Article Text

Abstract
Objectives Endothelial progenitor cells (EPCs) circulate with increased numbers in the peripheral blood of patients with highly-vascularised hepatocellular carcinoma (HCC) and contribute to angiogenesis and neovascularisation. We hypothesised that angiogenic EPCs, that is, colony forming unit-endothelial cells (CFU-ECs), and outgrowth EPCs, that is, endothelial colony-forming cells, may exert paracrine effects on the behaviours and metastatic capacities of human hepatoma cells.
Design Various molecular and functional approaches ranging from in vitro cell culture studies on molecular signalling to in vivo investigations on cell invasion and orthotropic transplantation models in mice and clinical specimens from patients with HCC were used.
Results Monocyte chemotactic protein-1 (MCP-1) was identified as a critical mediator released from CFU-ECs to contribute to the chemotaxis of Huh7 and Hep3B cells by inducing their microRNA-21 (miR-21) biogenesis through the C-C chemokine receptor-2/c-Jun N-terminal kinase/activator protein-1 signalling cascade. CFU-EC-induction of miR-21 in these cells activated their Rac1 and matrix metallopeptidase-9 by silencing Rho GTPase-activating protein-24 and tissue inhibitor of metalloproteinase-3, respectively, leading to increased cell mobility. MCP-1-induction of miR-21 induced epithelial-mesenchymal transformation of Huh7 cells in vitro and their intrahepatic metastatic capability in vivo. Moreover, increased numbers of MCP-1+ EPCs and their positive correlations with miR-21 induction and metastatic stages in human HCC were found.
Conclusions Our results provide new insights into the complexity of EPC-HCC interactions and indicate that anticancer therapies targeting either the MCP-1 released from angiogenic EPCs or the miR-21 biogenesis in HCC cells may prevent the malignant progression of primary tumours.
- Stem Cells
- Signal Transduction
- Liver Metastases
- Endothelial Cells
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Hepatocellular carcinoma (HCC) is an aggressive malignant tumour with high mortality rate that is attributable to a frequent occurrence of intrahepatic and distant metastases after resection or transplantation.
Owing to their ability of self-renewal and contribution to angiogenesis and neovascularisation, circulating endothelial progenitor cells (EPCs) have the potential to become a new diagnostic biomarker for patients with HCC and liver cirrhosis.
What are the new findings?
MCP-1 produced by human myeloid-derived angiogenic EPCs induces migration, invasion, epithelial-mesenchymal transformation, and intrahepatic metastasis of HCC cells through the induction of their miR-21, which activates Rac1 and MMP9 by silencing ArhGAP24 and TIMP3, respectively.
The CCR2/JNK/AP-1 signalling cascade is involved in EPC/MCP-1-induction of miR-21 in HCC cells.
The numbers of MCP-1+ EPCs are significantly increased in human HCC in comparison with benign liver tissues.
There is a positive correlation between the numbers of MCP-1+ EPCs and the expression levels of miR-21 in all examined human HCC (tumour node metastasis stages I and IV).
How might it impact on clinical practice in the foreseeable future?
Clinical application of ex vivo expanded EPCs delivering therapeutic drugs to the microvasculature in tumours or targeting EPC mobilisation to disrupt angiogenesis are potential therapeutic strategies against tumour development.
Targeting MCP-1/CCR2 intercellular signalling axis and miR-21 biogenesis in HCC show great potential for the development of specific EPC-based strategies for therapeutic intervention against HCC malignance.
Introduction
Hepatocellular carcinoma (HCC) is a common malignant tumour and a leading cause of death worldwide. Despite a great number of scientific advances, the high incidence of postsurgical recurrence and intrahepatic/extrahepatic metastasis remain serious problems.1 Accumulating evidence indicates that circulating endothelial progenitor cells (EPCs) can be recruited to non-vascularised tumour sites to facilitate the initial establishment of tumorous endothelium, contributing to angiogenesis by providing structural support to nascent vessels and producing proangiogenic cytokines.2 In clinics, the numbers of EPCs present are positively correlated with the advanced invasive stages of HCC.3 ,4 Moreover, EPCs serve as a critical regulator for tumour progression from micrometastases to macrometastases5; reduction of EPC population was shown to suppress liver tumour metastasis in an orthotropic liver tumour model.6 Thus, EPCs have the potential to serve as a diagnostic marker for HCC progression. However, the detailed mechanisms by which EPCs modulate the behaviours and metastatic capability of HCC cells remain unclear.
At present, there are two subpopulations of EPCs derived from circulation, namely, colony forming unit-endothelial cells (CFU-ECs) and endothelial colony-forming cells (ECFCs).2 Ex vivo expanded CFU-ECs, which are identified as myeloid-derived EPCs, are proangiogenic cells.7 In contrast, ECFCs, which are classified as vessel-derived EPCs, have robust proliferative and vessel-forming potential.7 ,8 Although there has been considerable evidence that EPCs can promote tumour progression, little is known about the paracrine effects of differential EPC subtypes on tumour cells and the mechanisms underlying these EPC effects on tumour.
The contributions of tumour cell mobility to metastasis require a coordinated sequence of multistep events, including tumour-host interactions and prometastatic molecular determinants.9 In clinical studies, many paracrine factors produced from tumour and stromal cells, such as vascular endothelial growth factor and stromal cell-derived factor-1, have been identified as critical players for EPC recruitment and tumour angiogenesis.4 These paracrine factors may modulate the expression of post-transcriptional regulators in tumour cells to modulate tumour malignance.10 For example, transforming growth factor-β modulates the expression of small non-coding RNAs in HCC.10 MicroRNAs (miRs), a type of small non-coding RNAs that act as a key prometastatic molecular determinant, have been reported to be aberrantly increased in malignant HCC cells in comparison with normal hepatocytes or tissues.11 Among those miRs, miR-21, miR-34a and miR-224 have been shown to be associated with proliferation, epithelial-mesenchymal transition (EMT) and metastasis of HCC.12 However, whether miRs are involved in the regulatory effects of EPCs on HCC cell chemotaxis and metastasis remain unclear.
In the present study ranging from in vitro cell culture studies on molecular signalling to in vivo investigations on cell invasion and orthotropic transplantation models in mice and clinical specimens from patients with HCC, we demonstrated that CFU-ECs, but not ECFCs, induce the biogenesis of miR-21 in non-metastatic HCC cells through the paracrine release of monocyte chemotactic protein-1 (MCP-1), with the consequent induction of HCC cell migration and invasion. We further demonstrated that CFU-EC-educated HCC cells exhibit high capabilities of EMT in vitro and invasion and intrahepatic metastasis in vivo. Our findings provide new insights into the mechanisms by which blood-derived proangiogenic EPCs contribute to the pathophysiology of HCC and may help the identification of EPC-associated targets for therapeutic intervention against HCC malignance.
Materials and methods
An expanded Materials and Methods section is given in online supplemental document.
Cell culture
Human circulating EPCs, including CFU-ECs and ECFCs, were isolated from fresh human peripheral blood from healthy volunteers and identified, as described.8 The investigations conformed to the principles outlined in the Declaration of Helsinki. Human non-metastatic Huh7 and Hep3B hepatoma cell lines and normal hepatocyte Chang liver cell line were obtained from American Type Culture Collection (ATCC) and grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Cell migration and invasion assays
The migration and invasion assays were performed in 24-well transwell chambers (transwell assay), modified IBIDITM Culture-Inserts (horizontal chemotactic mobility assay), and modified IBIDITM μ-slide plates (horizontal collagen gel invasion assay), as described.8
Pull-down analysis of Rho GTPase activity
The activities of Rho GTPases (RhoA, Rac1 and Cdc42) were assessed using a Rho activation assay kit (Upstate Biotechnology). Detailed procedures are described in online supplemental document.
Plasmid and lentiviral vector transfection
Dominant-negative and constitutively active mutants of Rac1, Cdc42 or RhoA (gifts from Dr Shu Chien, University of California, San Diego, California, USA), precursors (PreR21) or antagomirs (AMR21) of miR-21 or control (scrambled) miRs (Ambion, Austin, Texas, USA) were transfected into Huh7 or Hep3B cells using Lipofectamine 2000 (Invitrogen, Carlsbad, California, USA). MiR-21 inhibitor (LentimiRa-Off-hsa-miR-21 vector, mh30326) and control vector (LentimiRa-Off) were purchased from Abmgood (British Columbia, Canada). Short hairpin RNA-green fluorescence protein (GFP) lentiviral vector construct for C-C chemokine receptor-2 (CCR2) was purchased from Genedirex (Las Vegas, Nevada, USA). HCC cells were transfected with the lentivirus stocks and selected for stably-transduced cells (ie, Huh7GFP, CCR2-knockdown [CCR2KD] HuhGFP, and miR-21-off [miR-21off] Huh7GFP) by puromycin.
