Article Text

Abstract
Objective The technology for the growth of human intestinal epithelial cells is rapidly progressing. An exciting possibility is that this system could serve as a platform for individualised medicine and research. However, to achieve this goal, human epithelial culture must be enhanced so that biopsies from individuals can be used to reproducibly generate cell lines in a short time frame so that multiple, functional assays can be performed (ie, barrier function and host–microbial interactions).
Design We created a large panel of human gastrointestinal epithelial cell lines (n=65) from patient biopsies taken during routine upper and lower endoscopy procedures. Proliferative stem/progenitor cells were rapidly expanded using a high concentration of conditioned media containing the factors critical for growth (Wnt3a, R-spondin and Noggin). A combination of lower conditioned media concentration and Notch inhibition was used to differentiate these cells for additional assays.
Results We obtained epithelial lines from all accessible tissue sites within 2 weeks of culture. The intestinal cell lines were enriched for stem cell markers and rapidly grew as spheroids that required passage at 1:3–1:4 every 3 days. Under differentiation conditions, intestinal epithelial spheroids showed region-specific development of mature epithelial lineages. These cells formed functional, polarised monolayers covered by a secreted mucus layer when grown on Transwell membranes. Using two-dimensional culture, these cells also demonstrated novel adherence phenotypes with various strains of pathogenic Escherichia coli.
Conclusions This culture system will facilitate the study of interindividual, functional studies of human intestinal epithelial cells, including host–microbial interactions.
- EPITHELIAL CELLS
- E. COLI
- INTESTINAL EPITHELIUM
- CELL BIOLOGY
- DIFFERENTIATION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Conditioned medium containing Wnt3a, R-spondin 3 and Noggin supports the growth of mouse intestinal epithelial spheroids that are enriched for stem cells.
-
Proof-of-principle methods have demonstrated growth of human intestinal epithelial cells as organoids.
-
There is great interest in adapting human intestinal epithelial spheroid culture methods for the study of individual patients.
What are the new findings?
-
Spheroid lines can be reproducibly developed from biopsy material from healthy patients as well as from patients with inflammatory diseases in as little as 2 weeks.
-
The protocol for generation of epithelial lines is the same for all regions and donors.
-
All cultures showed robust growth and region-specific differentiation.
-
Adherence of specific pathogens to primary colonic epithelial cells was unexpected and enhanced compared to immortalised cells typically used as a system for host–pathogen interactions.
How might it impact on clinical practice in the foreseeable future?
-
This robust and reproducible culture system for human intestinal epithelial cells offers a novel platform to create individual-specific assays and to interrogate host–pathogen interactions.
Introduction
The intestinal epithelium is uniquely adapted to perform a variety of homeostatic functions, including barrier formation and maintenance, digestion and absorption of nutrients, modulation of the microbiome and participation in host immune responses.1 Perturbation of these functions is implicated in gastrointestinal disorders, including infections and IBD. It is difficult to dissect epithelial-intrinsic roles using in vivo models due to the complex interactions between epithelial and neighbouring cells, including stromal and haematopoietic cells as well as resident and transient intestinal microbes. To improve our understanding of epithelial cell function in health and disease, methods to study non-transformed epithelial cells in vitro are critical.
Recently, there have been substantial advances in mouse and human intestinal epithelial cell culture methods.2–16 These methods can be generally categorised as those that start cultures with embryonic stem cells or induced pluripotent stem (iPS) cells (induced organoid culture)6 ,13 or with dissociated crypts/stem cells from intestinal tissue (organoid or spheroid culture).2–5 ,7–9 ,11 ,12 ,14–16 While iPS-derived organoids are valuable for investigating embryonic development, the length of time that is required to generate mature, differentiated intestinal epithelial cells may limit their utility for patient-directed experimental approaches. In contrast, spheroid culture is particularly suited for individual-based medicine due to a relatively rapid differentiation rate. Accordingly, there is great interest in developing spheroid culture methods for the study of interindividual variation (attributed to genetics, age, gender, etc.) in human intestinal epithelial cell function. This would permit the use of an individual’s own intestinal epithelial cells to screen for novel therapies, define host–microbial/host–pathogen interactions and perform clinical testing to determine the efficacy of a particular therapy.17 ,18 Remaining challenges for spheroid culture include the ability to reproducibly isolate and culture material from endoscopic biopsies, a reduction in the technical complexity and cost associated with current methods and the ability to grow these cells in a robust and timely manner commensurate with patient care.
Both iPS cell-based and spheroid-based culture methods employ growth media that contain canonical Wnt ligand, R-spondin and Noggin, the critical factors that support intestinal epithelial stem cell growth.12 Because the cost of adding these components to the culture media as recombinant factors is very high, alternative strategies to deliver these factors have been employed, such as subepithelial myofibroblast feeder cells5 or conditioned media.7 ,8 We engineered an L-cell line to secrete Wnt3a, R-spondin 3 and Noggin (L-WRN), thereby creating a cost-effective conditioned medium (L-WRN CM) that can be collected and used to efficiently deliver these critical growth factors to cultured epithelial cells.7 ,8 The L-WRN CM supported the robust growth of mouse intestinal epithelial spheroids that were highly enriched for stem cells.7 Here, we overcame challenges associated with developing human intestinal spheroid culture protocols for patient-based assays by adapting our mouse method to the culture of human gastrointestinal epithelial cells.
