Article Text

Abstract
Background Data on genetic susceptibility to sporadic gastric carcinoma have been published at a growing pace, but to date no comprehensive overview and quantitative summary has been available.
Methods We conducted a systematic review and meta-analysis of the evidence on the association between DNA variation and risk of developing stomach cancer. To assess result credibility, summary evidence was graded according to the Venice criteria and false positive report probability (FPRP) was calculated to further validate result noteworthiness. Meta-analysis was also conducted for subgroups, which were defined by ethnicity (Asian vs Caucasian), tumour histology (intestinal vs diffuse), tumour site (cardia vs non-cardia) and Helicobacter pylori infection status (positive vs negative).
Results Literature search identified 824 eligible studies comprising 2 530 706 subjects (cases: 261 386 (10.3%)) and investigating 2841 polymorphisms involving 952 distinct genes. Overall, we performed 456 primary and subgroup meta-analyses on 156 variants involving 101 genes. We identified 11 variants significantly associated with disease risk and assessed to have a high level of summary evidence: MUC1 rs2070803 at 1q22 (diffuse carcinoma subgroup), MTX1 rs2075570 at 1q22 (diffuse), PSCA rs2294008 at 8q24.2 (non-cardia), PRKAA1 rs13361707 5p13 (non-cardia), PLCE1 rs2274223 10q23 (cardia), TGFBR2 rs3087465 3p22 (Asian), PKLR rs3762272 1q22 (diffuse), PSCA rs2976392 (intestinal), GSTP1 rs1695 11q13 (Asian), CASP8 rs3834129 2q33 (mixed) and TNF rs1799724 6p21.3 (mixed), with the first nine variants characterised by a low FPRP. We also identified polymorphisms with lower quality significant associations (n=110).
Conclusions We have identified several high-quality biomarkers of gastric cancer susceptibility. These data will form the backbone of an annually updated online resource that will be integral to the study of gastric carcinoma genetics and may inform future screening programmes.
- CANCER GENETICS
- GASTRIC CARCINOMA
- GENETIC POLYMORPHISMS
- META-ANALYSIS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Gastric cancer is a leading cause of death by cancer worldwide.
Germline genetic variation is believed to play a key role in cancer predisposition.
Hundreds of studies have linked specific gene polymorphisms to the risk of developing gastric carcinoma.
What are the new findings?
We built up the first comprehensive database collecting the published evidence on the association between genetic variants and risk of gastric carcinoma.
Data meta-analysis (using dedicated methodology) identified several high-quality biomarkers of gastric cancer susceptibility.
Subgroup analysis identified polymorphisms associated with specific subsets of tumour type (intestinal vs diffuse) and site (cardia vs non-cardia).
Much work is still to be done to identify gene–environment interactions.
How might it impact on clinical practice in the foreseeable future?
These data will form the backbone of an annually updated online resource that will be integral to the study of gastric carcinoma genetics and may inform future screening programmes.
Introduction
Gastric carcinoma is the fifth most common cancer and the third most frequent cause of cancer death worldwide,1 and despite some therapeutic advances its prognosis is often unfavourable.2 ,3 Although the cascade of molecular events leading to gastric carcinogenesis are still largely unknown,3–5 genetic background,6 ,7 behavioural factors (eg, alcohol consumption, smoking habit, diet)8 ,9 and infectious agents (ie, Helicobacter pylori (HP))10 have been associated with the risk of developing gastric carcinoma. Of note, its incidence shows remarkable geographic heterogeneity, the highest rates being observed in Asia, which has been at least in part attributed to the differences in genetic polymorphism distribution and prevalence of HP infection (or HP strains) in different populations.11 ,12
Germline variations of DNA sequence are believed to represent a key aspect of predisposition to most complex traits, such as cancer.13–15 As regards gastric cancer, a small fraction of the familial risk (approximately 3%) can be explained by rare mutations in high-penetrance genes such as CDH1 (encoding E-cadherin), which is associated with hereditary diffuse gastric cancer.16 Less than 15% of cases are believed to have familial clustering, most of these lacking an association with specific germline mutations,3 ,6 suggesting that most familial clustering of this tumour are due to a combination of multiple alleles with lower penetrance.3 ,6
Numerous studies (enrolling tens of thousands of subjects) have been performed to investigate the role of genetic variation in gastric carcinogenesis, and many polymorphisms have been identified as potential risk factors for the development of this disease. Interestingly, different DNA variants appear to be correlated with the risk of specific gastric cancer sites (cardia vs non-cardia) and histological types (intestinal vs diffuse). However, the findings of these studies are not always consistent, and no systematic review covering all tested polymorphisms has been published thus far.
The aim of this work is to fill this gap in the international medical literature by presenting the first systematic synopsis and meta-analysis of the available evidence in the field of DNA variation and the risk of sporadic gastric carcinoma, including the interaction of polymorphisms with both ethnicity, environmental factors (ie, HP infection) and tumour features (ie, primary cancer site and histological type).
Materials and methods
Search strategy, eligibility criteria and data extraction
We followed the principles proposed by the Human Genome Epidemiology Network (HuGeNet) for the systematic review of molecular association studies.17–19
Studies dealing with the association between any genetic variant and predisposition to gastric carcinoma in humans were considered eligible, provided that the raw data (necessary to calculate the risk) or summary data were available. Exclusion criteria were data published in abstract form only, minor allele frequency in controls <1% (rare variants) and a sample size with <10 cases or controls.