Protein array assay
Cell lysates were collected for the analysis of protein expression profiles using Human Cytokine Array C (Raybiotech, Parkway Lane, Norcross, Georgia, USA) that detects 120 cytokines, as described.8 ,13 Detailed procedures are described in online supplemental document.
Argonaute-2 (Ago2) pull-down assay
Cells were lysed for the Ago2 pull-down assay, as described.14 The purified RNA was analysed by quantitative real-time RT-PCR for Rho GTPase-activating protein 24 (ArhGAP24) and tissue inhibitor of metalloproteinase-3 (TIMP3) with primers provided in online supplementary table S1.
Mice
Eight-week-old BALB/c nude male mice were purchased from Charles River (Biolasco, Taipei, Taiwan) and housed in a pathogen-free animal facility. For all surgical procedures, mice received analgesia by Carprofen (5 mg/kg) and were anaesthetised by 2% isoflurane. All experiments were performed in accordance with the National Institutes of Health (NIH) guidelines and with the approval of the Animal Research Committees of National Health Research Institutes.
In vivo invasion assay
A total of 1×106 HCC cells mixed with matrigel were injected into the back flanks of nude mice, and tumours were allowed to grow for 4 weeks. The cylinder was filled with EPC-embedded matrigel EPCs (5×104 cells) or the cells mixed with anti-MCP-1 antibody or peptide nucleic acid-based microRNA inhibitor PNA-AMR21 (Panagene, Daejeon, Korea) or negative controls (IgG or PNA-control miRs). The cylinder was then inserted into the tumour mass. After 4 weeks, the cylinder was collected and photographed, and the invaded cells were retrieved from the explants and measured by flow cytometry.
Orthotropic transplantation model
Huh7GFP, CCRKD Huh7GFP, and miR21off Huh7GFP cells were co-cultured with CFU-ECs for 2 days, and then suspended in 40 µL DMEM/matrigel (1:1) for each nude mouse. Through an 8-mm transverse incision in the upper abdomen under anaesthesia, each nude mouse was orthotropically inoculated in the left hepatic lobe with a microsyringe. After 4 weeks, all mice were euthanised by intoxication with 100% carbon dioxide, and their livers and lungs were fixed with neutral formalin and prepared for histological examination.
Human HCC tissues
HCC specimens were obtained from Taiwan Liver Cancer Network (TLCN, National Health Research Institutes, Taiwan). Informed consent was obtained from each patient before surgery. Clinical parameters and pathological features were certified by TLCN. The use of the 15 HCC tissues with different tumour node metastasis (TNM) stages, paired non-tumour parts and hepatic haemangioma tissues (as control livers) in this study was approved by the Institutional Review Board and the TLCN User Committee of National Health Research Institutes.
Results
CFU-ECs, but not ECFCs, induce HCC cell chemotaxis through the Rac1 and MMP9 activations
We first assessed whether different EPCs, that is, CFU-ECs and ECFCs, exert differential effects on HCC cell chemotaxis. Transwell assays demonstrated that CFU-ECs, but not ECFCs, induce migration and invasion of human hepatoma Huh7 and Hep3B cells (figure 1A). As controls, these EPCs did not induce chemotactic movement of normal hepatocyte Chang liver cell line cells. The CFU-EC-inductions of Huh7 cell migration (figure 1B and see online supplementary movie S1) and invasion (figure 1C) were confirmed by using horizontal chemotactic mobility and collagen gel invasion assays, respectively. Co-culturing Huh7 and Hep3B cells with CFU-ECs induce increased activity of Rac1, but not RhoA and Cdc42, in these HCC cells (figure 1D and see online supplementary figure S1A). In contrast, co-culture with ECFCs did not have this inducibility. Transfections with constitutively active mutant of Rac1 (ie, Rac1V12), but not Cdc42 (ie, Cdc42V12) and RhoA (ie, RhoAV14), resulted in increases in the migrations of monocultured and CFU-EC-co-cultured Huh7 cells (figure 1E). In contrast, transfections with dominant-negative mutant of Rac1 (ie, Rac1N17) inhibited CFU-EC-induced Huh7 cell migration. Dominant-negative mutants of Cdc42 (ie, Cdc42N17) and RhoA (ie, RhoAN19) did not have this inhibitory effect. Moreover, co-culture with CFU-ECs, but not ECFCs, induced the expression and activity (figure 1F and see online supplementary figure S1B) of MMP9, but not MMP2 in HCC cells. Co-culture with these EPCs subtypes did not alter the cell cycle distribution and proliferation in HCC cells (see online supplementary table S2). Our data indicate that CFU-ECs induce HCC cell migration and invasion through increased activities of Rac1 and MMP9.


CFU-ECs activate Rac1 and MMP9 to elicit directed chemotaxis of HCC cells in vitro. Transwell (A), horizontal chemotactic mobility (B) and collagen gel invasion (C) assays were used to detect the in vitro migration and invasion of HCC cells and normal hepatocyte CHLs induced by co-culture with CFU-ECs and ECFCs, as described in online supplementary materials and methods.8 Monoculture controls contain only HCC cells without EPC co-culture (Ø). Schematic diagrams of experimental designs are shown in the panels. The cells were co-cultured for 1 day (B), 2 days (A) and 7 days (C). Representative photomicrographs in C show Huh7 cells labelled with CellTracker Orange CMRA (chloromethyl derivative of fluorescein, arrow) that invade into the collagen gel after co-culture. Bar=200 μm. (D) Rho GTPase activities were measured in the cell lysates of monocultured HCC cells and the cells co-cultured with different EPC subtypes by using a glutathione S-transferase (GST) pull-down assay. Controls are cell lysates preloaded with either GDP or GTP prior to conjugation with sepharose beads. (E) Huh7 cells transfected with dominant-negative (ie, Cdc42N17, RacN17 and RhoAN19) or constitutively active (ie, Cdc42V12, RacV12 and RhoAV14) mutants of Rho GTPases were kept as controls or co-cultured with CFU-ECs for 2 days, and their migration was assessed by transwell assay. (F) Huh7 and Hep3B cells were kept as controls or co-cultured with different EPCs for 1 day, and their enzymatic activities of MMP9 and MMP2 were determined by using gelatine zymography. Data in A–C and E are means±SEM from three independent experiments. *p<0.05 versus control Ø or mock. Results in D and F are representative of triplicate experiments with similar results. CFU-EC, colony forming unit-endothelial cell; CHL, Chang liver cell line; ECFCs, endothelial colony-forming cells; EPC, endothelial progenitor cell; HCC, hepatocellular carcinoma; GDP, guanosine diphosphate; GTP, guanosine triphosphate; MMP, matrix metallopeptidase.
