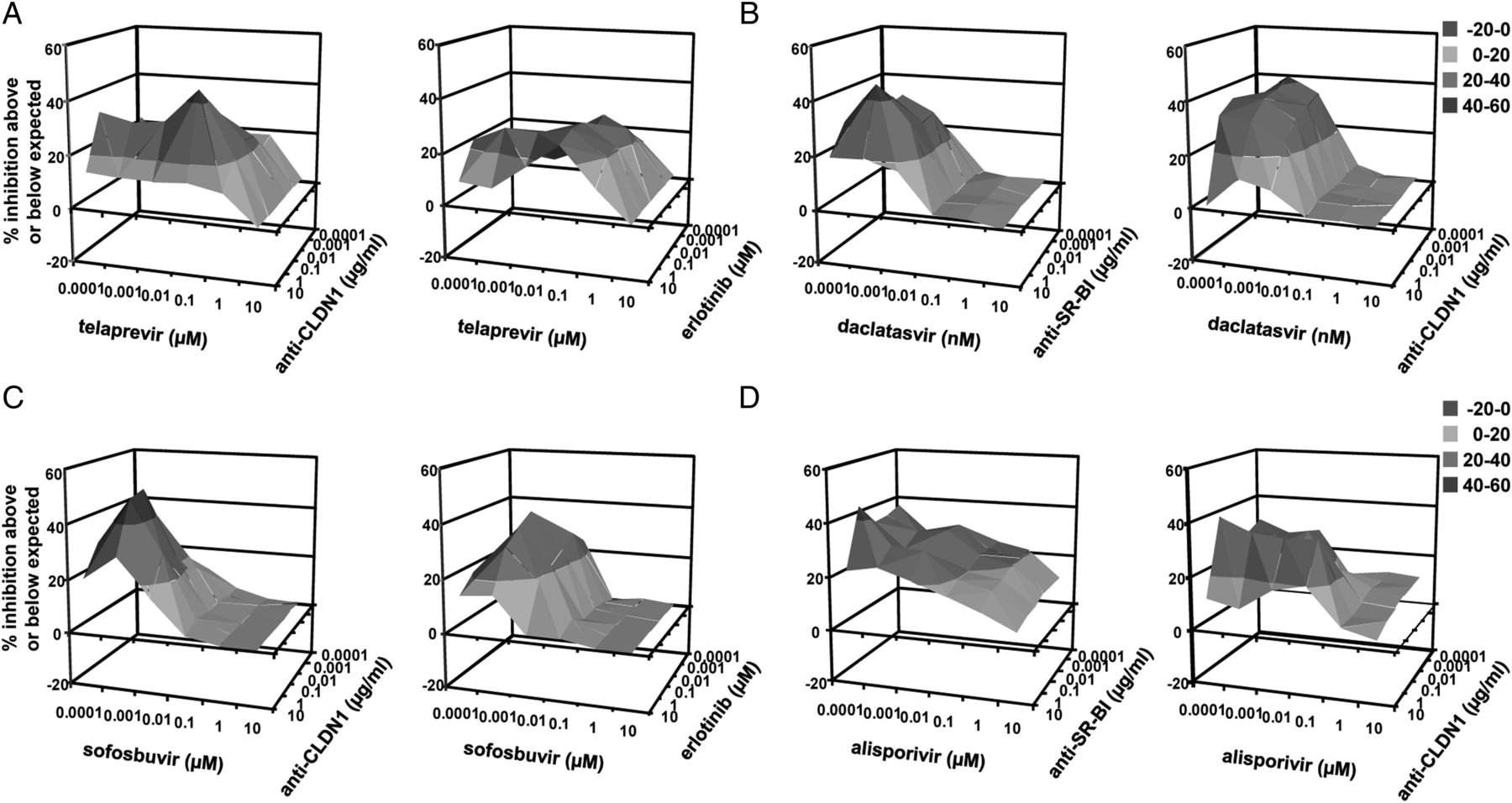
Combination of direct-acting antivirals (DAAs), host-targeting agents and entry inhibitors results in synergy in treatment of established HCV infection. Huh7.5.1 cells were infected for 5 days with HCVcc prior to treatment with DAAs or alisporivir and entry inhibitors as described in ‘Materials and methods’. Synergy between (A) telaprevir and anti-CLDN1 mAb (left) or erlotinib (right), (B) daclatasvir and anti-SR-BI mAb (left) or anti-CLDN1 mAb (right), (C) sofosbuvir and anti-CLDN1 mAb (left) or erlotinib (right), (D) alisporivir and anti-SR-BI (left) or anti-CLDN1 mAb (right) was assessed using the Prichard and Shipman method.25

Synergistic effect of combining direct-acting antivirals and entry inhibitors in treatment of HCV infection using persistently infected DMSO-differentiated hepatoma cells. HCV-infected cell culture was established by transfecting RNA encoding wild-type HCV Luc-Jc1 into Huh7.5.1 cells. The cells were then differentiated in the presence of 1% DMSO for 5 days before treatment with the combinations of (A) telaprevir+anti-CLDN1 mAb (left) or erlotinib (right), (B) daclatasvir+anti-SR-BI mAb (left) or anti-CLDN1 mAb (right), (C) sofosbuvir+anti-CLDN1 mAb (left) or erlotinib (right), (D) simeprevir+erlotinib (left) or anti-CLDN1 mAb (right). HCVcc infection was assessed by luciferase activity 5 days after treatment. Synergy was assessed using the Prichard and Shipman method.25
Entry inhibitors limit viral rebound from DAA therapy
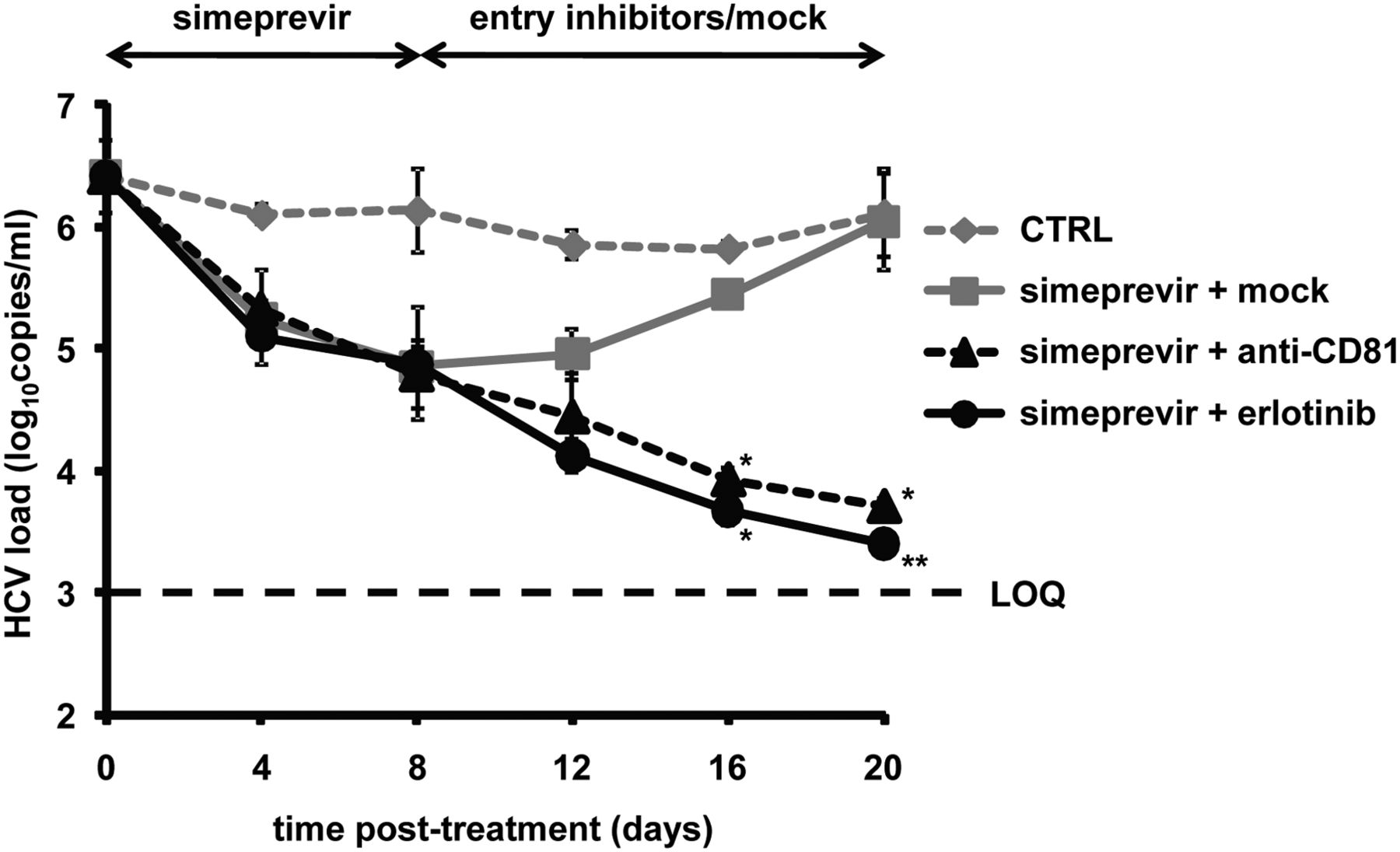