A two-step search strategy was adopted. First, a systematic review of original articles, reviews and meta-analyses analysing the association between any genetic variant and gastric cancer risk was performed by searching MEDLINE (via the PubMed gateway). The search included the following three groups of keywords: “cancer”, “tumor” and “carcinoma”; “gastric” and “stomach”; “polymorphism”, “single nucleotide polymorphism” and its acronym “SNP”, “variant”, “variation”, “risk” and “locus”. Searches were conducted using all combinations of at least one keyword from each group. Finally, in the light of the growing use of high-throughput platforms for the investigation of gene variants, the expression “genome wide association study” and its acronym “GWAS” were also used as a second version of the third group of keywords.
Once this search was completed, in the second phase, (A) each single polymorphism was used as a keyword to further refine the search; (B) cited references from eligible articles were also searched; (C) publicly available databases dedicated to genotype/phenotype associations were searched (ie, Database of Genotypes and Phenotypes [dbGaP], http://www.ncbi.nlm.nih.gov/gap; Genome Medicine DataBase of Japan [GeMDBJ], http://gemdbj.nibio.go.jp; and HuGeNet Phenopedia, http://www.hugenavigator.net/HuGENavigator/startPagePhenoPedia.do); and (D) authors were contacted whenever unreported data were potentially useful to enable the inclusion of the study into the systematic review or to rule out data published in different articles but regarding overlapping series.
For analysis purposes, the database, which will be updated annually and will be publicly available at http://www.mmmp.org,20 was frozen in May 2014.
Additional methodology details are available as online supplementary information.
Statistical analysis
OR and corresponding 95% CIs were used to assess the strength of association between each genetic variant and cancer risk. Per-allele OR were calculated for each study and each polymorphism, assuming an additive (co-dominant) genetic model. This approach was chosen for the following reasons: (A) for some studies (including GWAS), only per-allele OR were available; (B) the additive model is widely used as a conservative choice between the recessive and the dominant model; (C) one model does not require adjustment for multiple hypotheses (which is necessary when different models are tested); (D) methods that let the data dictate the genetic model (eg, the model-free approach21) require raw data on genotype distributions (which were unavailable for some studies); and (E) when large sample sizes are available (as expected in a systematic review and meta-analysis), non-additive models rarely identify high-quality associations unidentified by the additive model: therefore, the co-dominant model is largely used in genetic association synopses.22–24
Summary OR were calculated by performing random effects meta-analysis (using the inverse variance method), which reduces to a fixed effect meta-analysis when no between-study heterogeneity exists. The choice of this model was suggested mainly by the heterogeneity typically expected in genetic association studies.
For each variant, a meta-analysis was performed if at least three independent datasets were available. As regards GWAS, if data were available, discovery and replication phases were considered as separate datasets.
Subgroup analysis by ethnicity (Asian vs Caucasian/other), primary tumour site (cardia vs non-cardia), histological subtype (intestinal vs diffuse) and HP status (positive vs negative) was performed if data permitted.
Population-attributable risk (PAR) was calculated using the Levin's formula: P (RR−1)/[1+P(RR−1)], where P is the proportion of controls exposed to the genotype of interest and the relative risk (RR) was estimated using the summary estimates (OR) calculated by meta-analysis.
The α level of significance was set at 0.05, except for the Harbord test and Q-test (0.10), and the Hardy–Weinberg equilibrium test (0.01).
All statistical analyses were performed with STATA V.11.0/SE (StataCorp, College Station, Texas, USA).
Assessment of cumulative evidence
In order to evaluate the credibility of each nominally statistically significant association identified by meta-analysis, we applied the Venice criteria.18 ,19 Briefly, credibility level was defined based upon the grade (A=strong, B=moderate or C=weak) of three parameters:
Amount of evidence, roughly depending upon the study sample size, was graded by the sum of cases and controls expressing the risk allele: grade A, B or C were assigned for >1000, 100–1000 and <100, respectively.
Replication of the association was graded based on the amount of heterogeneity: grade A, B or C were assigned for I2 <25%, 25–50% and >50%, respectively.
Protection from bias was graded as A if there was no observable bias (bias was unlikely to explain the presence of the association), B if bias could be present or C if bias was evident. Assessment of protection from bias also considered the magnitude of association: a score of C was assigned to an association with a summary OR <1.15 (or >0.87 in case of protective effect).18
According to these criteria, the credibility level of the cumulative evidence was defined as high (A grades only), low (one or more C grades) or intermediate (all other combinations).
In addition to the Venice criteria, we assessed the noteworthiness of significant findings by calculating the false positive report probability (FPRP),25 which is defined as the probability of no true association between a genetic variant and disease (null hypothesis) given a statistically significant finding. FPRP is based not only on the observed p value of the association test but also on the statistical power of the test and on the prior probability that the molecular association is real (according to a Bayesian approach). We calculated FPRP values for two levels of prior probabilities: at a low prior (10E-3) that would be similar to what is expected for a candidate variant, and at a very low prior (10E-6) that would be similar to what would be expected for a random variant. To classify a significant association as ‘noteworthy’, we used a FPRP cut-off value of 0.2.25
For each non-significant meta-analysis, we assessed the quality of summary evidence for no association.22 To this aim, we calculated the power of detecting a non-negligible association (ie, OR ≥1.15 or ≤0.87) for a hypothetical study with sample size equal to the combined sample sizes of the studies included in the meta-analysis (with an α level of significance set at 5%). If the power were ≥90%, no between-study heterogeneity were found (I2<25%) and no bias were detected, we considered the level of cumulative evidence high (ie, sufficient to rule out any meaningful relationship between that variant and gastric cancer risk). Either power <80%, or significant between-study heterogeneity or detection of bias (or any combination of them) indicated a low level of evidence. In all other cases, the level of evidence for no association was considered intermediate.