MCP-1 released by CFU-ECs contributes to their induction of HCC cell migration and invasion
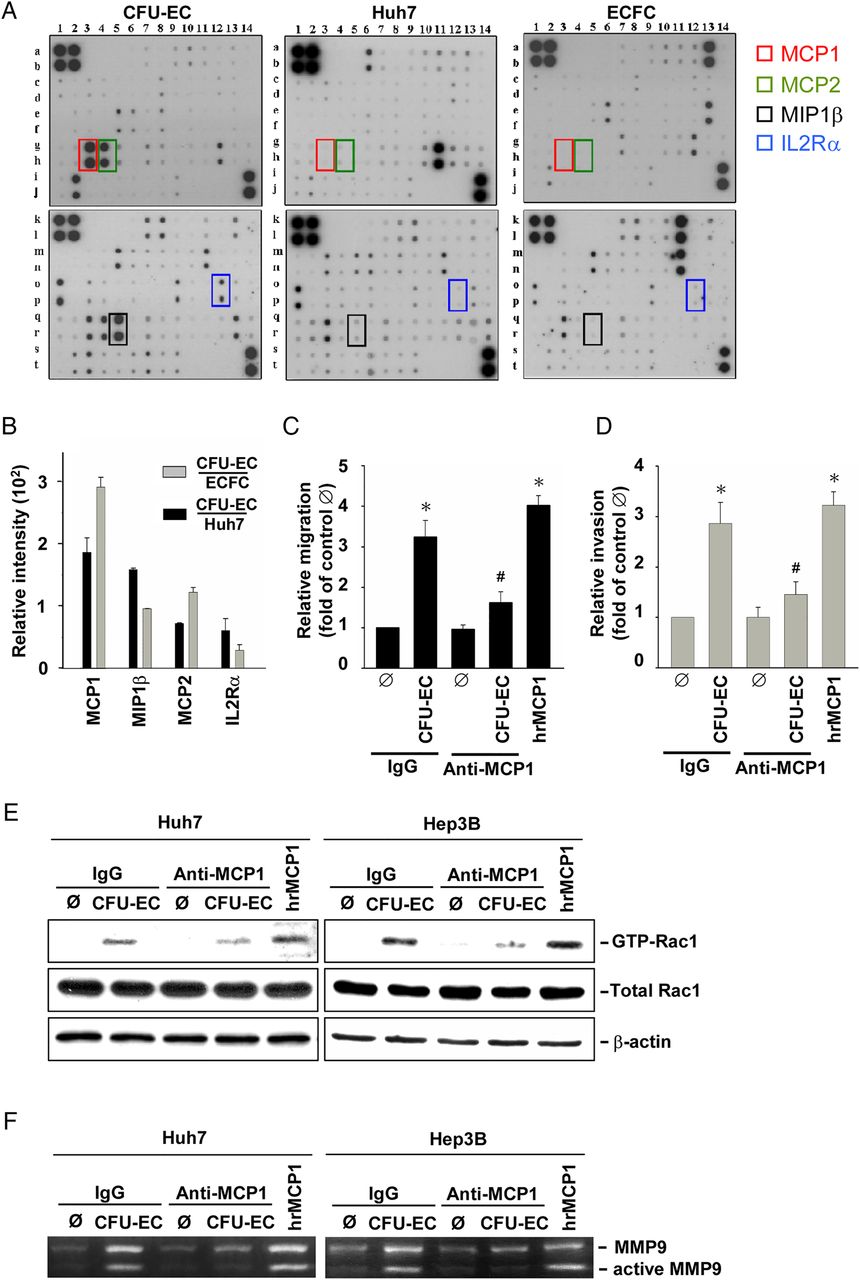
To examine which mediator(s) might be released from CFU-ECs to contribute to their induction of HCC cell migration and invasion, we analysed the cell lysates by using human cytokine arrays containing antibodies against 120 cytokines (see online supplementary table S3). MCP-1, macrophage inflammatory protein-1β, MCP-2, and interleukin-2 receptor-α are mediators whose expressions are significantly higher in CFU-ECs than in ECFCs and Huh7 cells (figure 2A and see online supplementary table S4). Among these proteins, MCP-1 showed the greatest expression in CFU-ECs in comparison with ECFCs and Huh7 cells (figure 2B). Pretreating CFU-ECs with an anti-MCP-1 neutralising antibody (compared with isotypic control IgG; 5 μg/mL each) for 1 h before co-culture resulted in inhibitions in CFU-EC-induced migration (figure 2C) and invasion (figure 2D) of Huh7 cells. As positive controls, human recombinant MCP-1 (hrMCP-1; 50 ng/mL) induced Huh7 cell migration and invasion. Moreover, treatments with an anti-MCP-1 neutralising antibody resulted in inhibitions in CFU-EC-induced Rac1 (figure 2E) and MMP9 (figure 2F) activities in Huh7 and Hep3B cells. These results indicate that MCP-1 released from CFU-ECs is a major mediator contributing to CFU-EC-induced migration and invasion of HCC cells.

HCC cell chemotaxis towards CFU-ECs is mediated by MCP-1 and its activations of Rac1 and MMP9. (A) Detection of protein levels of cytokines produced by CFU-ECs, ECFCs and Huh7 cells. Signal detection by enhanced chemiluminescence (ECL) shows that the levels of MCP-1 (red box), MCP-2 (green box), MIP-1β (black box) and IL-2Rα (blue box) produced by CFU-ECs are higher than those by ECFCs and Huh7 cells. (B) Quantitative analysis of the levels of MCP-1, MCP-2, MIP-1β and IL-2Rα produced by CFU-ECs relative to those by ECFCs and Huh7 cells. The migration (C) and invasion (D) of Huh7 or Hep3B cells and their activities of Rac1 (E) and MMP9 (F) induced by CFU-EC-co-culture were assessed. The cells were pretreated with control IgG or anti-MCP-1 neutralising antibody for 1 h. Positive controls are the cells stimulated with hrMCP-1 for 2 days. Data in B–D are means±SEM from three independent experiments. *p<0.05 versus control Ø. #p<0.05 versus IgG-treated cell. Results in A, E and F are representative of triplicate experiments with similar results. CFU-EC, colony forming unit-endothelial cell; ECFCs, endothelial colony-forming cells; HCC, hepatocellular carcinoma; IL-2Rα, interleukin-2 receptor-α; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein.
CFU-EC-induced HCC cell migration and invasion are mediated by MCP-1-induction of miR-21 in HCC cells
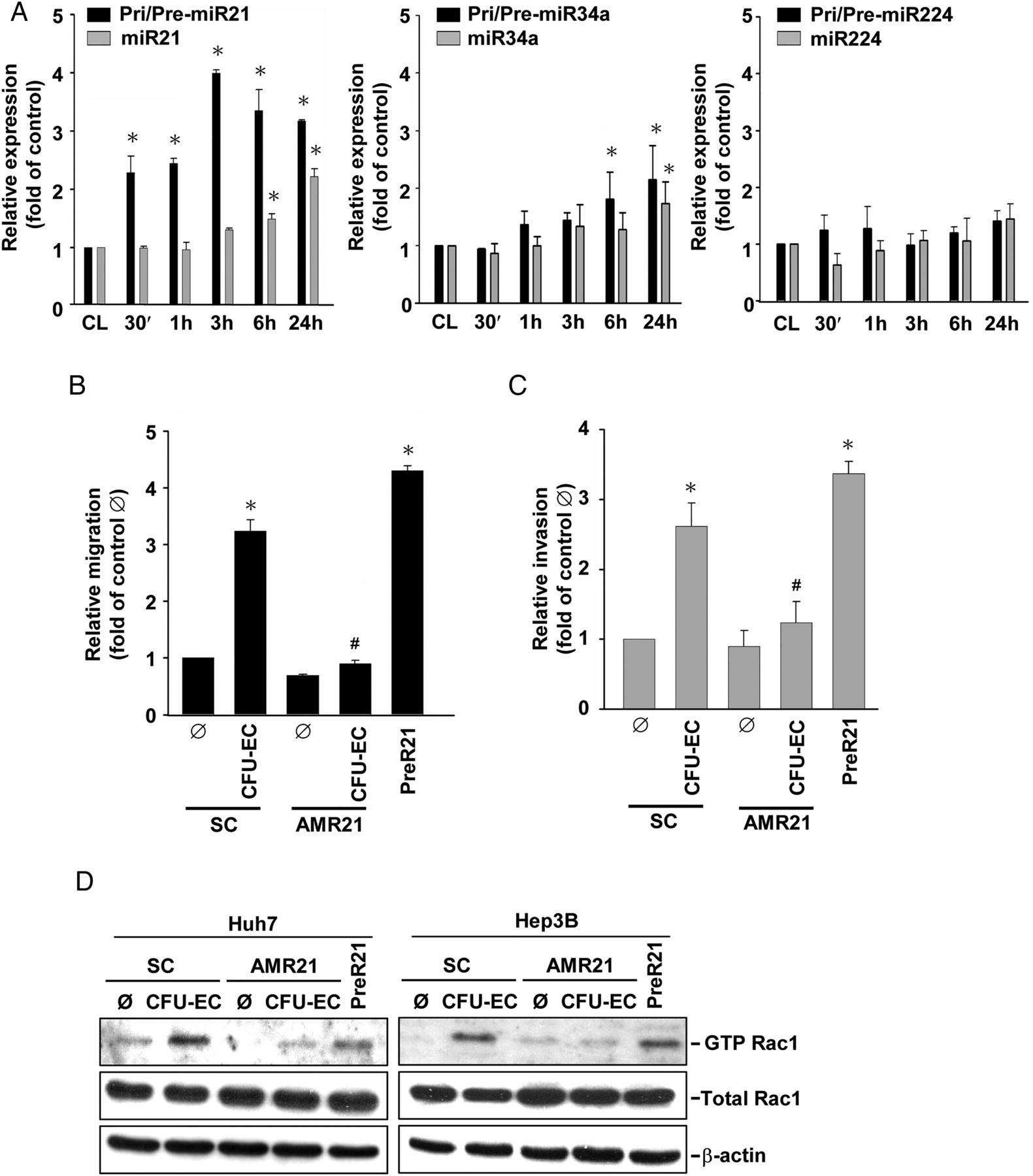
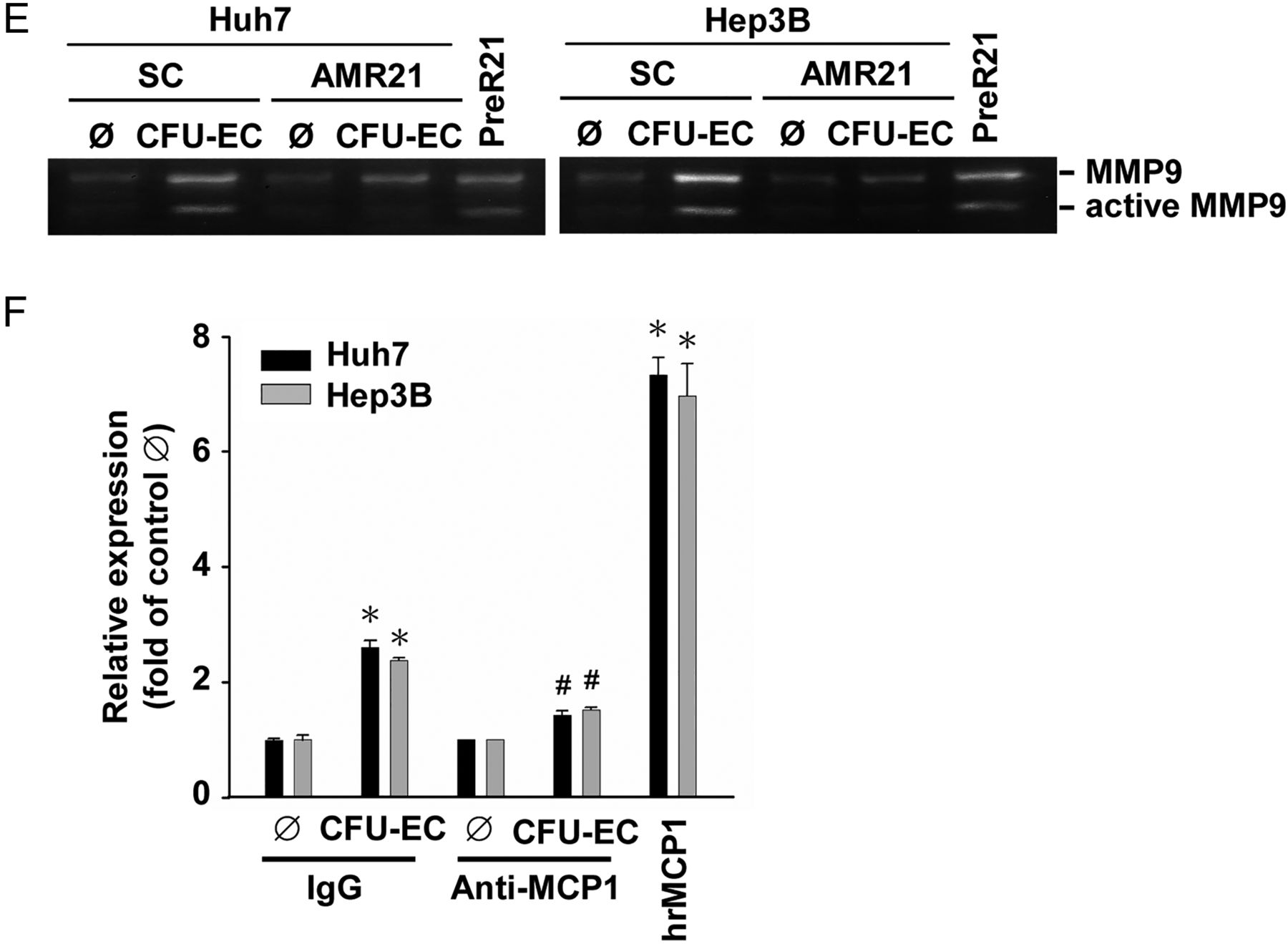
MiR-21, miR-34a and miR-224 have been shown to be onco-miRs, whose expression is positively correlated with the advanced invasive stages of HCC.12 ,15 Co-culturing Huh7 cells with CFU-ECs induced rapid increases in the expressions of primary/precursor (within 30 min) and mature (within 6 h) miR-21 in Huh7 cells (figure 3A). The induction of primary/precursor and mature miR-34a in Huh7 cells by CFU-ECs was later (within 6 h and 24 h after co-culture, respectively). Moreover, CFU-ECs had no effect on miR-224 expression in Huh7 cells. Since miR-21 showed the greatest responses to CFU-ECs among these examined miRs, we investigated whether miR-21 is involved in CFU-EC-induced HCC cell