Using the cell culture model system for chronic HCV infection, we studied whether entry inhibitors limit viral rebound following discontinuation of DAA treatment. As shown in figure 8, treatment of persistently HCV-infected cells with a well-characterised and recently FDA-approved protease inhibitor, simeprevir, resulted in a rapid reduction of viral load in a time-dependent manner. However, an increase in viral load was observed following withdrawal of simeprevir. In contrast, the addition of an entry inhibitor (anti-CD81 mAb or erlotinib) at the time of simeprevir withdrawal allowed to further decrease the viral load, indicating that entry inhibitors limit viral rebound from DAA therapy. Cell viability test at the end of the experiment demonstrated that the reduction of viral infection was not due to cytotoxicity (table 1D).
Absent toxicity of combinations of compounds in Huh7.5.1 cells and primary human hepatocytes

Entry inhibitors limit viral rebound from direct-acting antiviral therapy. Persistently HCV (Jc1) infected and DMSO-differentiated cells described in ‘Materials and methods’ were first treated with 500 nM simeprevir for 8 days followed by incubation with either 1% DMSO control medium (mock), 10 µg/mL anti-CD81 mAb or 10 µM erlotinib sequentially for another 12 days. Cells maintained in 1% DMSO throughout the experiment were used as control (CTRL). Viral load was assessed by HCV-specific RT-PCR every 4 days. The limit of quantification, indicated by a dashed line, was 103 copies/mL. Means±SD from a representative experiment performed in triplicate are shown. An asterisk indicates one out of three samples was HCV RNA negative; two asterisks indicate two samples were HCV RNA negative.
Combination of entry inhibitors and DAAs in vivo
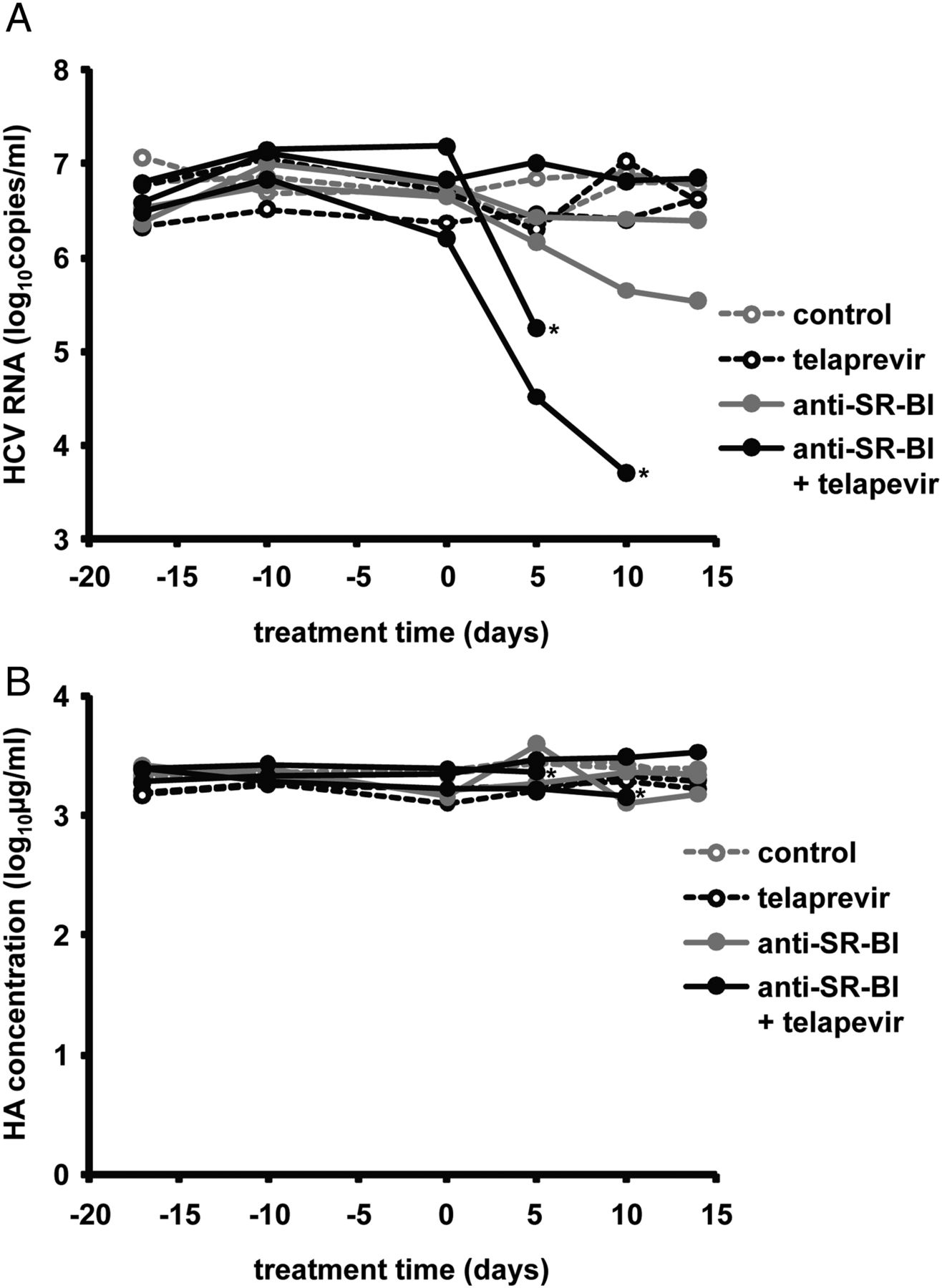
To assess the relevance of combining an entry inhibitor and a DAA for treatment of HCV infection and provide experimental evidence for a synergistic effect of entry inhibitors and DAAs in vivo, we used human liver-chimeric uPA/SCID mice persistently infected with HCVcc (Jc1). Nine