Results
Characteristics of eligible studies
The database with the main features and findings of all eligible studies is available in online supplementary table S1.
We identified 824 eligible articles (see online supplementary figure S1) comprising 2 530 706 subjects (cases: 261 386 (10.3%), range: 13–3632, mean: 316). More than 95% of these studies (788/824) were published after the year 2000 (with a steep positive trend over time; see online supplementary figure S2).
According to the prevalent ancestry (the race of at least 80% of the enrolled subjects), two-thirds of the studies were Asian (n=555, 67%).
Based on the design, the majority of the studies were hospital-based case–control studies (n=494, 60%), the remaining being population-based case–control studies, 24 of which were case–control studies within the frame of cohort prospective studies. Data from three GWAS were also available.26–28
Less than half of the eligible studies specified the histological subtypes (intestinal vs diffuse) of gastric carcinoma (n=329, 40%), the site (cardia vs non-cardia) of the primary tumour (n=338, 41%) and the status (positive vs negative) of gastric HP infection (n=102, 12%).
Overall, data on 2841 polymorphisms involving 952 distinct genes were available. Variation consisted mainly of SNP (n=2784; 97%), followed by insertions/deletions (n=34), variable number of tandem repeats (n=16) and polymorphisms revealed by a specific change in phenotype (n=7, such as presence/absence of enzymatic activity). These genetic variants were located in the DNA upstream the ‘relevant’ (meant as physically closest) gene (including the promoter region) (n=508), downstream the ‘relevant’ gene (n=215), in introns (n=1461), in exons (n=378), in the 5′-UTR (n=56) and the 3′-UTR (n=182). Among the exonic SNPs, the functional effects were generally missense (n=229) and synonymous coding changes (n=127), with the remaining being stop-gain, frameshift or splicing variations (n=5).
Distribution of variants (and significant associations with gastric cancer risk) across chromosomes is depicted in online supplementary figure S3. Of note, most of the variants tested are located on chromosome 1, no variant on chromosome Y has been ever investigated and only three variants in mitochondrial DNA have been studied.
Meta-analysis findings
The results of data meta-analysis are comprehensively reported in online supplementary table S2). At least three independent datasets were available for 156 variants across 101 genes, which allowed us to perform 456 meta-analyses. Of these, 164 were primary meta-analyses and 292 meta-analyses of subgroups as defined by ethnicity (Caucasian (n=44 meta-analyses) vs Asian (n=77)), cancer histological subtype (intestinal (n=41) vs diffuse (n=40)), primary tumour site (cardia (n=34) vs non-cardia (n=36)) and HP infection status (positive (n=12) vs negative (n=8)).
The number of datasets meta-analysed ranged from 3 to 68, the mean number being 7. Based on the number of datasets, the 10 most studied variants were the following: IL1B rs16944 (datasets, n=68), IL1RN rs2234663 (n=60), GSTM1 wild/null (n=50), IL1B rs1143627 (n=47), GSTT1 wild/null (n=40), TNF rs1800629 (n=38), TP53 rs1042522 (n=33), MTHFR rs1801133 (n=29), IL10 rs1800896 (n=29) and IL10 rs1800872 (n=25).
The number of subjects (cases plus controls) enrolled in the 456 meta-analyses ranged from 477 to 1 496 612 (median: 3704). Based on the number of subjects, the 10 most studied variants were the following: ABO rs8176719/rs8176746/rs8176747 (n=1 496 612 individuals; however, nearly 99% of these subjects were controls), PSCA rs2294008 (n=42 274), PLCE1 rs2274223 (n=33 219), IL1B rs16944 (n=29,789), PRKAA1 rs13361707 (n=25 319), GSTM1 wild/null (n=25 222), IL1RN rs2234663 (n=24 930), MUC1 rs4072037 (n=23 024), GSTT1 wild/null (n=22 008) and IL1B rs1143627 (n=20 922).
Of the 456 meta-analysis performed, 121 (26%) resulted nominally statistically significant (p<0.05), the remaining 335 being non-significant. Among the significant associations identified by meta-analysis, the level of summary evidence was high, intermediate or low in 11 (9%), 38 (31%) and 72 (60%) analyses, respectively. Between-study heterogeneity was the most frequent single cause of non-high-quality level of evidence (12/38 intermediate level, 56/72 low level). Considering all meta-analyses, FPRP was optimal (<0.2) at the 10E-3 and 10E-6 level for 28/121 and 14/121 contrasts, respectively. Among the high-quality associations, FPRP was optimal at the 10E-3 and 10E-6 level for 9/11 and 5/11 contrasts, respectively.
The details of significant associations characterised by a high or intermediate level of summary evidence are reported in table 1. These polymorphisms, arranged upon their gene main function, are shown in figure 1. Combinations of variants to predict gastric cancer risk are reported in table 2.