migration and invasion. Transfecting Huh7 cells with AMR21 (compared with scramble controls, 10 nM each) inhibited CFU-EC-induced miR-21 expression in Huh7 cells (see online supplementary figure S2), which was accompanied by the reductions in CFU-EC-induced migration (figure 3B) and invasion (figure 3C) of these cells. As positive controls, transfection with PreR21 (10 nM) resulted in increases in miR-21 expression in Huh7 cells and hence their migration and invasion. The molecular signalling involved in the modulations of HCC cell migration and invasion by AMR21 and PreR21 included their concomitant modulations in Rac1 (figure 3D) and MMP9 (figure 3E) activities. Pretreating HCC cells with an anti-MCP-1 neutralising antibody resulted in inhibitions in CFU-EC-induction of miR-21 in these cells (figure 3F). As positive controls, hrMCP-1 stimulation induced miR-21 expression in these HCC cells. These results indicate that CFU-EC-induced HCC cell migration and invasion are mediated by MCP-1-induction of miR-21 and its modulations in Rac1 and MMP9 activities in HCC cells.


CFU-ECs induce miR-21 expression to regulate Rac1 and MMP9 activities in HCC cells through MCP-1. (A) Huh7 cells were kept as controls or co-cultured with CFU-ECs for the indicated times, and their expression of primary/precursor and mature forms of miR-21, miR-34a, and miR-224 was analysed by quantitative RT-PCR. (B–E) Huh7 or Hep3B cells were transfected with AMR21 or scramble controls (SC) for 1 day, and then kept as controls or co-cultured with CFU-ECs for an additional 1 day. Positive controls are cells stimulated with PreR21 for 1 day. The migration (B) and invasion (C) of these cells and their activities of Rac1 (D) and MMP9 (E) were assessed by using the transwell, GST pull-down, and gelatine zymographic assays, respectively. (F) HCC cells were pretreated with anti-MCP1 neutralising antibody versus control IgG for 1 h, and then kept as controls or co-cultured with CFU-ECs for 2 days. Positive controls are cells stimulated with hrMCP-1 for 2 days. The expression levels of miR-21 were determined by quantitative RT-PCR. Data in A–C and F are means±SEM from three independent experiments. *p<0.05 versus control cell or Ø. #p<0.05 versus SC-treated or IgG-treated cell. Results in D and E are representative of triplicate experiments with similar results. AMR21, antagomirs of miR-21; CFU-EC, colony forming unit-endothelial cell; HCC, hepatocellular carcinoma; hrMCP, human recombinant MCP; MCP, monocyte chemotactic protein; miR-21, microRNA-21; PreR21, precursors of miR-21.
The CCR2/c-Jun N-terminal kinase (JNK)/activator protein-1 (AP-1) cascade is involved in CFU-EC/MCP-1-induced miR-21 biogenesis in HCC cells
We next sought to elucidate the signalling pathways involved in CFU-EC-induction of miR-21 in HCC cells. Co-culture with CFU-ECs induced the phosphorylation of JNK, but not extracellular signal-regulated kinases, in Huh7 cells over the 2 h-period tested (figure 4A). This CFU-EC-activation of JNK in Huh7 cells was abolished by transfecting with CCR2-specific siRNA (figure 4B). For downstream signals, co-culture with CFU-ECs induced phosphorylation of c-Jun, which is an AP-1 component, in Huh7 cells (figure 4C). This response was inhibited by transfecting Huh7 cells with CCR2-specific and JNK-specific siRNAs. As positive controls, hrMCP-1 stimulation induced JNK and c-Jun phosphorylations in Huh7 cells. Knockdowns of CCR2 and JNK inhibited CFU-EC-induced migration and invasion of Huh7 cells (see online supplementary figure S3).