persistently infected mice were grouped randomly to receive either telaprevir monotherapy, anti-SR-BI mAb monotherapy, combination of telaprevir and anti-SR-BI mAb or the control treatment for 2 weeks as described in ‘Materials and methods’. In line with previous reports,44 telaprevir only slightly reduced viral load of HCV genotype 2-infected mice, while anti-SR-BI mAb-treated mice showed a reduction of HCV RNA levels of approximately 1 log10 at the end of the treatment. Interestingly, the combination of telaprevir and anti-SR-BI mAb resulted in a more potent reduction of viral load at each tested time point than the calculated sum of both monotherapies (at d10 HCV load for the control treatment is 6.86±0.06 log10; for telaprevir monotherapy 6.72±0.42 log10; for anti-SR-BI mAb 6.03±0.54 log10; for combination therapy in the responder mouse 3.71 log10), reaching more than 2 log10 in one of the mice after 10 days (figure 9A). One mouse treated with telaprevir and anti-SR-BI mAb did not respond to treatment (figure 9A). Stable levels of HA in the blood of mice from different treatment groups (figure 9B) suggested that their liver function was not affected by the treatment, indicating that telaprevir or/and anti-SR-BI mAb combination therapy did not result in major hepatotoxicity in vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of combination of an entry inhibitor and direct-acting antiviral on HCV infection in vivo. Human liver-chimeric uPA/SCID mice engrafted with primary human hepatocyte were inoculated with HCVcc (Jc1) 8 weeks before the experiment as described in ‘Materials and methods’. Nine mice were grouped randomly into control group (two mice), telaprevir monotherapy group (two mice), scavenger receptor B type I (SR-BI)-specific mAb (NK8-H5-E3) monotherapy group (two mice) and