Meta-analysis results: genetic variants significantly associated with gastric cancer risk with a high or intermediate level of summary evidence
Combination of independent polymorphisms (pairwise r2<0.1) to predict risk of gastric carcinoma

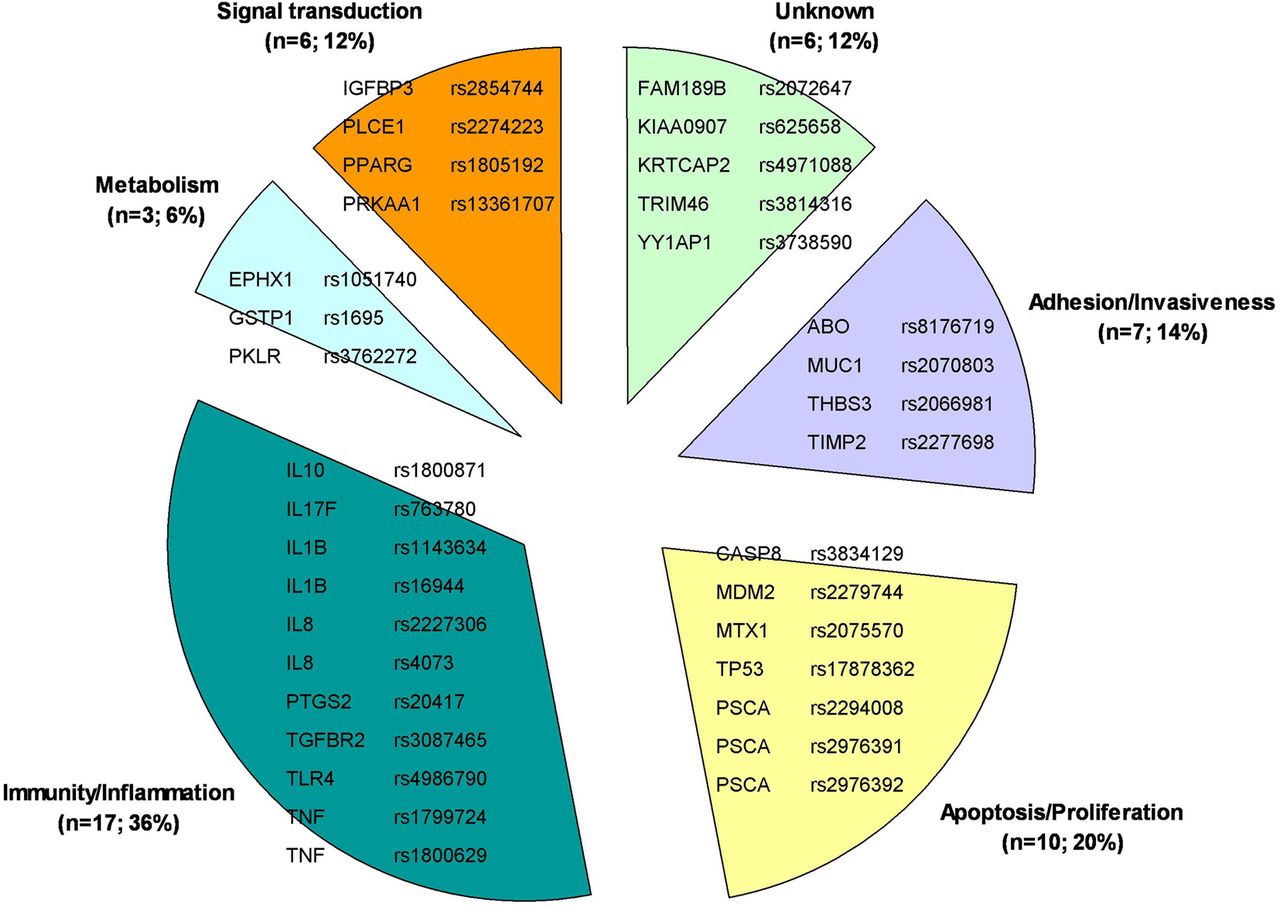{kind=link}
Genetic variants statistically significantly associated with risk of gastric carcinoma (with a high or intermediate level of evidence according to the Venice criteria) arranged by (main) gene function.
High-quality significant associations emerging from data meta-analysis are discussed below.
MUC1 rs2070803
Located on chromosome 1q22, rs2070803 is a G>A SNP downstream of both MUC1 and TRIM46. MUC1 codes for mucin-1, a cell surface glycoprotein that has been repeatedly reported to be involved in the carcinogenesis of different tumours,29 including gastric cancer,30 ,31 and is also widely used in the clinical setting as a tumour marker known as CA15.3. TRIM46 codes for the tripartite motif-containing protein 46, a zinc-finger containing protein of still uncertain function recently associated with serum urate concentrations.32 MUC1 is located downstream of the TRIM46 gene: these two genes are part of a cluster of genes (including also KRTCAP2, THBS3, MTX1, PKLR and HCN3) located in a region of strong linkage disequilibrium (LD) and are transcribed in opposite directions (ie, convergently). Since TRIM46 is not expressed in gastric mucosa,33 rs2070803 (whose functional effect is unknown) might be a tagging SNP for variants in other genes located in this LD block, such as MUC1, which are associated with gastric carcinogenesis.
The primary meta-analysis (including 8789 subjects) showed a statistically highly significant association between this SNP and gastric cancer risk (summary OR: 0.66, 95% CI 0.59 to 0.74, p=2.55E-13); however, the level of evidence was intermediate due to between-study heterogeneity. When subgroups were considered, the meta-analysis of six datasets (five deriving from two GWAS) including 7279 subjects (all of Asian ancestry) demonstrated that the rare A allele was associated with a reduced risk of developing the diffuse subtype of gastric carcinoma (summary OR: 0.585, 95% CI 0.526 to 0.650, p=2.24E-23), with a high level of cumulative evidence as per the Venice criteria and a low FPRP at both prior probability levels (10E-3 and 10E-6) (see table 1). Noticeably, this SNP did not result to be associated with intestinal subtype gastric carcinoma (summary OR: 0.759, 95% CI 0.568 to 1.012, p=0.061), suggesting that rs2070803 might be specifically linked to diffuse gastric cancer.