CFU-ECs induce miR-21 transcription in HCC cells through the CCR2/JNK/AP-1 pathway. Huh7 cells were kept as controls or co-cultured with CFU-ECs for the indicated times (A), 2 h (B) or 1 day (C–F). In some experiments, Huh7 cells were transfected with specific siRNA of CCR2 (siCCR2) or JNK (siJNK) or control siRNA (siCL) (25 nM each) before co-culture. Positive controls are the cells stimulated with hrMCP-1. The phosphorylations of ERK, JNK, and c-Jun were determined by western blot analysis. (D) Huh7 cells were transfected with a series of luciferase reporter vectors bearing the full-length or truncated promoter fragments of miR-21 with the wild type putative AP-1 binding sites or mutated binding sites. These cells were then co-cultured with or without CFU-ECs and subjected to a luciferase activity assay. (E) Huh7 cells were co-transfected with JNK-specific or CCR2-specific siRNA and a luciferase reporter bearing four copies of AP-1 sites (TRE×4-luc), and then were kept as controls or co-cultured with CFU-ECs. The luciferase activities were assessed. (F) Huh7 cells were kept as controls or co-cultured with CFU-ECs, and the association of c-Jun with the promoter regions of miR-21 (miPPR21) was analysed by ChIP assay. Data in A, D and E are means±SEM from three independent experiments. *p<0.05 versus control cell, mock, Ø. #p<0.05 versus AP-13-transfected or siCL-transfected cell, δp<0.05 versus AP-12-transfected cell. Results in B, C and F are representative of triplicate experiments with similar results. AP-1, activator protein-1; CCR2, C-C chemokine receptor-2; CFU-EC, colony forming unit-endothelial cell; ChIP, chromatin immunoprecipitation; ERK, extracellular signal-regulated kinases; HCC, hepatocellular carcinoma; hrMCP, human recombinant monocyte chemotactic protein; JNK, Jun N-terminal kinase; miR-21, microRNA-21.
Since the promoter region of miR-21 contains three AP-1 binding sites,14 we investigated whether AP-1 regulates CFU-EC-induction of HCC cell miR-21 at the transcriptional level. CFU-EC-co-culture of Huh7 cells transfected with a wild type promoter region of miR-21 (compared with empty control) induced ≈fivefold increases in miR-21 promoter activity in these Huh7 cells (AP-13 in figure 4D). This CFU-EC-induced miR-21 promoter activity was reduced when Huh7 cells were transfected with a mutant with a deletion of a AP-1 binding site between nt −410 and −246 (AP-12 in figure 4D). Further deletion between nt −246 and −150 (AP-11 in figure 4D) or mutation at the AP-1 binding site between nt −195 and −185 (AP-1x in figure 4D) caused additional decreases in CFU-EC-induced luciferase activity. Moreover, further mutation at the two AP-1 binding sites (nt −195 to −185 and −95 to −74; AP-1xx in figure 4D) abolished CFU-EC-induced luciferase activity. Reporter assays using a luciferase reporter driven by four copies of the AP-1 site (TRE×4-luc) demonstrated that co-culture with CFU-ECs induces AP-1 binding activity in Huh7 cells through the MCP-1/CCR2/JNK cascade (figure 4E). Chromatin immunoprecipitation assay demonstrated that CFU-ECs induce in vivo binding of c-Jun to the promoter region of miR-21 in Huh7 cells through the MCP-1/CCR2/JNK cascade (figure 4F). These results indicate that CFU-ECs induce miR-21 expression in HCC cells at the transcriptional level through their production of MCP-1 and its induction of the CCR2/JNK/AP-1 cascade in HCC cells.
CFU-EC-induction of miR-21 activates Rac1 and MMP9 in HCC cells by silencing ArhGAP24 and TIMP3, respectively
By using three bioinformatic algorithms PicTar, microRNA.org and TargetScan 4.2, we predicted that the MMP inhibitors TIMP1, TIMP3, reversion-inducing-cysteine-rich protein with kazal motifs (RECK), the negative regulator of Rho GTPases ArhGAP24, and the tumour suppressor phosphatase and tensin homologue (PTEN) may be target genes of miR-21. Co-culturing Huh7 cells with CFU-ECs or transfecting with PreR21 reduced the mRNA expressions of ArhGAP24 and TIMP3, but not TIMP1, RECK and PTEN, in Huh7 cells (figure 5A). These CFU-EC-reductions of ArhGAP24 and TIMP3 were rescued, at least in part, by transfecting Huh7 cells with AMR21. Similar results were obtained in the protein levels of ArhGAP24 and TIMP3 (figures 5B, C). Analysis of miRNA-induced silencing complexes (miRISCs) immunoprecipitated with an anti-Ago2 antibody showed increased levels of ArhGAP24 (figure 5D) and TIMP3 (figure 5E) mRNAs and miR-21 (see online supplementary figure S4) in CFU-EC-co-cultured or PreR21-transfected Huh7 cells. Transfecting Huh7 cells with AMR21 inhibited CFU-EC-induced ArhGAP24 and TIMP3 mRNAs and miR-21 expressions in miRISCs. Transfecting Huh7 cells with ArhGAP24-specific and TIMP3-specific siRNAs (30 nM each), which reduced the expression of the respective mRNAs by 70–80% compared with control siRNA (see online supplementary figure S5), resulted in increases in their Rac1 and MMP9 activities, respectively (figures 5F, G). Moreover, knockdowns of ArhGAP24 and TIMP3 inhibited CFU-EC-induced Rac1 and MMP9 activities in Huh7 cells, respectively. These results indicate that ArhGAP24 and TIMP3 are regulated by miR-21 at the post-transcriptional level through their binding to Ago2 in the miRISCs, which consequently modulate Rac1 and MMP9 activities in Huh7 cells, respectively.

CFU-EC-induction of miR-21 silences ArhGAP24 and TIMP3 expressions to activate Rac1 and MMP9 in HCC cells, respectively. (A and B) Huh7 cells were transfected with AMR21 or scramble controls (SC) for 1 day, and then were kept as controls or co-cultured with CFU-ECs for an additional 1 day. The expression of indicated mRNAs (A) and proteins (B) was examined by RT-PCR and western blot analysis, respectively. Positive controls are the cells transfected with PreR21. (C) Immunofluorescence staining of ArhGAP24 or TIMP3 in monocultured or CFU-EC-co-cultured Huh7 cells transfected with AMR21, PreR21 or scramble controls for 1 day. Bar=50 μm. (D and E) Ago2 pull-down assay was performed to determine the expressions of ArhGAP24 (D) and TIMP3 (E) in Ago2-immunoprecipitated miRISCs by quantitative RT-PCR. (F and G) Huh7 cells were transfected with ArhGAP24-specific (F) or TIMP3-specific siRNA (G) or control siRNA, and then kept as controls or co-cultured with CFU-ECs for 1 day. The activities of Rac1 (F) and MMP9 (G) were determined by GST pull-down and gelatine zymographic assays, respectively. Data in D and E are means±SEM from three independent experiments. *p<0.05 versus control Ø. #p<0.05 versus SC-transfected cell. Results in A–C, F, and G are representative of triplicate experiments with similar results. AMR21, antagomirs of miR-21; ArhGAP24, Rho GTPase-activating protein 24; CFU-EC, colony forming unit-endothelial cell; HCC, hepatocellular carcinoma; miR-21, microRNA-21; miRISCs, miRNA-induced silencing complexes; TIMP3, tissue inhibitor of metalloproteinase-3.