combination therapy group (three mice). Mice received telaprevir monotherapy (300 mg/kg PO BID), anti-SR-BI mAb monotherapy (500 µg IP twice a week), combination treatment of telaprevir and anti-SR-BI mAb or control treatments as described in ‘Materials and methods’. Indicated by an asterisk, two mice from the group treated with combination of telaprevir and anti-SR-BI mAb had to be sacrificed before the end of the experiment due to bleeding and injury. (A) HCV RNA from mouse sera was measured by RT-PCR every 5–10 days. Viral load of each individual mouse is shown. (B) Human albumin (HA) concentration in the blood of human liver-chimeric uPA/SCID mice. HA was measured during the treatment as described in ‘Materials and methods’.
Absent toxicity of combinations in PHHs and hepatoma cells
Noteworthy, the uncovered combinations did not exhibit any detectable toxicity in PHH nor Huh7.5.1 cells, neither in short-term or long-term infection experiments (table 1). In contrast, anti-Fas antibody or the well-characterised kinase inhibitor flavopiridol resulted in easily detectable toxicity (table 1). The 50% cytotoxic concentrations (CC50) of entry inhibitors in long-term experiments were more than 10-fold or 100-fold higher than the therapeutic concentrations used in treatment experiments (table 1C). Taken together, these data demonstrate that the uncovered combinations have a favourable safety profile in cell culture models and primary cells providing the rationale for their further development.
Discussion
There is an unmet medical need for novel strategies for the prevention of HCV graft infection following LT, and a need to develop more efficient and better tolerated combination therapies for chronic infection for certain patient subgroups and patients with resistance.1 ,2 Many DAAs are restricted