Article Text

Abstract
Objective Eosinophils reside in the colonic mucosa and increase significantly during disease. Although a number of studies have suggested that eosinophils contribute to the pathogenesis of GI inflammation, the expanding scope of eosinophil-mediated activities indicate that they also regulate local immune responses and modulate tissue inflammation. We sought to define the impact of eosinophils that respond to acute phases of colitis in mice.
Design Acute colitis was induced in mice by administration of dextran sulfate sodium, 2,4,6-trinitrobenzenesulfonic acid or oxazolone to C57BL/6J (control) or eosinophil deficient (PHIL) mice. Eosinophils were also depleted from mice using antibodies against interleukin (IL)-5 or by grafting bone marrow from PHIL mice into control mice. Colon tissues were collected and analysed by immunohistochemistry, flow cytometry and reverse transcription PCR; lipids were analysed by mass spectroscopy.
Results Eosinophil-deficient mice developed significantly more severe colitis, and their colon tissues contained a greater number of neutrophils, than controls. This compensatory increase in neutrophils was accompanied by increased levels of the chemokines CXCL1 and CXCL2, which attract neutrophils. Lipidomic analyses of colonic tissue from eosinophil-deficient mice identified a deficiency in the docosahexaenoic acid-derived anti-inflammatory mediator 10, 17- dihydroxydocosahexaenoic acid (diHDoHE), namely protectin D1 (PD1). Administration of an exogenous PD1-isomer (10S, 17S-DiHDoHE) reduced the severity of colitis in eosinophil-deficient mice. The PD1-isomer also attenuated neutrophil infiltration and reduced levels of tumour necrosis factor-α, IL-1β, IL-6 and inducible NO-synthase in colons of mice. Finally, in vitro assays identified a direct inhibitory effect of PD1-isomer on neutrophil transepithelial migration.
Conclusions Eosinophils exert a protective effect in acute mouse colitis, via production of anti-inflammatory lipid mediators.
- MUCOSAL IMMUNOLOGY
- IBD BASIC RESEARCH
- IMMUNOREGULATION
- INFLAMMATION
- LIPID MEDIATORS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Video abstract
Significance of this study
What is already known on this subject?
IBD is a disease characterised by increased numbers of eosinophils.
The exact role of eosinophils during the onset of inflammation and in chronic disease remains unclear.
What are the new findings?
In the absence of eosinophils, mice succumb to a greater colonic inflammation during acute experimental models of colitis.
Eosinophil-deficient mice develop an exuberant and compensatory neutrophilic inflammation of the colon, accompanied by greater increases in levels of proinflammatory molecules.
Eosinophil-deficient mice have diminished levels of arachidonate-15-lipoxygenase, the lipid biosynthetic enzyme, and the proresolving lipid mediator protectin D1 during acute colonic inflammation, and administration of protectin D1-isomer to eosinophil-deficient mice attenuates inflammatory indices of colitis, including neutrophilic infiltration.
How might it impact on clinical practice in the foreseeable future?
Since eosinophils are normal residents of the colonic mucosa, further studies are necessary to determine eosinophil's role in health and disease. Protective factors released or regulated by eosinophils may be beneficial to colonic health.
Introduction
Eosinophils synthesise and release a broad range of biologically active mediators, including antimicrobial, proremodelling or anti-inflammatory associated factors (reviewed in1 ,2). An increasing body of evidence suggests that eosinophils play a primary role in a number of diseases such as asthma3–5 and eosinophilic GI diseases.6–8 In these diseases, eosinophils increase within the mucosal surfaces where they may participate in processes that often contribute to tissue damage9 ,10 and/or remodelling,11 either way leading to eventual organ dysfunction.
Acute inflammatory/immune reparative responses are necessary self-limited processes that participate in maintaining host health and tissue function. Mucosal surfaces use and amplify acute restorative anti-inflammatory activities through a number of mechanisms including anti-inflammatory lipid mediator synthesis.12 ,13 In this regard, omega-3 polyunsaturated fatty acid (PUFA)-derived resolvins and protectins are potent anti-inflammatory lipid mediators. For example, mice ingesting chow rich in endogenous omega-3 fatty acids have less severe colitis.14 By contrast, increased consumption of dietary proinflammatory omega-6 PUFA correlates with an increase in IBD incidence.15 ,16 Thus, dietary omega-3 fatty acid-derived anti-inflammatory molecules have emerged as potent options for therapeutic intervention.17–20 Docosahexaenoic acid (DHA), an omega-3 PUFA abundant in fish oils, is converted to hydroxydocosahexaenoic acids (HDoHE), by enzymatic oxidation. Protectin D1 (PD1) (10,17- dihydroxydocosahexaenoic acid (DiHDoHE)) is biosynthesised by 15-lipoxygenase (LOX) in humans and 12/15-LOX in mice, and present in inflamed tissues.21–24 Recent reports show that both human and mouse eosinophils express high levels of leucocyte-type 15-LOX and 12/15-LOX, respectively, and are capable of producing PD1 when activated in vitro.24 ,25 Other cells with the potential to express 12/15-LOX include dendritic cells, tissue-resident macrophage, mast cells and epithelial cells.21 While the presence of the enzyme is necessary, it is not always sufficient for PD1 biosynthesis, as expression may be regulated by environmental factors, such as cytokines or cell differentiation/activation states.21 Administration of exogenous PD1 to mouse models of inflammation protects tissues from inflammatory damage, expediting phagocytic resolution of acute peritonitis and retinal injury models, attenuating inflammation and airway hyper-responsiveness in mouse asthma models, while mouse eosinophils contribute to the resolution of acute peritonitis in a 12/15-LOX-dependent manner.12 ,22–24 ,26 To date, the receptor and, therefore, cellular target for PD1 has not been identified.