Materials and methods
Collection of human tissue samples and spheroid culture
Biopsy tissues were obtained from 47 adults during routine endoscopy at the Washington University School of Medicine in collaboration with the Washington University Digestive Diseases Research Core Center (DDRCC) BioSpecimens Core. Biopsies were collected using standard endoscopic biopsy forceps (Radial Jaw 4 Large Capacity Jaw O.D 2.4 mm, Boston Scientific, Natick, Massachusetts, USA) and immediately placed in 5 mL of ice-cold washing medium (DMEM/F12 with HEPES (Sigma D6421) supplemented with 10% fetal bovine serum (Sigma), 2 mM L-glutamine, 100 units/mL penicillin and 0.1 mg/mL streptomycin (Sigma)). The study was approved by the Institutional Review Board of Washington University School of Medicine. Written informed consent was obtained from all donors.
Procedures for establishing and maintaining human spheroid cultures are based on our previous publications7 ,8 ,19 ,20 with additional details provided in online supplementary methods. In brief, to isolate crypt/gland units, biopsies were minced with fine scissors and digested in 1 mL of collagenase solution (2 mg/mL collagenase type I (Invitrogen, Grand Island, New York, USA) and 50 μg/mL gentamicin (Invitrogen) in washing media) for ∼20–30 min with pipetting every 5–10 min. Crypts/glands were filtered through a 70 µm strainer, incubated with 9 mL of ice-cold washing media and pelleted by centrifugation at 20–50 g for 5 min. Frequently, a second incubation with 5 mL washing medium followed by centrifugation at 50–150 g for 5 min was performed. Pelleted crypts/glands were then suspended in 1 mL washing medium, transferred to a 1.5 mL tube and pelleted by centrifugation at 200 g for 5 min. The supernatant was carefully removed by pipette, and the pellet was suspended in Matrigel (BD Biosciences, San Jose, California, USA; 15 µL/well). The Matrigel mixture was plated into 24-well tissue culture plates on ice. Plates were flipped upside down (prevents epithelial cells from attaching to the plastic) and incubated at 37°C to polymerise the Matrigel and 400 µL of 50% L-WRN CM,7 ,8 a 50/50 mix of L-WRN CM and fresh primary culture media (Advanced DMEM/F-12 (Invitrogen) supplemented with 20% fetal bovine serum, 2 mM L-glutamine, 100 units/mL penicillin and 0.1 mg/mL streptomycin), supplemented with 10 µM Y-27632 (ROCK inhibitor; Tocris Bioscience, R&D Systems, Minneapolis, Minnesota, USA) and 10 µM SB 431542 (TGFBR1 inhibitor; Tocris Bioscience, R&D Systems) was added to each well. Lower percentages of L-WRN CM were obtained by further dilution with primary culture medium. Differentiation media (with or without 5 µM DAPT (EMD Millipore)) included Y-27632, but not SB 431542. Experiments used the spheroid lines bolded in online supplementary table S1 at passages 6–40. Live spheroids were imaged with a Pupil Cam (Ken-a-Vision, Kansas City, Missouri, USA) fixed to a phase microscope (Fisher Scientific). Cell growth was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cell Proliferation Assay Kit (ATCC, Manassas, Virginia, USA).
Gene expression analysis
RNA was isolated with the NucleoSpin RNA II kit (Macherey-Nagel, Bethlehem, Pennsylvania, USA), which includes DNase treatment. Reverse transcriptase reactions (50 µL) used 1 µg RNA and the Iscript cDNA synthesis kit (Bio-Rad, Hercules, California, USA). Eukaryotic quantitative PCR (qPCR) reactions were performed with SYBR Advantage qPCR Premix (Clontech, Mountain View, California, USA) and the primers in online supplementary table S2. Expression levels were determined with triplicate assays per sample and normalised to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which was expressed at similar levels in all samples.