to genotype 1 and their use in difficult-to-treat patients could be hindered by significant and potentially harmful drug–drug interactions.7 Among the most promising DAAs are polymerase and NS5A inhibitors, which demonstrated potent pan-genotypic antiviral activity with a high barrier for resistance when combined with SOC or RBV.45 ,46 Nevertheless, in vivo emergence of resistant mutants conferring cross-resistance to polymerase inhibitors sofosbuvir and mericitabine has been described,47 and combination of daclatasvir with protease inhibitors was associated with significant viral breakthrough rates.3 ,37 Recent phase III studies indicate that sofosbuvir and RBV had only limited efficacy against genotype 3 compared with SOC, with lowest efficacy in cirrhotic patients.40 ,41 Thus, strong antiviral efficacy and high genetic barrier to resistance will remain key requirements for IFN-free regimens to avoid early treatment failure and the optimal combination for defined patient groups and viral genotypes still needs to be defined.
To address the limitations of SOC and DAAs, we evaluated the antiviral effect of combining entry inhibitors with IFN-α, DAAs or HTAs. Our results uncover novel combinations of entry inhibitors and IFN-α, DAAs or HTAs highlighted by marked synergy in vitro (figures 1⇑⇑⇑⇑⇑–7, online supplementary figure S1 and tables S1–S3). Antiviral activity of compounds in the HCVcc model correlates well with clinical antiviral activity in patients,36 and synergy was robust and significant at IC50, IC75 and IC90, at all concentrations tested and similar in experimental prevention and treatment designs. Furthermore, we also demonstrate evidence for a synergistic effect of the combination of an entry inhibitor with a DAA in vivo using the human liver-chimeric uPA/SCID mouse model (figure 9).