In our synopsis, data on a second MUC1 variant (rs4072037) were meta-analysed, but the association resulted significant only with a low level of evidence.
MTX1 rs2075570
This intronic A>G SNP in the MTX1 gene (on chromosome 1q22) is in strong LD with the previously described MUC1 rs2070803. MTX1 encodes metaxin-1, a mitochondrial outer membrane protein involved in apoptosis.34 ,35
According to the primary meta-analysis (including 8189 subjects), there was a highly significant association, although the level of evidence was intermediate due to between-study heterogeneity. Upon subgroup meta-analysis, data from five datasets (all deriving from two GWAS) including 6902 subjects (all of Asian ancestry) showed that the G allele was associated (with a high level of evidence) with a reduced risk of developing the diffuse subtype of gastric carcinoma (summary OR: 0.587, 95% CI 0.525 to 0.655, p=3.00E-21), with a low FPRP at both prior probability levels. This SNP was not significantly associated with the intestinal subtype of gastric carcinoma (summary OR: 0.848, 95% CI 0.643 to 1.117, p=0.241), suggesting that rs2075570 might be specifically linked to diffuse gastric cancer.
PKLR rs3762272
This intronic A>G SNP of the PKLR gene (chromosome 1q22) codes for pyruvate kinase (liver and red blood cell isoenzyme). This enzyme catalyses the transphosphorylation of phosphoenolpyruvate into pyruvate and ATP, the rate-limiting step of glycolysis36 that is the key metabolic event underlying the Warburg effect in malignant cells.37
Upon primary meta-analysis (including 5750 subjects) the association with this SNP was statistically marginal with a high degree of heterogeneity that made the level of evidence low. Instead, the meta-analysis of three datasets (all deriving from one GWAS) including 4463 subjects (all of Asian ancestry) showed that the G allele is associated (with a high level of evidence) with a reduced risk of developing the diffuse subtype of gastric carcinoma (summary OR: 0.710, 95% CI 0.631 to 0.799, p=1.42E-08), with a low FPRP at the higher prior probability level (10-3). Noticeably, this SNP did not result to be associated with intestinal subtype gastric carcinoma (summary OR: 0.927, 95% CI 0.717 to 1.197, p=0.559), suggesting that rs3762272 might be specifically linked to diffuse gastric cancer.
PSCA rs2294008 and rs2976392
The C>T SNP rs2294008 is located in exon 1 of PSCA (chromosome 8q24.2) and leads to a frameshift variation that causes a change in the starting codon and is associated with reduced gene transcription.26 PSCA was first identified as a prostate-specific antigen overexpressed in prostate cancer,38 but was then found to also be expressed by other tumour types as well as some normal tissues (including stomach and bladder epithelial cells).39 PSCA protein product can inhibit proliferation of gastric cancer cells,26 but its role in carcinogenesis appears complex and is still debated.40
The primary meta-analysis of 15 datasets (42 274 subjects) revealed a highly significant association with disease susceptibility; however, the level of summary evidence was low due to between-study heterogeneity. When subgroup analysis was performed, a similar situation occurred with ancestry and histotype subgroups. In contrast, seven datasets (none from GWAS) including 12 665 subjects (both Asian and Caucasian ancestry) showed that the T allele is associated with a high level of evidence with an increased risk of developing the non-cardia subtype of gastric carcinoma (summary OR: 1.329, 95% CI 1.254 to 1.408, p<1.00E-20), with a low FPRP at both prior probability levels. SNP rs2294008 was not associated with cardia subtype gastric carcinoma (summary OR: 1.119, 95% CI 0.933 to 1.342, p=0.225), suggesting that this SNP might be specifically linked to non-cardia gastric cancer.
Other meta-analyses have recently confirmed the association between gastric cancer and rs2294008,41–44 a variant also associated with the risk of bladder cancer.41 ,45
The intronic A>G polymorphism rs2976392 (whose functional effect is unknown) is in strong LD with the previously discussed PSCA rs2294008.
Primary meta-analysis (16 338 subjects) revealed a highly significant association, but as with SNP rs2294008, the resultant level of evidence was low due to between-study heterogeneity. Upon subgroup meta-analysis, data from four datasets (two deriving from a GWAS) including 7045 subjects (only Asian ancestry) showed that the G allele is associated (with a high level of evidence) with a decreased risk of developing the intestinal subtype of gastric carcinoma (summary OR: 0.808, 95% CI 0.751 to 0.870, p=1.42E-08), with a low FPRP at the higher prior probability levels. This variant was also associated with disease risk in patients with diffuse subtype stomach carcinoma (summary OR: 0.619, 95% CI 0.549 to 0.697, p=4.22E-15), although for this subgroup the level of evidence was low due to between-study heterogeneity.
We could meta-analyse the data regarding a third PSCA polymorphism (rs2976391) that resulted significantly associated with stomach cancer risk with an intermediate level of summary evidence.