Analysis of primary epithelial cell monolayers
Transwell culture methods were adapted from our recently published method for mouse colonic spheroids.19 Human spheroids (∼1 well of a 24-well plate per Transwell) were dissociated, strained through a 40 µm filter, seeded onto Transwell membranes (Fisher Scientific, CoStar 3470) coated with 0.1% gelatin (earlier experiments) or Matrigel diluted 1:40 in phosphate buffered saline (PBS) (later experiments) and provided 5% L-WRN CM (10 µM Y-27632 was included O/N and then removed during daily media changes). Transepithelial resistance (TER) measurements19 and mucus layer analyses21 were performed as previously described. Z-stack images (1.1 µm, with an optimal interval of 0.55 µm) were generated with a Zeiss LSM510 Meta laser scanning confocal microscope (Carl Zeiss Inc, Thornwood, New York, USA) equipped with Argon (Ex. 488 Em. BP 505–530) and HeNe1 (Ex. 543 Em. BP 560–615) lasers, a 63×, 1.4 numerical aperture Zeiss Plan Apochromat oil objective and LSM software. Rectal and ileal spheroid lines were infected with recombinant lentiviruses expressing an enhanced green fluorescent protein (EGFP) under the hPGK promoter7 ,8 using a described protocol.8
Bacterial adherence assays
Epithelial cells grown on glass chamber slides (Lab-Tek; coated with 1:40 Matrigel diluted in PBS) were washed in antibiotic-free medium followed by incubation with 0.3 mL of Escherichia coli-containing media (∼109 colony forming units/mL) for 1 h (37°C, 5% CO2), washing three times with media to remove non-adherent bacteria and returned to the incubator. After 3 h, cells were washed five times with ice-cold PBS (to further remove non-adherent bacteria) and either fixed/stained (cold methanol/0.5% crystal violet) or lysed for gDNA isolation (DNeasy Tissue kit; Qiagen). Qualitative adherence assays were performed in duplicate (n=2–4 assays/human cell line). qPCR was performed using human GAPDH and bacterial malB primers (see online supplementary table S2) (n=4 assays/E. coli strain/human cell line). The relative adherence index (RAI) for each wild-type E. coli is the ratio of malB to GAPDH divided by this ratio for the non-adherent strain ORN172.
Statistical analyses
A Student’s t test or one-way analysis of variance (ANOVA) followed by a Dunnett’s multiple comparisons or Tukey post-test were performed as indicated in the figure legends with Prism GraphPad V.6 software. p Value<0.05 was considered to be significant.
Results
Establishment of gastrointestinal epithelial spheroid cultures from endoscopic biopsies
To reproducibly grow intestinal epithelial cells from individual patients, we adapted a system developed for mouse intestinal epithelial cells. To initiate cultures, intestinal crypts were isolated by collagenase digestion from biopsy specimens (4–18 mm2 each; 2–3 biopsies per culture) collected during routine endoscopy (figure 1A). Isolated crypts were embedded in basement membrane matrix (Matrigel) and incubated in a 50/50 mix of L-WRN CM and fresh primary culture media (referred to herein as 50% L-WRN CM). We found that ROCK (Y-27632) and TGFBR1 (SB 431542) inhibitors were required to maintain the human intestinal epithelial cell cultures for >3 passages (see online supplementary figure S1), which is a modification from our mouse culture system. Within 24 h of plating, small spheroids formed22 (figure 1A). Typically, the cultures could be sufficiently expanded to prepare frozen stocks (10 vials/line) within 4–6 passages (15–25 days) (see online supplementary figure S2).

Establishment of human epithelial spheroid cultures from multiple regions of the gastrointestinal tract. (A) Representative images showing the procedure for establishment of intestinal spheroid culture. Ruler lines above biopsy samples indicate 1 mm. Bars, 100 µm. (B) Diagram illustrating the numbers of spheroid and gastric lines established to date. The first number reports the total number of lines established from the indicated region, and the number in parenthesis indicates the number of those lines established from patients with IBD.
This method to establish human intestinal spheroid lines was successful for all endoscopically accessible sites of the gastrointestinal tract including individuals who were either healthy or had inflammatory disease (ie, Barrett’s oesophagus and IBD; figure 1B). We also established epithelial lines in patients with prior intestinal surgery from tissue sites that included neo-terminal ileum (post-bowel resection surgery) and the ileal pouch portion of an ileal pouch-anal anastomosis. To date, we generated 65 human spheroid lines from 47 individuals, including 25 patients with IBD (see figure 1B and online supplementary table S1). Thus, this culture system can support growth of epithelial cells from multiple sites of the gastrointestinal tract and from patients with or without disease.
Robust growth of epithelial spheroids from human rectum and ileum
Rectal and ileal spheroids, regardless of donor status (healthy control or IBD), grew rapidly in 50% L-WRN CM (figure 2A,B). Over a 2-day period, the rectal and ileal spheroids exhibited a robust increase in cell number (2.4-fold and 2.9-fold increase, respectively; figure 2B). This result suggested that the spheroids grown in 50% L-WRN CM were enriched for a highly proliferative stem and/or progenitor cell population. Accordingly, rectal and ileal cell lines abundantly expressed leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5), a well-established stem cell marker in mouse intestine23 (figure 2C). The spheroid lines expressed similar levels of villin 1 (VIL1), demonstrating that they retained their intestinal epithelial identity (figure 2C). Expression of vimentin (VIM) was not detected in any line after passage 7 (the earliest passage analysed for this marker), confirming that mesenchymal cells were eliminated during initial passages (figure 2C). Thus, in contrast to other culture techniques that contain cells of mesenchymal origin,5 ,10 ,13 ,24 this system allows for the study of isolated epithelial cells.