A challenge of current and future therapies remains the countering of the development of drug-resistant variants.1–3 Compared with the high variability of viral proteins targeted by DAAs, the variability of host factors targeted by HTAs is low.8 Therefore, host-targeting entry inhibitors may impose a higher genetic barrier to resistance than DAAs. Supplementary to this concept, we and others have shown that entry inhibitors potently inhibit highly infectious escape variants of HCV that are resistant to host neutralising antibodies.9 ,10 ,13–15 ,22 Interestingly, the disease outcome for HIV-infected individuals has significantly improved with the development of antiretroviral drugs targeting different steps of the viral life cycle including viral entry.48 Although viral variants resistant to HIV entry inhibitors have been described, there is no evidence of cross-resistance between different classes of antivirals.48 In contrast to HIV, co-receptor tropism/switch has not been described for HCV as a potential mechanism for viral escape and successful antiviral therapy can definitively eradicate HCV from infected patients. Combination of entry inhibitors with compounds targeting complementary steps of the viral life cycle may open a perspective to overcome antiviral resistance, allow shortening treatment schedules, lower the risk for adverse effects and reduce the doses of the single compounds.
Given that HTAs interfere with host targets, there is theoretically a greater risk of cellular toxicity than with DAAs. Host targets CD81, SR-BI, CLDN1 and EGFR described in this study are expressed in various tissues and play an important role in cell adhesion, lipid metabolism or signalling. Noteworthy, the large majority of licensed drugs in clinical use (eg, in cardiovascular, inflammatory disease or oncology) targets host proteins and side effects are not necessarily more pronounced than in drugs targeting the virus.8 Indeed, the EGFR inhibitor erlotinib, a clinically licensed drug for non-small-cell lung cancer, has a favourable safety profile and is well tolerated in lung cancer patients.49 Moreover, clinical licensed HIV host-targeting entry inhibitors do not necessarily display more adverse events than antiretroviral drugs.48 Nevertheless, safety is an important issue and needs to be carefully addressed for both DAAs and HTAs. Host-targeting antibodies will need to be humanised prior to clinical development.
A pan-genotypic, highly efficient, IFN-free regimen is the ultimate goal for HCV therapy. Compared with many DAAs that act in genotype-dependent manner, host-targeting entry inhibitors exhibit a broad pan-genotypic activity: examples include CLDN1-,22 SR-BI-14 ,15 and CD81-specific16 ,17 mAbs, ITX-5061,18 erlotinib,13 ezetimibe,12 flavonoids,19 ,20 lectins,21 phosphorothioate oligonucleotides50 and silymarin.51 ,52 Thus, combination of entry inhibitors with DAAs may define novel options for patients with non-genotype 1 infections and simplify treatment regimens.
Entry inhibitors are ideally suited for prevention of HCV liver graft infection. Currently, there is no option to protect HCV-negative grafts from reinfection while the clinical use of hepatitis B immune globulin (a well-characterised HBV entry inhibitor) in combination with nucleos(t)ide analogues has essentially eliminated HBV recurrence in LT patients.5 Since HCV entry is a key mechanism for liver graft infection,9 ,10 the observed synergy between DAAs and entry inhibitors uncovers a promising opportunity for prevention of graft infection. The combination of host-targeting entry inhibitors with DAAs may be particularly attractive since the high variability of HCV has so far hampered the development of efficient cross-neutralising antienvelope antibodies.53
Interfering with HCV cell entry offers several targets at different steps of viral entry: blocking virus–target cell interaction during attachment, interfering with postbinding events such as CD81-CLDN1 associations or viral fusion.8 Interestingly, combination of different entry inhibitors, including CLDN1-specific mAb and kinase inhibitors, also showed synergy on HCV entry and infection (figure 5A). While CLDN1-specific antibodies and PKIs both interfere with CD81-CLDN1 coreceptor interactions, PKIs also interfere with glycoprotein-dependent viral fusion.13 This additional and complementary MOA on membrane fusion of PKIs is most likely responsible for the synergistic effect. This concept is confirmed by the finding that combination of anti-CD81 and anti-CLDN1 mAbs, which target a similar step in the viral entry process, only exhibits an additive effect (figure 5A). Thus, these results define novel combinations of entry inhibitors for prevention of graft infection.