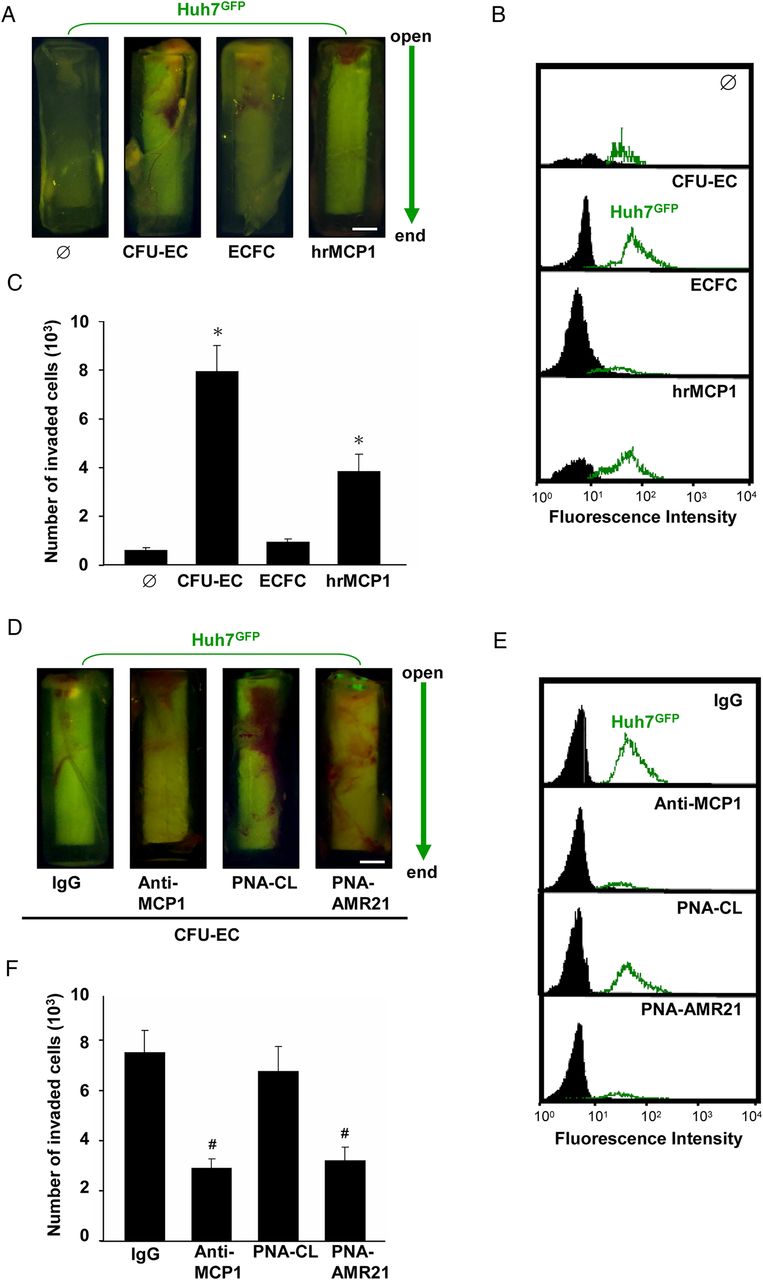
MCP-1 released from CFU-ECs induces HCC cell invasion through miR-21 in vivo
We further characterised the in vivo role of EPCs in regulating HCC cell invasion using a modified cell invasion assay (see online supplementary figure S6). Huh7 cells were labelled with GFP and injected into nude mice for 4 weeks to allow growing to a tumour mass (≈1 cm3). A cylinder tube, which contains matrigel embedded with CFU-ECs, ECFCs or hrMCP-1, was grafted into the tumour mass. Over 4 weeks, the invaded Huh7 cells into the cylinders were photographed (figure 6A) and extracted for analysis (figures 6B,C). Co-incubation with CFU-ECs or hrMCP-1 induced Huh7 cell invasion into the cylinders (figure 6A–C). In contrast, ECFCs did not have this inducibility. This CFU-EC-induced Huh7 cell invasion was inhibited by co-incubating Huh7 cells with anti-MCP-1 neutralising antibody or PNA-AMR21 in the cylinders (figure 6D–F). These results indicate that MCP-1 and miR-21 play important roles in modulating CFU-EC-induced HCC cell invasion in vivo.

MiR-21 regulates HCC cell invasion induced by CFU-EC-released MCP-1 in vivo. CFU-ECs (106 cells), ECFCs (106 cells), or hrMCP-1 (50 ng/mL) were premixed with matrigel in a silicone tube and then grafted into the Huh7GFP tumour mass for 4 weeks (A–C). In some experiments, Huh7GFP cell masses were premixed with anti-MCP-1 neutralising antibody (5 μg/mL), PNA-antagomir-21 (PNA-AMR21, 200 nM), control IgG or PNA-control (PNA-CL), and then transplanted into subcutaneous injection of Nu/Nu mice and placed into the opening of the CFU-EC-embedded tubes for 4 weeks (D–F). (A and D) Representative photographs indicate the movement of Huh7GFP cells (top) towards the bottom sides of the tubes. (B, C, E, and F) The numbers of invaded Huh7GFP cells in different tubes were analysed by fluorescence-activated cell sorting (FACS). Bar=1 mm. Data in C and F are means±SEM from three independent experiments. *p<0.05 versus control Ø. #p<0.05 versus IgG-treated or PNA-CL-treated cell. Results in A, B, D, and E are representative of triplicate experiments with similar results. AMR21, antagomirs of miR-21; CFU-EC, colony forming unit-endothelial cell; ECFCs, endothelial colony-forming cells; GFP, green fluorescence protein; HCC, hepatocellular carcinoma; hrMCP, human recombinant monocyte chemotactic protein; miR-21, microRNA-21.
CFU-EC-educated HCC cells exhibit EMT phenotype with increased stemness marker expression in vitro and have intrahepatic metastatic capability in vivo. CCR2KD and miR-21off Huh7 stable clones were established (see online supplementary figure S7) to investigate the effects of CFU-ECs on HCC cell EMT in vitro and intrahepatic metastasis in vivo. Co-culturing Huh7 cells with CFU-ECs resulted in increased expressions of N-cadherin, vimentin, Snail1 and Twist, and decreased expression of E-cadherin in these cells (figure 7A), with morphological changes from cuboidal-like epithelial towards elongated, fibroblast-like mesenchymal phenotypes (see online supplementary figure S8A). These CFU-EC-regulations of EMT-related genes in Huh7 cells can be mimicked by treating with rhMCP-1 and transfecting with PreR21, and were reduced in CCR2KD and miR-21off Huh7 cells co-cultured with CFU-ECs (figure 7A). Knockdowns of Rac1 and MMP9 also inhibited these CFU-EC-induced gene regulations in Huh7 cells (figure 7B and see online supplementary figure S9). CFU-EC-culture induced the expression of stemness markers CD133, Nanog, Sox2, EpCAM and CXCR4 in Huh7 and Hep3B cells (see online supplementary figure S8B), with increased percentage of cell subpopulation double positive for CD133 and EpCAM, as compared with monocultured cells (see online supplementary figure S8C). Anchorage-independent growth assays showed that CFU-EC-co-culture increases the numbers of Huh7 cell spheres (figure 7C) and colonies (figure 7D), and these responses are reduced when CCR2KD and miR-21off Huh7 cells are used.