Robust growth of cultured intestinal spheroids in conditioned medium containing Wnt3a, R-spondin 3 and Noggin. (A) Representative images demonstrating the qualitative growth of an individual spheroid imaged daily over a 3-day period. Bars, 100 µm. (B) Spheroids were seeded (day 0) into 96-well format and growth was assessed at 24 h intervals by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay on days 1, 2 and 3. The reduction of MTT (yellow colour) to formazan (purple colour) by metabolically active cells was measured by absorbance at 595 nm with a plate reader. The background subtracted results for three rectal (gray bars) and three ileal (red bars) cell lines are presented as fold change (mean±SEM) compared with day 1 for each region (n=9 per line at each time point). ***p=0.004 by Student’s t test. (C) qPCR gene expression analysis for (A) LGR5, (B) VIL1 and (C) VIM in rectal and ileal epithelial cell cultures. Fresh biopsy samples were used as a positive control (white bars). Data are presented as fold change relative to the biopsy samples (mean±SEM; n=3 samples per bar). n.d., not detected.
Human spheroids retain a region-specific differentiation programme
Because the spheroid cultures appear to be enriched for stem cells, it is important to identify conditions that permit differentiation. Wnt signalling is a well-established pathway that contributes to intestinal stem cell activity and maintenance in vivo.25–29 We have previously shown that the 50% L-WRN CM is the major source of Wnt stimulation in our culture system and that its dilution results in diminished Wnt signalling activity in mouse intestinal epithelial spheroids.7 Similar results were produced using the human spheroids (figure 3A). Moreover, addition of the Notch inhibitor N-[N-(3,5-Difluorophenacetyl-L-alanyl)]-(S)-phenylglycine t-butyl ester (DAPT), which promotes the differentiation of mouse and human spheroids,4 ,10 ,11 ,19 ,20 rapidly reduced the proportion of cells in S-phase in rectal and ileal lines (see figure 3B and online supplementary figure S3). Thus, by downregulating Wnt and Notch signalling, cell cycle exit was quickly induced.

Human intestinal epithelial spheroids grown in low percentages of conditioned medium containing Wnt3a, R-spondin 3 and Noggin (L-WRN CM) exhibit decreased Wnt signalling activity. (A) Human rectal and ileal spheroids (n=3 lines per region) were cultured for 2 days in 50%, 20%, 10%, 5% or 0% L-WRN CM. Gene expression of AXIN2, LGR5, OLFM4, MKI67 and MYC was determined by qPCR (n=3 experiments per line). A dose-dependent decrease was observed for AXIN2 (canonical Wnt target), mouse intestinal stem cell markers LGR5 and olfactomedin 4 (OLFM4), and proliferative markers MKI67 and MYC. Both 0% and 5% L-WRN CM showed a significant decrease for all of these markers compared with 50% L-WRN CM. As some assays required cells with some potential to divide, we used cells grown in 5% L-WRN CM for downstream applications. Data were presented as fold change (mean±SEM) compared with 50% L-WRN CM for each experiment. *p<0.05; **p<0.01; ****p<0.0001 by one-way analysis of variance (ANOVA) followed by a Dunnett's post-test with 50% L-WRN CM set as the control group: F=17.58, p<0.0001 (A, AXIN2); F=449.60, p<0.0001 (A, LGR5); F=113.70, p<0.0001 (A, OLFM4); F=56.13, p<0.0001 (A, MKI67); F=87.09, p<0.0001 (A, MYC); F=49.48, p<0.0001 (B, AXIN2); F=1570.00, p<0.0001 (B, LGR5); F=156.70, p<0.0001 (B, OLFM4); F=63.81, p<0.0001 (B, MKI67); F=125.20, p<0.0001 (B, MYC). (B) Spheroids were passaged (day 0) and allowed to recover overnight in 50% L-WRN CM. Media was replaced daily, with some cultures changed to 5% or 5% L-WRN CM plus 5 µM DAPT for the indicated number of days. On day 3, the spheroids were treated with EdU for 2 h, dissociated, stained and sorted by flow cytometry based on EdU incorporation and SYTOX AAD (DNA content marker). An average of 22.2% of rectal cells and 16.8% of ileal cells were in S-phase when grown in 50% L-WRN CM. One day of culture in 5% L-WRN CM was not sufficient to reduce the proportion of cells in S-phase; however, after 2 days, a decrease in the proportion of cells in S-phase was observed for both rectal and ileal lines. Addition of 5 µM DAPT to the 5% L-WRN CM rapidly decreased the proportion of S-phase cells within 1 day and led to greatly diminished proportions of S-phase cells (1.6–2.9%) after 2 days.**p<0.01; ***p<0.001; ****p<0.0001 by one-way ANOVA followed by a Dunnett's post-test with 50% L-WRN CM set as the control: F=43.01, p<0.0001 (rectal); F=54.13, p<0.0001 (ileal).