Expression of these lipid mediators, association with colonic eosinophils and their role in resolution responses during colonic inflammation is incompletely understood. The aim of our study was to determine the role(s) of eosinophils in mouse models of acute colitis. Here we provide evidence for a novel protective role of eosinophils and concurrently show that this protection also correlates with the induced levels of 12/15-LOX-derived anti-inflammatory lipid mediators. Results from these studies have broad implications on potential therapeutic options for patients with IBD.
Materials and methods
Mice
All studies were performed with female C57BL/6J mice (wild type, WT) and PHIL.27 Eosinophil depletion studies were completed using a monoclonal antibody specific for interleukin-5 (TRFK-5; Cayman Chemical, Ann Arbor, Michigan, USA). Age and sex-matched C57BL/6J littermate mice were used as controls. The University of Colorado and Mayo Clinic Arizona IACUC approved studies.
Dextran sodium sulfate, 2,4,6-trinitrobenzenesulfonic acid and Oxazolone-induced colitis
Colitis was induced by administering dextran sodium sulfate (DSS) (2.5%–3.5%(w/v)) 36 000–50 000 kDa; (MP Biomedicals, Santa Ana, California, USA), trinitrobenzenesulfonic acid (TNBS) or oxazolone (OXA) (Sigma, St Louis, Missouri, USA) to 10-week old mice for up to 6–8 days. Daily assessments of disease activity index (DAI) (ie, weight loss, stool consistency and faecal blood) and postnecropsy colon lengths were measured as previously described.28
In some studies, the commercially available DHA-derived PD1 isomer (10S, 17S-diHDoHE)23 ,29 (hereafter referred to as PD1-isomer) (0.05 mg/kg) (Cayman Chemical, Ann Arbor, Michigan, USA) was administered to mice.
Generation of bone marrow chimeric mice
Bone marrow chimeric mice were generated by transferring isolated bone marrow from PHIL (PHIL->WT) or WT (WT->WT) donor mice into the retro-orbital plexus of sublethally irradiated WT recipients, as described previously.30
Tissue processing and histological analysis
Whole length colons were removed, processed and a global inflammatory score was developed from H&E stained sections modified to account for area of tissue affected from that described previously.28
Immunohistochemical assessment of eosinophil, myeloperoxidase or F4/80 cellular inflammation
Eosinophils were localised in mouse and human tissues (Institutional Review Board approved) by eosinophil major basic protein-1 (MBP-1) and eosinophil peroxidase immunohistochemistry as previously described.11 ,31 Neutrophils were localised in mouse tissues by myeloperoxidase (MPO) immunohistochemistry. Macrophage and other F4/80+ cells were localised in mouse tissues by F4/80 immunohistochemistry.
In situ hybridisation assessment of CXCL1 production
Formalin-fixed paraffin-embedded sections were subjected to in situ hybridisation with probes targeted to the CXCL1 mRNA as per manufacturer’s recommendations (Advanced Cell Diagnostics, Hayward, California, USA). Slight modifications are detailed in online supplementary methods. Probes directed towards Ubc and DapB were used as positive and negative controls, respectively.
Colonic lamina propria and bone marrow leucocyte isolation for flow cytometry
Leucocytes were isolated from colonic mucosa by collagenase digestion as previously described.11 ,30 ,32 Bone marrow cells were isolated from femurs and tibias.33 Viable single cell suspensions were subjected to flow cytometric analysis as previously described.32
RNA isolation and real-time RT-PCR
Taqman gene expression assays (Applied Biosystems, Foster City, California, USA) were performed on whole colon RNA, processed as previously described.11 Data were normalised to 18S and calculated as relative quantity (2−ΔΔCt, where Ct is cycle threshold for each sample).
Colon lipidomic analysis
Colon tissues were harvested and processed for liquid chromatography tandem mass spectrometry (LC-MS/MS) based lipidomics analyses as previously described.12 MS/MS analyses were conducted in negative ion mode, and fatty acid metabolites were identified and quantified by multiple reaction monitoring.
Cell culture and in vitro transmigration assay
Caco-2 human intestinal epithelial cells were cultured and polymorphonuclear (PMN) cell transmigration towards a gradient of 100 nM Formyl–Methionyl–Leucyl–Phenylalanine (FMLP; Sigma, St Louis, Missouri, USA) was performed as previously described.34 ,35 PMNs were added in the presence of 0.2 or 20 nM of PD1-isomer or vehicle alone.