PRKAA1 rs13361707
This T>C SNP (whose functional effect is unknown) is located in the first intron of PRKAA1 (chromosome 5p13), a gene encoding the catalytic α subunit of AMP-activated protein kinase. This enzyme is a cellular energy sensor that maintains energy homeostasis46 and might be involved in cancer development since its activation is associated with decreased phosphorylation of mammalian target of rapamycin and S6 kinase, which in turn causes a general reduction in mRNA translation and protein synthesis.47 ,48
The primary meta-analysis (25 319 subjects) demonstrated a highly significant association between this variant and gastric cancer, although the level of evidence was low due to between-study heterogeneity. Nevertheless, restricting the meta-analysis to five datasets (three from a GWAS, all of Asian ancestry) comprising 15 478 subjects supported with a high level of evidence the link between the C allele of rs13361707 and an increased risk of non-cardia subtype gastric cancer (summary OR: 1.406, 95% CI 1.334 to 1.482, p<1.00E-20) with a low FPRP at both prior probability levels. rs13361707 resulted associated also to cardia subtype gastric cancer, although this evidence was of intermediate level due to potential leading study bias (linked to a study49 identified by the leave-one-out sensitivity analysis).
PLCE1 rs2274223
This A>G SNP located in exon 26 of the PLCE1 gene (chromosome 10q23) causes a missense variation (His>Arg) in the protein product named phospholipase-C epsilon-1. This enzyme catalyses the hydrolysis of phosphatidylinositol-4,5-bisphosphate to generate two second messengers (ie, inositol 1,4,5-triphosphate and diacylglycerol) that subsequently regulate various processes affecting cell growth, differentiation and gene expression.50 Notably, recent investigations have demonstrated that PLCE1 might have a role in gastric carcinogenesis as it is overexpressed in precancerous chronic atrophic gastritis tissues and stomach carcinoma (as compared with normal gastric tissues)51 and its inhibition has therapeutic potential in a xenograft model.52 Nevertheless, the ultimate function of PLCE1 in cancer development is still debated since this molecule is downregulated in other tumours and shows a tumour suppressor activity in some animal models.53
When all available datasets were included in the meta-analysis (n=11, subjects: 33 219; both Asian and Caucasian ancestry), the G allele was significantly associated with an increased risk of gastric cancer (summary OR: 1.219, 95% CI 1.081 to 1.373, p=0.001). However, the level of evidence was low (due to a high degree of between-study heterogeneity) and FPRP was high at both prior probability levels.
In contrast, the meta-analysis of five datasets (two from a GWAS) composed of 21 366 subjects (Asian ancestry only) showed the G allele of rs2274223 to be associated (with a strong level of evidence) with an increased risk of cardia subtype gastric cancer (summary OR: 1.565, 95% CI 1.486 to 1.648, p<1.00E-20) with a low FPRP at both prior probability level. When the only Caucasian study is included,54 this association is still highly significant (summary OR: 1.539, 95% CI 1.386 to 1.709, p value: 8.88E-16) but the level of evidence becomes low due to between-study heterogeneity. Interestingly, rs2274223 was not associated with non-cardia gastric cancer with a high level of evidence, which suggests that this SNP is specifically linked to the carcinomas originating in the cardia region of the stomach.
In the present work, data on other four PLCE1 variants were meta-analysed (rs11187842, rs3765524, rs3781264 and rs753724), with only one of them significantly associated with disease risk (rs3765524), although with a low level of summary evidence.
TGFBR2 rs3087465
This G>A SNP is located in the promoter region of TGFB2 (chromosome 3p22), which encodes the transforming growth factor β receptor 2, a transmembrane Ser/Thr protein kinase protein that forms a heterodimeric complex with TGFBR1 and binds TGFB ligands. In turn, this mediates a variety of physiological and pathological processes including cell cycle arrest in epithelial and haematopoietic cells, control of mesenchymal cell proliferation and differentiation, wound healing, extracellular matrix production, immunosuppression, epithelial–mesenchymal transition, as well as carcinogenesis in different tumour models,55 ,56 including gastric cancer.57 ,58
The meta-analysis of the four available datasets (subjects: 4670; Asian ancestry only) showed a highly significant association between the minor allele (A) of rs3087465 and a decreased risk of gastric cancer (summary OR: 0.691, 95% CI 0.620 to 0.769, p=1.54E-11). This evidence was of a high level and FPRP was low at the higher prior probability level. No subgroup analysis could be performed due to the lack of permissive data.
GSTP1 rs1695
This A>G SNP is located in exon 5 of GSTP1 (chromosome 11q13) and causes a missense variation (Ile>Val) in the encoded protein glutathione S-transferase pi-1, which leads to a lower activity of the enzyme. GSTP1 belongs to a family of enzymes that play a key role in detoxification of a variety of endogenous and exogenous compounds by catalysing their conjugation with reduced glutathione.59 Given its importance in the maintenance of integrity of different cell components (eg, membranes and DNA), GSTP1 has been extensively investigated in the field of carcinogenesis.60
The primary meta-analysis of all 20 datasets (subjects: 11 258; both Asian and Caucasian ancestry) showed no association between the minor allele (G) of rs1695 and gastric cancer risk, with low-level evidence (due to between-study heterogeneity). However, when the meta-analysis was performed separately in Asian and Caucasian subjects, a significant association of high quality was observed only in the former subgroup (summary OR: 1.191, 95% CI 1.092 to 1.299, p value: 7.88E-05), with a low FPRP at the higher prior probability level, with no association being demonstrated in the other subgroups.
CASP8 rs3834129
This variant consists of a 6-nucleotide deletion in the promoter region of CASP8 (chromosome 2q33), which leads to a reduced gene transcription. The gene product, named caspase-8, is a cysteine-aspartic acid protease and is a pivotal mediator of programmed cell death, In brief, once activated by death receptors, it activates both caspase-3 (which in turn activates enzymes with DNAse activity) and Bid (which initiates the mitochondrial apoptotic pathway).61 Given the importance of malignant cell escape from apoptosis, the role of CASP8 in carcinogenesis has been extensively investigated in different tumour models.62–65
The meta-analysis of the three datasets (n=1701; both Asian and Caucasian ancestry) revealed that the minor allele (deletion) of rs3834129 is associated, with a high level of evidence, with a decreased risk of gastric cancer (summary OR: 0.732, 95% CI 0.617 to 0.869, p value: 3.48E-04), although FPRP was high at both prior probability levels. Available data did not enable us to perform subgroup analysis.