We next examined mRNA expression of differentiated cell markers in rectal, ileal and duodenal spheroids grown in 50% L-WRN CM or 5% L-WRN CM with or without DAPT. As expected, LGR5 expression was greatly decreased and expression of differentiated cell markers was generally increased in spheroids cultured in 5% L-WRN CM with or without DAPT as compared with 50% L-WRN CM (see figure 4 and online supplementary figures S4 and S5). In accordance with their in vivo expression patterns, carbonic anhydrase 1 (CA1) was specifically induced in rectal spheroids and sucrase-isomaltase (SI) was specifically induced in ileal spheroids when grown in 5% L-WRN CM with DAPT (figure 4A, B). The addition of DAPT was important for inducing expression of secretory cell markers, including transcription factors atonal homologue 1 (ATOH1) and neurogenin 3 (NEUROG3), goblet cell markers mucin 2 (MUC2) and trefoil factor 3 (TFF3) and endocrine cell marker chromogranin A (CHGA) (see figure 4C and online supplementary figures S4 and S5). Expression of the Paneth cell marker α-defensin 5 (DEFA5) (see figure 4D and online supplementary figure S5) was strongly induced only in small bowel spheroids when grown in 50% L-WRN CM with DAPT, in accordance with the in vivo distribution and the critical role of Wnt signalling in the formation of Paneth cells.30 ,31 Region-specific induction of small bowel markers was also observed. Transient receptor potential cation channel, subfamily V, member 6 (TRPV6; calcium channel) and solute carrier family 10, member 2 (SLC10A2; bile acid transporter), were preferentially expressed in duodenal and ileal spheroids, respectively (figure 4E, F). These data suggest that regional specification of epithelial cells is, at least in part, cell-intrinsic, as previously suggested.32

Region-specific differentiation of rectal and ileal spheroids. (A–D) qPCR gene expression analysis for (A) CA1, (B) sucrase-isomaltase (SI), (C) MUC2, (D) DEFA5, (E) TRPV6 and (F) SLC10A2 in rectal (gray bars), ileal (red bars) or duodenal (blue bars) spheroids (n=3 lines per region). RNA was collected from spheroids that had been passaged, allowed to recover overnight in 50% conditioned medium containing Wnt3a, R-spondin 3 and Noggin (L-WRN CM) and then treated with 50% or 5% L-WRN CM with or without the addition of 5 µM DAPT for an additional 2 days, with media replaced daily. Data are presented as fold change relative to rectal or duodenal 50% L-WRN CM, except for DEFA5, which is presented as fold change relative to ileal 50% L-WRN CM (mean±SEM; n=3 per line). n.d., not detected. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 by one-way analysis of variance followed by a Tukey's post-test: F=7.23, p=0.0008 (A, rect); F=3.73, p=0.0210 (A, ile); F=3.53, p=0.0256 (B, rect); F=6.24, p=0.0019 (B, ile); F=4.99, p=0.0060 (C, rect); F=6.16, p=0.0020 (C, ile); F=9.42, p=0.0001 (D, ile); F=9.05, p=0.0002 (E, duod); F=0.67, p=0.5777 (E, ile); F=17.33, p<0.0001 (F, duod); F=4.39, p=0.0151 (F, ile).
Development of a Transwell system for human primary intestinal epithelial cells
The growth of intestinal epithelial cell monolayers is necessary for in vitro studies of physiological processes, such as analyses of cellular transport or secretion or for co-culture experiments that will likely be used in patient-based assays. Transwell assays have been difficult to perform using other culture methods for primary intestinal epithelial cells, likely due to the inability to generate sufficient numbers of cells. We recently developed a method to form functional epithelial monolayers with mouse colonic epithelial cells.19 We adapted this method to grow human intestinal epithelial cells on Transwell membranes (figure 5). Within 3 days of seeding, both rectal and ileal epithelial cells formed polarised monolayers, as evidenced by H&E staining and co-immunostaining for the apical brush border protein VILLIN 1 and the tight junction protein ZO-1 (figure 5A, B). Goblet (MUC2-positive) and endocrine (CHGA-positive) cell populations (figure 5A, B) were present in proportions similar to those observed in colonic surface or ileal villus epithelium, with goblet cells accounting for 8.4% and 8.7% and endocrine cells accounting for 0.4% and 1.0% of total epithelial cells in the rectal and ileal monolayers, respectively. UEA-1 lectin can be used to distinguish both mouse and human Paneth cells33 (see online supplementary figure S6). We observed rare UEA-1 reactive cells in ileal, but not in rectal, monolayers (figure 5B and data not shown).