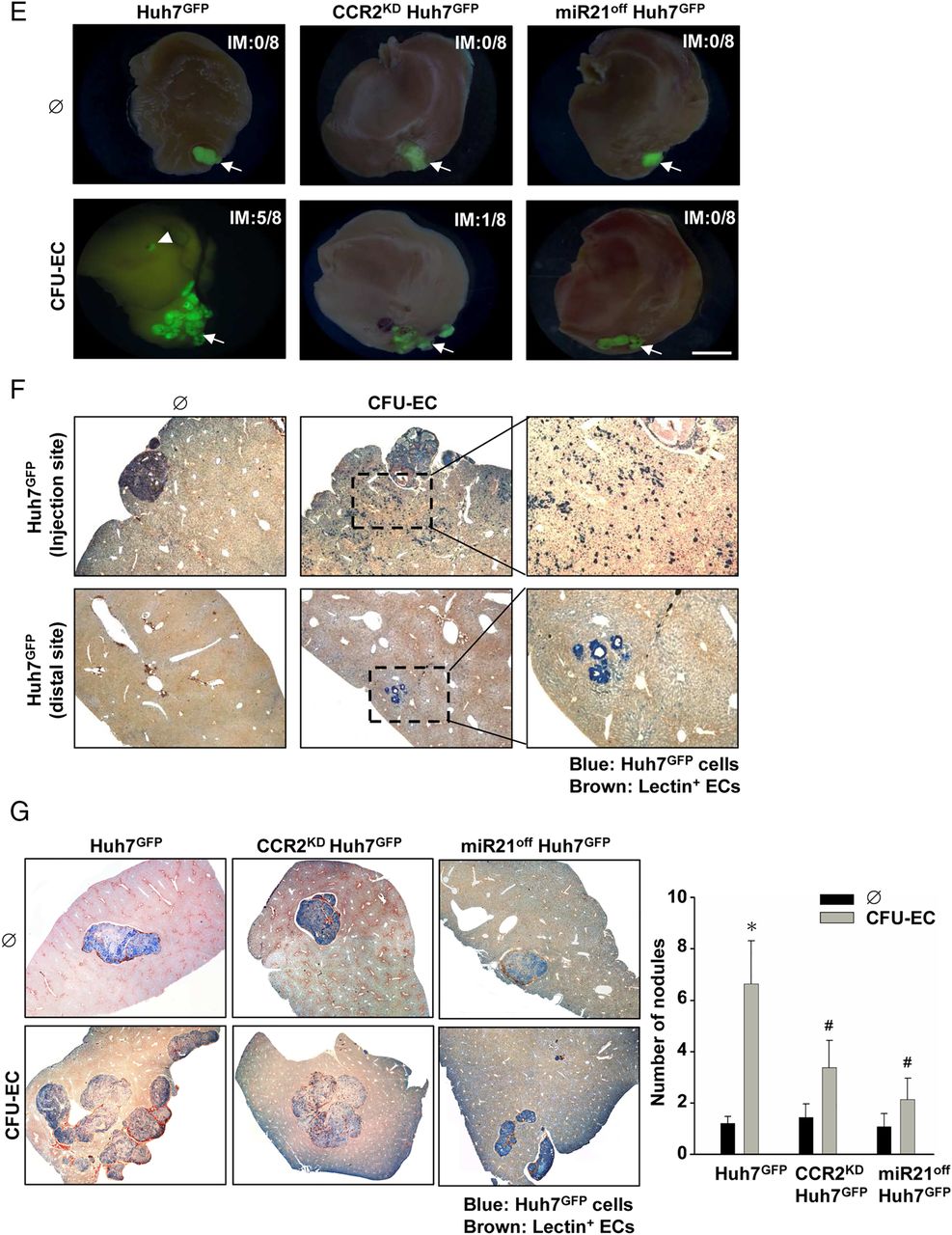
CFU-EC-stimulated HCC cells exhibit the EMT phenotype in vitro and possess the intrahepatic metastasis potential in vivo. (A) The mRNA levels of indicated EMT-related genes in mono-cultured Huh7GFP, CCR2KD Huh7GFP, and miR21off Huh7GFP cells, and the cells co-cultured with CFU-ECs for 3 days were examined. Positive controls are the cells treated with hrMCP-1 and PreR21. In some experiments, Huh7 cells were transfected with Rac1-specific or MMP9-specific siRNA or control siRNA for 1 day before co-culture (B). (C and D) Cells were co-cultured with CFU-ECs for 2 days; the spheres (C) and colonies (D) were photographed after 4 weeks. Bar=200 μm. (E–G) Orthotropic tumours were formed in the livers of nude mice after implantation of conditioned Huh7 cells for 4 weeks (n=8). These conditioned Huh7 cells included control Huh7GFP, CCR2KD Huh7GFP, and miR21off Huh7GFP cells co-cultured with or without CFU-ECs for 2 days. (E) Gross examination of tumour-bearing livers. Injection site of Huh7GFP cells (arrow) and the intrahepatic tumours generated at distal sites (arrowhead) were photographed after 4 weeks. IM indicates the ratio of intrahepatic metastasis (n=8). Bar=5 mm. (F and G) Eosin and immunohistochemical staining was performed by using the anti-GFP antibody (blue) and EC marker isolectin (brown) in xenograft tumours at the injection and distal sites 7 days after implantation. The numbers of intrahepatic nodules were quantified (G). Data in A–D and G are means±SEM from three to eight independent experiments. *p<0.05 versus control Ø. #p<0.05 versus Huh7GFP cell or siCL-transfected cell. CCR2, C-C chemokine receptor-2; CFU-EC, colony forming unit-endothelial cell; EMT; epithelial-mesenchymal transformation; GFP, green fluorescence protein; HCC, hepatocellular carcinoma; hrMCP, human recombinant monocyte chemotactic protein; miR-21, microRNA-21.
To investigate whether CFU-EC-educated HCC cells can metastasise in vivo, Huh7 cells (5×106) co-cultured with CFU-ECs for 2 days were orthotropically transplanted into the left lobes of nude mice. The results showed that unstimulated Huh7 tumour growth was constrained to the inoculation site (figure 7E). In contrast, CFU-EC-educated Huh7 cells spread out to form multiple nodules near the implanted sites; some tiny intrahepatic tumours were visualised by GFP (figure 7E–G). Interfering with CCR2 and miR-21 expressions in Huh7 cells inhibited intrahepatic metastasis (IM), as manifested by the decreases in the incidence of intrahepatic metastasis from 37.5% to 12.5% and 0%, respectively (figure 7E). Similarly, knockdowns of CCR2 and miR-21 expressions in Huh7 cells reduced the numbers of tumour nodules induced by CFU-EC-co-culture (figure 7G). However, no distant metastasis to other organs was observed. These results indicate that CFU-ECs can induce HCC cell EMT with increased stem cell marker expression in vitro and intrahepatic metastatic capability in vivo through the MCP-1/CCR2/miR-21 signalling cascade.
Increased numbers of MCP-1+ EPCs and their correlation with miR-21 expression in human HCC
To investigate the clinical relevance of our findings to human HCC, the numbers of MCP-1+ EPCs and their correlation with miR-21 expression in human HCC without and with metastatic capacities (ie, TNM stage I and stage IV, respectively) were examined. Serial sections of TNM stages I and IV HCC specimens (n=5 each) were co-immunostained for MCP-1 and the EPC marker inhibitor of DNA binding 1 (ID1), MCP-1 and the HCC marker glypican-3 (GPC3), and CCR2 and GPC3, and were counterstained with 4',6-diamidino-2-phenylindole (DAPI). MiR-21 expression was examined by in situ hybridisation method. The results showed that the numbers of MCP-1+ID1+ EPCs are significantly increased in human HCC (TNM stages I and IV) in comparison with benign liver tissues (figure 8A). The number of MCP-1+ID1+ EPCs in TNM stage IV HCC was higher than that in TNM stage I HCC (figure 8A–C). Interestingly, there was a positive correlation between the numbers of MCP-1+ID1+ EPCs and the expression levels of miR-21 among all examined HCC specimens (figure 8B). Moreover, the expression level of miR-21 in TNM stage IV CCR2+GPC3+ HCC cells was higher than that in TNM stage I HCC specimens (figure 8C). These results indicate that EPCs and their released MCP-1 in tumour niches may act as critical clinicopathological parameters for human HCC, with positive correlation with increased expression of miR-21.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MCP-1+ID1+ EPCs and miR-21 are clinicopathological parameters for metastatic stages of human HCC. (A) The numbers of EPCs stained double-positively for MCP-1 and ID1 were examined in human HCC tissues at TNM stages I (TI) and IV (TIV) and benign liver tissues (BL) (n=5 each). (B) Scatter plots showing a positive correlation between the average fluorescence intensities (arbitrary unit, A.U.) of in situ hybridisation for miR-21 and the numbers of MCP-1+ID1+ EPCs in the examined HCC sections (n=25, r=0.495, p<0.001). p Values and correlation coefficients (r) were calculated using the Spearman correlation test. (C) Serial sections of HCC at TNM stages I and IV (n=5 each) were co-immunostained for MCP-1 and ID1, MCP-1 and GPC3, and CCR2 and GPC3, and were counterstained with DAPI. miR-21 expression was examined by in situ hybridisation method. Arrows indicate the cells stained double-positively for MCP-1 and ID1 in the TNM stage I (upper panel) and stage IV (lower panel) HCC. B, blood. Bar=20 μm. (D) Schematic diagram showing that proangiogenic EPCs (ie, CFU-ECs) promote the malignance of HCC cells through EPC production of MCP-1 and its induction of miR-21 biogenesis in HCC. CCR2, C-C chemokine receptor-2; CFU-EC, colony forming unit-endothelial cell; EPC, endothelial progenitor cell; GPC3, glypican-3; HCC, hepatocellular carcinoma; ID1, inhibitor of DNA binding 1; MCP, monocyte chemotactic protein; miR-21, microRNA-21; TNM, tumour node metastasis.