TNF rs1799724
This C>T SNP is located in the promoter region of the TNF gene (chromosome 6p21.3), which encodes a TNF superfamily member known as tumour necrosis factor. This is a proinflammatory cytokine that plays an important role in the immune response to infectious agents but is also involved in the pathogenesis of autoimmune diseases66 and presents pleiotropic functions in cancer development and progression.67 ,68
Primary meta-analysis of the 10 available datasets (n=5953; both Asian and Caucasian ancestry) showed a significant association between the minor allele (T) of rs1799724 and an increased risk of gastric cancer (summary OR: 1.173, 95% CI 1.047 to 1.314, p value: 0.006), characterised by a high level of evidence but also by a high FPRP at both prior probability levels. This association remained significant when the meta-analysis was restricted to Asian subjects (intermediate level of evidence), but not when it was restricted to the other two subgroups (Caucasian subjects and non-cardia subtype) for which data were available (low level of evidence).
Another four TNF polymorphisms (rs1800629, rs1799964, rs1800630 and rs361525) have been meta-analysed in this synopsis, but only one (rs1800629) was significantly associated to gastric cancer risk with an intermediate level of evidence.
Non-significant associations
Among 335 non-significant primary or subgroup meta-analyses, the level of evidence for lack of association was high, intermediate or low for 5 (1.5%), 21 (6.3%) and 309 (92.2%) analyses, respectively (see online supplementary table S2). The most frequent single cause of non-high level of evidence was insufficient statistical power (130/330), followed by between-study heterogeneity (71/330).
The details of non-significant associations characterised by a high or intermediate level of evidence are reported in table 3.
Meta-analysis results: variants with non-significant association to gastric cancer risk with a high or intermediate level of summary evidence
Discussion
In this article, we describe the results of the first systematic synopsis and meta-analysis (with evaluation of the quality of the cumulative evidence) in the field of genetic predisposition to sporadic gastric carcinoma. We found that out of 2841 variants investigated 11 polymorphisms in 10 genes were associated with susceptibility to gastric cancer in general or to the susceptibility of specific subgroups identified by ancestry (Asian vs Caucasian), primary tumour site (cardia vs non-cardia) and histological subtype (intestinal vs diffuse), with a high level of evidence. Of note, for 7 of these 11 variants (MUC1 rs2070803, MTX1 rs2075570, PKLR rs3762272, PRKAA1 rs13361707, TGFBR2 rs3087465, CASP8 rs3834129, TNF rs1799724) this is the first time that a meta-analysis confirms their association with risk of developing gastric carcinoma.
These findings provide investigators with a robust platform of genetic information useful to elucidate the molecular mechanisms underlying the pathogenesis of gastric carcinoma. For instance, our data support the hypothesis that the genetic information contained in chromosome 1 might play a predominant role in the susceptibility of this disease: in fact, the highest number of statistically significant contrasts with a high or intermediate level of evidence are located in this chromosome (16/49, 32.6%). Interestingly, these 12 variants are located in the long chromosomal arm and 9 of them belong to a region (chromosomal band 1q21–22) repeatedly reported to be affected by chromosomal aberrations (eg, copy number variations) in a range of tumours (such as breast,69 ,70 ovarian,70 hepatocellular,71 ,72 lung,73 oesophageal,74 cervical75 and oral76 carcinomas, neuroblastoma77 and liposarcoma78), including stomach sporadic carcinoma.79 Moreover, GWAS have recently identified this region as a susceptibility locus for cutaneous melanoma80 and testicular germ cell tumour.81 Therefore, this region of the genome represents a promising target for more extensive investigations in the field of gastric cancer research. On the other side, this synopsis also points out DNA regions scarcely investigated in the literature and thus worth new efforts.
By arranging our data upon (main) gene function, another interesting observation is that the majority of statistically significant results with high or intermediate level of evidence regard variants of genes involved in immunity/inflammation or adhesion/invasiveness (24/49, 48.9%). This finding is in line with the pivotal role of HP infection in the development of gastric cancer.10 In particular, our data draw the attention to polymorphisms that might explain the still enigmatic relationship between the high prevalence of HP infection in the general population (up to 70%82) and the relatively low gastric cancer incidence (up to 50 cases/100 000/year1).11 Unfortunately, the scarcity of published data on the interaction between HP status and DNA variants only allowed us to perform 20 HP-specific subgroup meta-analyses (out of 456 meta-analyses) and did not enable us to yield any meaningful results.
The 11 common low-penetrance variants identified by our meta-analysis for their strong evidence of association with gastric cancer could also be used to set up prediction algorithms for stratifying disease risk within the frame of targeted screening programmes.83–87
Since it is well recognised that each genetic locus confers a small contribution to the overall risk of a polygenic disease such as cancer, it is daunting to consider any single polymorphism as a test for routine clinical use.83 In our synopsis, the mean PAR of the 11 high-quality biomarkers was 18%. Nevertheless, their combination can raise the fraction of the disease burden attributable to genetic variation and thus improve the predictive power of tests based on genetic profiles, as suggested for other tumours such as breast,84 colorectal85 and prostate cancer.86
In this regard, haplotype studies have already shown the potential of polymorphism combination in the identification of subjects at high risk of gastric carcinoma88–90 by showing ORs >2 (or <0.5) and PAR up to 60%. However, the published data did not enable us to perform any meta-analysis since a minimum of three independent datasets was not available for any of the haplotypes studied so far.