Dissociated spheroid cells form polarised monolayers on Transwell membranes. (A and B) Spheroids were dissociated and seeded onto coated Transwell membranes to form epithelial monolayers. After 3 days, monolayers were used for measurement of transepithelial resistance (TER) and/or fixed and paraffin-embedded. Paraffin sections from rectal (A) and ileal (B) epithelial monolayers were stained for H&E or immunostained for VIL1 and ZO-1, MUC2 or chromogranin A (CHGA) (with UEA-1 lectin for ileal cells) to visualise differentiated cell types. (a) and (b) denote insets. Bars, 10 µm. (C) TER measurements are shown as the resistance × area product (Ω cm2) for three lines per intestinal region (mean±SEM; n=4 Transwells per line). (D) Representative confocal Z-stack images of ileal monolayer (most epithelial cells are GFP-positive, green) and fluorescent beads (red). The top image is a transverse view with white dashed lines denoting the apical surface of the epithelial layer and range of suspended beads and a yellow solid line indicating the measured mucus thickness. The lower image shows a tilted view of the transverse image. Bars, 10 µm. (E) Graph of mucus thickness measurements in rectal and ileal monolayers before (−) and after (+) disruption of the mucus layer by pipetting (mean±SEM; n=4–7 Transwells per bar). **p<0.01; ***p<0.001 by Student’s t-test comparing mucus thickness before and after disruption from the same region.
We tested the barrier function of the epithelial cells grown on Transwells. Rectal and ileal monolayers exhibited an average TER of 395 and 400 Ω cm2, respectively, indicating barrier integrity (figure 5C). We also demonstrated a mucus layer using a recently reported method for mouse tissue explants.21 For these experiments, spheroid cells labelled with EGFP by lentiviral infection (see online supplementary figure S7) were used to generate epithelial monolayers. Red fluorescent beads (1-µm diameter) were allowed to sediment into the mucus layer. The distance between the apical surface of the epithelial cells and the beads, which become suspended in the mucus, was determined using Z-stack imaging by confocal microscopy (figure 5D). We observed a mucus layer reproducibly covering the apical surface of the epithelial monolayers (36 and 26 µm thick in rectal and ileal monolayers, respectively; figure 5E). Aspiration by pipetting was reported to remove the outer mucus layer in mouse tissue explants.21 ,34–36 After aspiration by pipetting, we found a significant decrease in mucus thickness in both ileal and rectal lines (figure 5E), suggesting that the mucus layers were disrupted by mechanical dissociation.
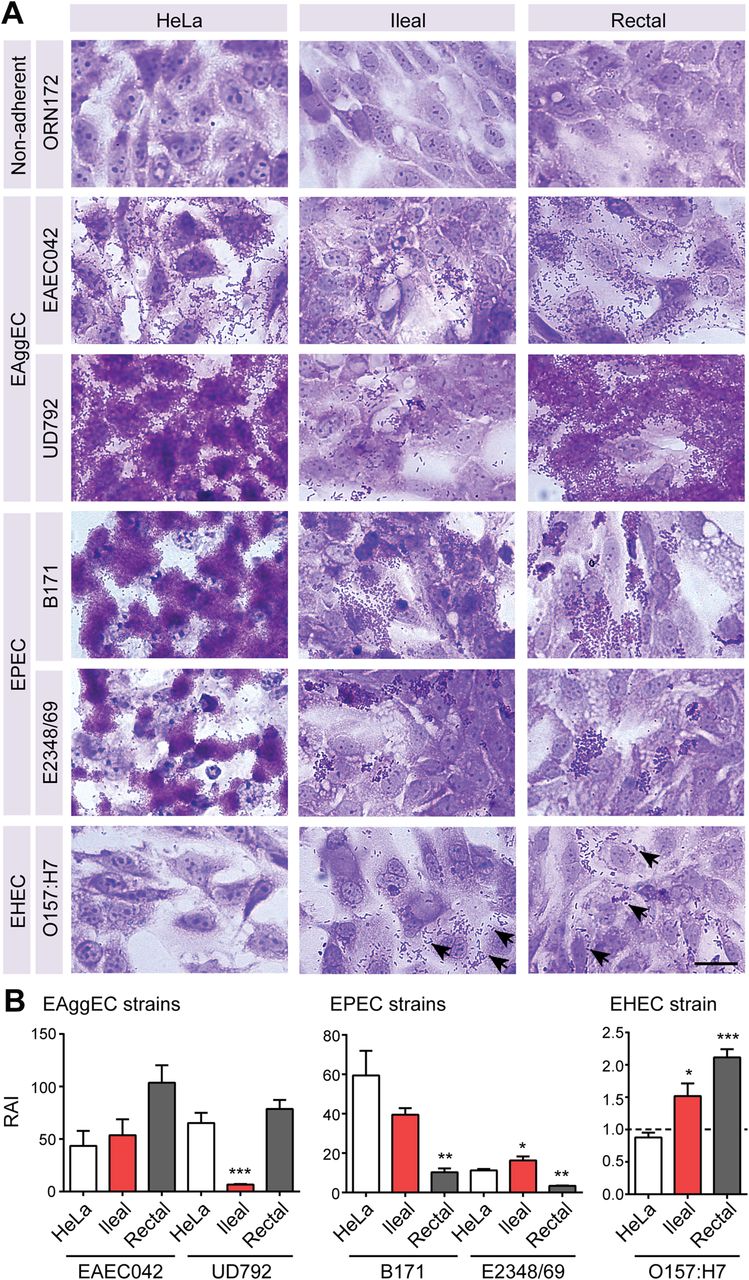
E. coli adherence to human primary intestinal epithelial cells
As a proof of concept that host-based assays with cultured intestinal epithelial cells are feasible and offer data not already known from standard cell line assays, we compared the adherence of various diarrheagenic E. coli strains to primary intestinal epithelial lines and HeLa cells (a conventional immortalised cell line used currently for this assay37). We first performed adherence assays using wild-type strains with enteroaggregative (EAggEC; 042 and UD792)38 ,39 or enteropathogenic (EPEC; B171 and E2348/69)40 ,41 E. coli phenotypes previously demonstrated with HeLa cells (figure 6). These strains adhered robustly to both rectal and ileal epithelial cells with patterns similar to those observed with HeLa cells (ie, more diffuse adherence for EAggEC vs dense microcolonies of localised adherence for EPEC). Similar adherence patterns were observed with epithelial lines derived from different individuals (n=3 per region). The non-adherent laboratory strain E. coli ORN172,42 as expected, did not adhere to