Discussion
Angiogenesis and metastasis of de novo tumours are two causes of poor prognosis of HCC. In clinics, EPCs have been shown to act as angiogenic switches in tumour metastasis.2 In this study, we demonstrated that myeloid-derived EPCs (ie, CFU-ECs), but not outgrowth EPCs (ie, ECFCs), can produce high levels of MCP-1 to induce the chemotaxis of non-metastatic HCC cells and promote their EMT and intrahepatic metastasis though the induction of miR-21 biogenesis (summarised in figure 8D). This MCP-1-induction of miR-21 can silence its targets ArhGAP24 and TIMP3 to activate Rac1 and MMP9, respectively, and hence promote HCC cell migration and invasion. Thus, in addition to physical contribution to angiogenesis,5 our study advances the new notion that these CFU-ECs in proximity to HCC cells may act as a new metastatic activator to induce chemotaxis and EMT of HCC cells and their intrahepatic metastatic capability in vivo.
It has been hypothesised that EPCs play a dual role in angiogenesis by providing structural support and producing paracrine angiogenic factors once recruited to tumour site.2 The contributions of EPCs to neovasculature vary from poorly to well-differentiated tumours in animal models,16 and depend on the tumour grades in patients with HCC with or without cirrhosis.17 These observations raise a question of whether non-metastatic tumour cells can experience phenotypical changes proximal to EPCs or EPC-generated humoral environments. Recent studies by Giannoni et al18 reported that in prostate carcinoma, direct contact between EPCs and mesenchymal tumour cells induces the EphA2/ephrinA1 signalling to elicit a mesenchymal amoeboid transition in tumour cells, allowing them to attain a metastatic advantage. Moreover, EPCs have been shown to promote melanoma metastasis through the secreted protein acidic and rich in cysteine-driven cell-cell interactions and endocytosis.19 In concert with these previous studies, our results indicate that EPCs recruited to the tumour sites may represent a critical component of ‘soil’ to induce phenotypical switch of a non-metastatic tumour towards one with an increased potential for metastasis.
Clinical studies have shown a strong upregulation of MCP-1 in serum of patients with different liver diseases.20 There is a significant correlation between serum MCP-1 and well-differentiated tumours and lymph node metastasis in patients with cancer, as compared with healthy patients.21 In addition to promoting angiogenesis in tumour,21 the MCP-1/CCR2 signalling has been shown to recruit inflammatory monocytes to facilitate breast-tumour metastasis.22 Interestingly, the expression of MCP-1 was generally lower in human hepatoma cells than other stromal cells (ie, myofibroblast).23 Our data provide the first demonstration that MCP-1 is a key factor released from angiogenic CFU-ECs to induce the chemotaxis and metastatic potential of non-metastatic hepatoma cells. However, it cannot be excluded that the effects of CFU-ECs on HCC cells may be contributed by the synergistic effects of multiple mediators, rather than MCP-1 alone, although MCP-1 is a major mediator contributing to CFU-EC-modulation of HCC cell functions.
Recent studies have demonstrated the importance of miRs in regulating hepatocarcinogenesis.10 MiR-21 upregulation has been linked to liver disease and attracted intense interest.10 ,11 ,24 However, there is a lack of studies on the mechanisms by which miR-21 modulates gene expression and function in HCC cells and their interactions with stromal microenvironment. Several studies have indicated that the post-transcriptional expression of miR-21 is triggered by the JNK/AP-1 pathway in inflammation, colorectal cancer and HCC.25 In addition, there have been several genes, including PTEN, RECK and TIMP3, reported to be targeted by miR-21 to regulate tumour metastasis.24 In this study, we demonstrated that miR-21 biogenesis in HCC cells can be induced by CFU-EC-released MCP-1 through its downstream CCR2/JNK/AP-1 signalling cascade to act as a tumour onco-miR for hepatocarcinogenesis, particularly for EMT and intrahepatic metastasis. Moreover, in addition to TIMP3, our study identified a RhoGAP protein ArhGAP24 as a new target that can be silenced by miR-21. ArhGAP24 has been shown to inhibit Rac1-mediated migration, invasion and metastasis of tumour cells.26 While miR-21 is highly expressed in HCC, most of the RhoGAP proteins are downregulated in HCC in vivo.27 These results implicate an opposite correlation between miR-21 expression and RhoGAP activity in HCC in vivo. Our findings support this notion that CFU-EC-induction of miR-21 can silence the expression of ArhGAP24, with the consequent inductions of Rac1 activity in HCC cells and their migration and invasion. Although we found that miR-21 is highly responsive to MCP-1/CFU-ECs, it cannot be excluded that other miRs may also be responsive. A screening for MCP-1 responsive miRs in HCC remains an important issue that warrants further investigation.
MiR-21 has been shown to be an important component of cellular signalling circuitry that regulates the EMT programme.24 ,28 MiR-21 overexpression contributes to prostate cell transformation coupled with the appearance of EMT features,24 whereas miR-21 antagonism in malignant breast cancer cells reverses their EMT.29 However, the mechanisms by which miR-21 regulates EMT remain unclear. Our data suggest that CFU-EC-released MCP-1 and its induction of miR-21 in non-metastatic HCC cells may induce their EMT through the upregulation of Snail and Twist transcriptions. Recent studies indicate that tumour cells undergoing EMT may gain stem cell-like properties to give rise to tumour-initiating cells, with high proliferative capacity.30 MCP-1 has been shown to induce self-renewed expansion of cancer stem cells (CSCs) to promote in vivo tumourigenesis.31 In the present study, we found that co-culturing Huh7 cells with CFU-ECs can induce the expression of stemness markers CD133, Nanog, Sox2, EpCAM and CXCR4 in HCC cells and increase their EpCAM+CD133+ cell subpopulation. Thus, our findings suggest that CFU-ECs may be able to induce migration, invasion and EMT of HCC cells, and can generate the CSC-like subpopulation in HCC cells.
The clinical relevance of EPC-associated MCP-1 and miR-21 inductions to human HCC has been revealed by our study on human HCC tissues at different TNM stages (figure 8). There is increasing evidence that MCP-1 may exert protumour and antitumour effects. Anti-MCP-1 neutralising antibodies and MCP-1-specific anti-inflammatory compounds, which interfere with the MCP-1/CCR2 signalling axis, have been shown to inhibit the dissemination and metastasis of cancer cells in mouse xenograft models.32 ,33 However, total absence of MCP-1/CCR2 signalling in MCP-1−/− and CCR2−/− mice resulted in increased metastatic disease.34 In addition, MCP-1/CCR2 in patients with HCC has been shown to be linked to better prognosis.35 Thus, MCP-1 may play a dual role in early tumour immunosurveillance and sustaining the growth and progression of established neoplasms.34 ,36 Our data on human HCC suggest that EPCs and their MCP-1 and positive correlation with miR-21 induction may act as critical clinicopathological parameters in human HCC.
Recent reports have indicated that mobilised circulating EPCs may serve as biomarkers to predict HCC progression and liver cirrhosis.37 Disrupting the recruitment of these circulating EPC spikes by antiangiogenic drugs resulted in marked reductions in tumour rim size and blood flow in neovessels.38 As EPCs are endowed with multifunctional capacities, ex vivo-manipulated EPCs might be used as cellular vehicles to deliver therapeutic genes or vascular targeting drugs.2 Our in vivo experimental animal studies using invasion and orthotropic transplantation models demonstrated that MCP-1-induced miR-21 biogenesis is a connecting link for the chain of events leading to EPC-induced chemotaxis, EMT and intrahepatic metastasis of HCC cells. Targeting EPC-tumour interactions and their associated signalling axis may be developed as promising strategies for therapeutic intervention against tumour development. Our findings highlight an important mechanism by which EPC-tumour interactions link EPC secretomes to epigenetic control of tumour pathobiology, which consequently influence tumour progression and metastasis.
Acknowledgments
The authors thank Taiwan Hsinchiu Blood Donation Center for providing steady-state human blood samples, and also NHRI Core Facilities and Taiwan Liver Cancer Network for technical support with human specimens.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table S1
- Data supplement 4 - Online table S2
- Data supplement 5 - Online table S3
- Data supplement 6 - Online table S4
- Data supplement 7 - Online video
Footnotes
Contributors Y-TS: designed the study, performed the experiments, and wrote the manuscript. M-CW: performed the experiments. H-HP and JZ: wrote the manuscript and provided financial support to the study. D-YL: performed the data analysis. J-JC: designed the study, wrote the manuscript and provided financial support to the study. All authors reviewed the manuscript.
Funding This work was supported by Taiwan NSC grant 101-2321-B-400-001.
Competing interests None.
Patient consent Obtained.
Ethics approval National Health Research Institutes.
Provenance and peer review Not commissioned; externally peer reviewed.