To provide readers with an idea of the predictive potential of combining multiple polymorphisms, we calculated four joint-PAR (using seven variants unrelated to one other as defined by a pairwise correlation coefficient r2<0.1), whose values ranged from 21.3% to 51.2%. Of note, these joint-PAR would lead to a number needed to screen (number of people who must be tested to prevent one case of gastric carcinoma91) that varies from 22 to 990, depending also on the lifetime risk of disease (range: 1–5%) (table 2).
In this synopsis, other interesting results came from subgroup meta-analysis. First, we confirmed the difference between the results obtained in populations of different ancestry (see online supplementary information and supplementary figures S4 and S5), which support the hypothesis that the molecular pathway to gastric cancer susceptibility is not necessarily the same across different ethnicities and underscore the need for race-specific risk prediction tools. Moreover, our results also show some evidence as regards the genetic pathways specific for some gastric cancer subtypes (see online supplementary information and supplementary figures S6 and S7). Considering the different epidemiology and aggressiveness of different subsets of the disease,8 ,92 these subgroup-specific findings might help elucidate the molecular mechanisms underlying not only the development but also the progression of gastric cancer subtypes.
As regards the already existing literature, the past decade has witnessed the publication of a growing number of systematic reviews and meta-analyses addressing genetic predisposition to a variety of tumour types.93 ,94 In the field of stomach cancer, we found 243 such type of publications, which almost always take into consideration single polymorphisms, polymorphisms of single genes or variants of genes belonging to the same cell pathway. In a single article, published in 2009, the authors extend their analysis to 225 variants across 95 genes by collecting the data from 203 primary studies and report the findings of 61 primary or subgroup meta-analyses (the only subgrouping variable being ethnicity).95 A previous similar work, published in 2002, gathered a much lower number of primary studies (n=31).96 Finally, other investigators have reviewed the meta-analyses regarding the association between genetic variations and gastric cancer97 or different tumour types including gastric cancer.14
The present work—which outnumbers the previously published literature by number of articles retrieved, variants investigated, meta-analyses performed, subgroups considered and subjects enrolled—is characterised not only by a truly comprehensive search of the literature in this field but also by the effort to grade the quality of the cumulative evidence based on criteria specifically set up by a panel of international experts (ie, Venice criteria). Moreover, for each statistically significant meta-analysis, we calculated FPRP in order to further increase the information provided to readers on the reliability of the findings generated by our analysis. This way, we provide an updated and more critical summary of the available evidence compared with the already existing literature in this field.
We must also recognise the limits of this synopsis. First, despite our efforts to be comprehensive and accurate, we might have missed some relevant articles and may have included partially overlapping series.
Second, some series have been used for more than one meta-analysis, which potentially raises the statistical issue of type I error inflation. However, following the implementation of the FPRP method, this occurrence should be minimised; moreover, even after Bonferroni correction for multiple testing, all 11 high-quality biomarkers identified by our meta-analysis would remain statistically significant.
Third, we considered only one genetic model (ie, additive, which generates per-allele ORs), as explained in the 'Materials and methods' section. The aim of this work was not to define the best genetic model for some specific well-established biomarkers but rather to quantitatively summarise the evidence regarding hundreds of variants, a job that is best done using a conservative approach such as the additive model. In addition, no genotype data are available for a large number of variants. Also, the use of different models in primary and subgroup meta-analyses would have approximately tripled the number of meta-analyses, which would have in turn amplified the issue of type I error inflation.
Fourth, as mentioned previously, the paucity of data on gene–environment interactions did not allow us to thoroughly investigate potential sources of between-study heterogeneity, with special regard to HP infection status. For other potential effect modifiers, such as smoking, alcohol and dietary habits (though they are less important than HP infection8 ,9), available data were so scarce that we could not even take them into consideration. Although some articles reported OR values adjusted by one or more confounding factors, this was not always the case. The lack of data, coupled with the fact that adjusting covariates of multivariable logistic regression analysis differed in most circumstances, prevented us from implementing this information in our analyses, which are exclusively based on raw (unadjusted) ORs.
Finally, we must remind readers that some polymorphisms (eg, those located on chromosome 1q22) we found associated with gastric cancer risk are in strong LD with each other, which might imply they actually represent a single signal; moreover, some of our result interpretation—such as the clues on genes involved in the molecular pathways leading to gastric carcinogenesis—rely on the a priori assumption that the relevant gene of extra-genic variants is the nearest gene. Clearly, fine mapping and functional studies are required to identify the most likely causal variants and the genes they control before any predictive genetic tool is set up and any therapeutic target is considered, which highlights once again the burden of work still to be done in this field.
In conclusion, we performed the first systematic review and meta-analysis of the evidence linking DNA variation to the development of gastric carcinoma. We hope that this unprecedented collection of data might represent a useful platform for investigators involved in this research field not only for designing future studies but also to create a continuously updated databank dedicated to the genetic basis of this disease, a long-standing gap in the initiatives of the international scientific community.
Acknowledgments
We thank Dr Marta Briarava for setting up and managing the database dedicated to the collection of such a large amount of data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
Footnotes
Contributors SM: design, literature search, data extraction, data analysis and manuscript writing. DV and DN: design, literature search and data extraction. KAP: manuscript writing.
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.