any cell line by visual assessment (figure 6A). A quantitative assay to determine the RAI (see legend) for each bacterial strain corroborated the visual assessment of EPEC and EAggEc adherence, with RAI values for rectal and ileal epithelial cells exceeding those of ORN172 by up to two orders of magnitude (figure 6B). These data also confirmed that the EAggEC UD792 strain adhered more robustly to rectal cells as compared with ileal cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Development of a bacterial adherence assay for human intestinal spheroid cells. Rectal and ileal spheroids were grown on glass chamber slides for 3 days to promote two-dimensional cell growth prior to assessment, which allows for the visualisation of adherent bacteria. For adherence assays, rectal and ileal spheroid-derived epithelial cells were incubated with the indicated strains for 1 h followed by several washes to remove non-adherent Escherichia coli. Similarly treated HeLa cells served as a positive control for adherence. (A) Visual assessment of adherence phenotype was assessed by crystal violet staining (adherent bacteria are dark purple). Clustered and scattered adherence of the O157:H7 strain was observed with spheroid-derived epithelial cells (arrows). Bar, 30 µm. (B) qPCR was used to determine the relative adherence index (RAI) for the enteroaggregative E. coli (EAggEC), enteropathogenic E. coli (EPEC) and enterohemorrhagic (EHEC) strains (mean±SEM; n=4 per strain per cell type). The RAI for each wild-type E. coli is the ratio of malB to glyceraldehyde 3-phosphate dehydrogenase divided by this ratio for ORN172 (represented by dashed line in EHEC strain graph), which is a non-adherent control. *p<0.05; **p<0.01; ***p<0.001 compared with HeLa cells by one-way analysis of variance followed by a Tukey's post-test: F=4.41, p=0.0463 (EAEC042); F=26.73, p=0.0002 (UD792); F=10.66, p=0.0042 (B171); F=27.35, p=0.0001 (E2348/69); F=19.04, p=0.0006 (O157:H7).
We next assessed adherence of the enterohemorrhagic E. coli O157:H7 strain 86–24 (figure 6).43 ,44 Whereas exceptionally rare HeLa cells exhibited any adherent bacteria (see figure 6 and online supplementary figure S8), diffuse adherence of E. coli O157:H7 was readily observed in the rectal and ileal epithelial cells (figure 6A). These visual observations were supported by our quantitative assay, where significantly greater RAI values were observed for rectal and ileal epithelial cells than for HeLa cells (figure 6B).
Discussion
Here, we describe a culture system for human gastrointestinal epithelial cells with great potential for use in patient-specific assays. Key advantages of this system include use of endoscopic biopsy tissue as starting material and the rapid expansion of the spheroids, which allows for line establishment from an individual patient within a time frame that is commensurate with patient care (∼2–3 weeks). By eliminating the requirement for surgical specimens, spheroid cultures from a wide array of patient populations can be analysed, including healthy controls or patients preselected based on age, genotype, diagnosis, disease features, etc.
Another advantage of this spheroid culture system is that the protocol is relatively simple. The same protocol can be used to establish and maintain spheroid lines from any accessible gastrointestinal site and there is no requirement for cell sorting schemes, such as those described by other protocols.4 ,11 ,15 We have had near 100% success in establishing cell lines from particular patients as long as the biopsy contains a sufficient number of crypts. This high level of reproducibility is important because spheroid lines will need to be produced following a single endoscopic procedure in order to perform individualised assessments.