Interestingly, two entry inhibitors erlotinib (targeting EGFR and CD81-CLDN1 co-receptor associations) and ITX-5061 (targeting HCV-SR-BI interactions) have reached clinical development to determine the safety and efficacy in patients with HCV infection (ClinicalTrials.gov identifier NCT01835938 and NCT01292824). Moreover, a significant number of compounds tested in this study are already FDA-approved for treatment of chronic HCV infection: telaprevir, boceprevir, sofosbuvir and simeprevir. Erlotinib and dasatinib have been approved for cancer treatment exhibiting a good safety profile.54 It will thus be interesting to investigate the synergistic effect of these drugs in patients with chronic hepatitis C. Taken into account the efficacy, safety, pharmacokinetics, stage of development combined with the synergy data of the various compounds (figures 1⇑⇑⇑⇑⇑–7), our data suggest that combinations of simeprevir34 or sofosbuvir 40 with erlotinib could be a starting point to assess synergy in randomised clinical trials. Combining sofosbuvir and anti-CLDN1 or anti-SR-BI mAb might be promising for prevention of liver graft infection following the clinical development of the monoclonal antibodies. Collectively, novel combinations based on synergy uncovered in this study may widen the therapeutic arsenal against HCV infection for prevention of liver graft infection, treatment of difficult-to-treat-patients and provide alternatives for patients with contraindications to particular compounds of SOC or future IFN-free regimens.
Acknowledgments
We thank Dr R Bartenschlager (University of Heidelberg, Germany) for providing plasmids for production of HCVcc, Dr FV Chisari (The Scripps Research Institute, La Jolla, CA) for the gift of Huh7.5.1 cells, Dr CM Rice (Rockefeller University, New York, NY), Dr JA McKeating (University of Birmingham, UK), Dr D Samuel (Inserm U785, Paul Brousse Hospital, Villejuif, France), Dr JM Pawlotsky (Inserm U955, Henri Mondor Hospital, Créteil, France), Dr T Berg (University Hospital, Leipzig, Germany), Dr RT Chung (Massachusetts General Hospital, Boston) and Dr M Dandri (University Medical Center, Hamburg, Germany) for helpful discussions. We thank L Heydmann, S Durand, T Wu and N Brignon (Inserm U1110) for excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
FX and IF contributed equally.
-
Contributors TFB initiated, designed and supervised research. FX, IF, CT, LM, RA, ER, PL, JN, MBZ and TFB performed research. FX, IF, CT, LM, RA, ER, PL, JN, MBZ and TFB analysed data. NM and MS performed statistical analyses. FH and MD provided reagents. FX, IF, MBZ and TFB wrote the paper.
-
Funding This work was supported by Inserm, University of Strasbourg, the European Union (ERC-2008-AdG-233130-HEPCENT, INTERREG-IV-Rhin Supérieur-FEDER-Hepato-Regio-Net 2009 and 2012, EU FP7 HepaMab), ANRS (2011/132, 2012/239, 2012/318, 2013/108), Laboratoire d'excellence LabEx HEPSYS (Investissement d'Avenir; ANR-10-LAB-28), the Institut Hospitalo-Universitaire Mix-Surg and the Direction Générale de l’Offre de Soins (A12027MS). This work has been published under the framework of the LABEX ANR-10-LABX-0028_HEPSYS and benefits from a funding from the state managed by the French National Research Agency as part of the investments for the future programme. The group in Leuven was funded by a grant from the Fund for Scientific Research (FWO) (G.0728.09N) and KULeuven GOA 10/014.
-
Competing interests Inserm and the University of Strasbourg have filed patent applications on the synergy of interferons, direct-acting antivirals and entry inhibitors as well as SR-BI-specific antibodies, erlotinib, dasatinib for prevention and treatment of HCV infection. Inserm, University of Strasbourg and Genovac/Aldevron have filed a patent application for claudin-1–specific antibodies for prevention and treatment of HCV infection. TB serves as scientific advisor for Biotest, Vironexx and Gilead.
-
Ethics approval All animal experiments in this study were performed in accordance with local laws and ethical committee approval (AL/02/19/08/12 and AL/01/18/08/12).
-
Provenance and peer review Not commissioned; externally peer reviewed.