Biopsy material has also been successfully applied in explant/organ culture systems.24 ,45–48 Cultured explants enable study of interactions between epithelial cells and neighbouring cells, such as resident immune cells, whereas spheroid culture enables study of epithelial-specific responses. An important advantage of the spheroid culture system is the ability to establish and store lines from specific patients, such that multiple experiments can be performed with the same cell line over time. Indeed, we observed that experiments repeated over time with the same lines produced similar results. In addition, the rapid growth and differentiation capacity of the cultured spheroids allowed us to obtain the critical cell mass needed to perform a variety of functional assays to assess the characteristics of stem/progenitor cells or differentiated cells. Such assays could easily be adapted for the analysis of other epithelial functions, in vitro screening and other patient-based assays.
The mucus layer is a critical component of the physical barrier separating the host from the luminal environment, thus providing additional protection from potential pathogens.49 We found that the spheroid-derived intestinal epithelial cells produced a mucus layer that could be mechanically dissociated. This disruption was more prominent with ileal mucus than rectal mucus, a finding in agreement with in vivo studies that show a single loose mucus layer in the ileum that can be mostly removed by pipetting and a more complex and resistant mucus layer in the colon.21 ,34–36
The adherence of EPEC to rectal epithelial cells was unexpected as these organisms are generally considered to be small bowel pathogens based on extensive cell biology that has focused on the attaching and effacing lesions that result when they adhere to microvilli50 and the classic association of EPEC with non-bloody diarrhoea. However, some reports have described patients infected with EPEC or EAggEC who have bloody diarrhoea,38 ,51 and EPEC have been noted to adhere to the colon during human infections,52 ,53 suggesting that there may be a role for these bacteria in colonic pathology. Additionally, E. coli O157:H7 strain 86–24 adhered well to both rectal and ileal epithelial cells, which contrasts with the extremely sparse adherence to HeLa cells,43 ,44 a phenotype we confirmed. These data suggest that primary human epithelial cells are more appropriate for studying the adherence phenotypes of diarrheagenic E. coli than immortalised cell lines. These phenotypes also suggest new lines of investigation into intestinal, and especially colonic, receptors for bacterial adhesins. Interestingly, even though there is considerable colonic pathology in E. coli O157:H7 human infections,54 there is little direct evidence of the ability of this pathogen to adhere to the mucosa lining this organ55 and a lack of clarity regarding direct bacterial–colon interaction.56 We also present a new way to quantify E. coli adherence to epithelial cells using unified technology (single locus copy number of bacteria and host cells by DNA amplification) to normalise bacterial adherence to eukaryotic cells. This technique extends prior work using lung epithelial cells57 and qPCR of enteropathogens adherent to manually enumerated intestinal cell lines.58
The microbiome plays an important role in the pathogenesis of Crohn’s disease (CD).59–61 The CD microbiome is enriched for Enterobacteriaceae (including E. coli),62–64 with several studies reporting higher proportions of adherent and invasive E. coli.65–68 These findings suggest that overgrowth of specific bacteria, such as adherent-invasive E. coli, may be a driver of inflammation in CD.62 An advantage of the spheroid culture system is that it allows for preselection of donors with particular genotype and, thus, would permit the study of potential CD susceptibility genotype–phenotype relationships that have been hypothesised to affect the sensing of and response to microbes by intestinal epithelial cells.69 Thus, we propose that the primary intestinal epithelial cell culture system described here will provide an informative experimental platform to test potential inter-host variations that might affect the colonisation potential of these bacteria.
Acknowledgments
The authors thank Dr Bill Stenson for comments on the manuscript and Drs Chien-Huan Chen, Themistocles Dassopoulos, Dayna Early, Alexandra Gutierrez, Kevin Korenblat, Deborah Rubin, Shelby Sullivan, Sandeep Tripathy and Jean Wang for collection of biopsy tissues. We thank Kelly Monroe for assisting with patient recruitment and consent, Wandy Beatty for assistance with confocal imaging and Dr. James Nataro for providing strain EAEC042.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors KLV, HM, CM, NS, PIT, MAC and TSS contributed to the study concept and design. JMM and MAC contributed to the review of endoscopy procedures and clinical records. KLV and JMM contributed to establishment of cell lines. KLV performed epithelial cell experiments. NS performed E. coli adherence assays. KLV, HM, NS, PIT, MAC and TSS contributed to the analysis and interpretation of the data. KLV and TSS drafted the manuscript. All authors contributed to the critical revision of the manuscript. MAC and TSS obtained funding and supervised the study.
-
Funding This work was supported by the NIH (grants DK071619, DK089016, DK100737, DK90251 and DK102557; Mucosal Immunology Studies Team Translational Pilot Project and Young Investigator Pilot Award, Parent Grant AI095550) and the Crohn’s and Colitis Foundation of America (CCFA) Genetics and Microbiome Consortiums. KLV was supported by an NIH training grant (T32 AI007163) and a CCFA Research Fellowship Award. The Washington University Digestive Disease Research Core Center is supported by a grant from the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) (P30DK052574).
-
Competing interests None.
-
Ethics approval Institutional Review Board of Washington University School of Medicine.
-
Provenance and peer review Not commissioned; externally peer reviewed.