Statistical analysis
Statistical analyses of data outcomes were performed by Student t test, one-way analysis of variance or Log-rank (Mantel–Cox) test (conservative) survival curve where appropriate. Data are expressed as means±SEM. A p value of ≤0.05 was considered as statistical significance although in some cases higher levels of significance are noted and described in the figure legends where applicable: *†p≤0.05, ††p≤0.01, †††p≤0.001.
Further detail can be found in online supplementary materials and methods.
Results
Protective role for eosinophils in three models of acute colitis
Analysis of WT-mice and human tissues identified eosinophils as a significant component of the acute inflammatory response in experimental DSS-colitis and ulcerative colitis (figure 1A–C). Additionally, analysis of molecular responses identified that eosinophilic chemokines were significantly increased in experimental DSS-colitis (figure 1D). Except for the absence of eosinophils, the normal colonic mucosa of WT-mice and eosinophil-less (PHIL)-mice appeared the same (figure 2A). We induced DSS-colitis and monitored disease activity to determine the impact of eosinophils in acute experimental colitis. Upon harvest, colonic length was found to be significantly shorter (48.4 vs 53.8 mm, PHIL-DSS vs WT-DSS; p<0.001), and in congruence, tissue injury was significantly greater in PHIL-DSS compared with WT-DSS challenged mice, as evidenced by histological scoring of ulceration, oedema and leucocytic infiltration (19.5 vs 12.1, PHIL vs WT; p<0.001) (figure 2B, C). As shown in figure 2D, PHIL-mice experienced a greater mortality compared with WT-mice (PHIL-DSS—29% death vs WT-DSS—0% at 7 days). Further studies, with lower DSS concentrations, determined that PHIL-DSS mice developed significantly greater weight loss than WT-DSS mice in a time-dependent fashion (8.2% greater loss day 6, p<0.001; 5.9% greater loss day 7, p<0.01; 6.4% greater loss day 8, p<0.05; figure 2E). PHIL-DSS mice also had a higher DAI compared with WT-DSS mice (4.4 points greater day 6, p<0.001; 2.5 points greater day 7, p<0.01; 3 points greater day 8, p<0.05; figure 2F).

Eosinophilic inflammation in acute colitis. (A) H&E, and major basic protein immunohistochemistry on mouse colons following dextran sodium sulfate (DSS)-colitis. (B) Eosinophil peroxidase immunohistochemistry on human colons from normal and ulcerative colitis subjects. (C) Wild type whole mouse colon analysis by flow cytometry for infiltrating myeloid leucocytes. (D) Colon mRNA analysis for eosinophil-associated chemokines CCL11, CCL24 and CCL5. Scale bars represent 100 and 50 μM, respectively. Data are expressed as means±SEM of 3–7 individual mice per group and represent >3-independent experimental repeats following 6 days of colitis. *p≤0.05.

Protective role for eosinophils in acute colitis. Dextran sodium sulfate (DSS) or water-vehicle was administered to wild type (WT)-mice and PHIL-mice. (A) Representative H&E-stained sections of colons from WT-DSS and PHIL-DSS mice (100× magnification), (B) Colon lengths were assessed upon sacrifice and (C) A histological scoring tool combining injury and inflammation was applied to H&E-stained sections following 6 days of colitis. (D) Kaplan–meier survival curve comparing PHIL-DSS and WT-DSS littermates. (E) Colitis-induced weight loss as a percentage of day zero and (F) overall disease activity index calculated in PHIL compared with WT controls following 8 days of colitis. Data are expressed as means±SEM of 15 individual control mice and seven individual mice per DSS group and represent >3-independent experimental repeats. *p≤0.05, **p≤0.01, ***p≤0.001.
We next induced TNBS-colitis and monitored disease activity as described above. Results from these studies were remarkably similar to those found in DSS-colitis. PHIL-mice compared with WT-mice, experienced a greater rate of weight loss (7% greater loss day 2, p<0.05; 7% greater loss day 3, p<0.05), significantly shorter colon length (50 mm vs 61 mm, PHIL-TNBS vs WT-TNBS; p<0.001) and greater histological measure of disease (9.8 vs 3.5, PHIL-TNBS vs WT-TNBS; p<0.01) (see online supplementary figure S1).
In a third method of experimentally inducible colitis, WT and PHIL mice were treated with OXA-colitis. The death rate was 89% in PHIL-OXA and 20% in WT-OXA mice at day 4 postinstillation, p<0.0005 vs WT-OXA. No vehicle-treated mice of either genotype died. At 3 days postinstillation, maximum weight loss was observed (WT-OXA 88.29±2.3% vs PHIL-OXA 78.833±3.9% (p=0.07)). By 4 days postinstillation, most PHIL mice had died. Thus, consistent with findings in DSS and TNBS models of colitis, PHIL mice were more susceptible than their WT littermate counterparts and did not survive the induction of colitis using the OXA methodology.
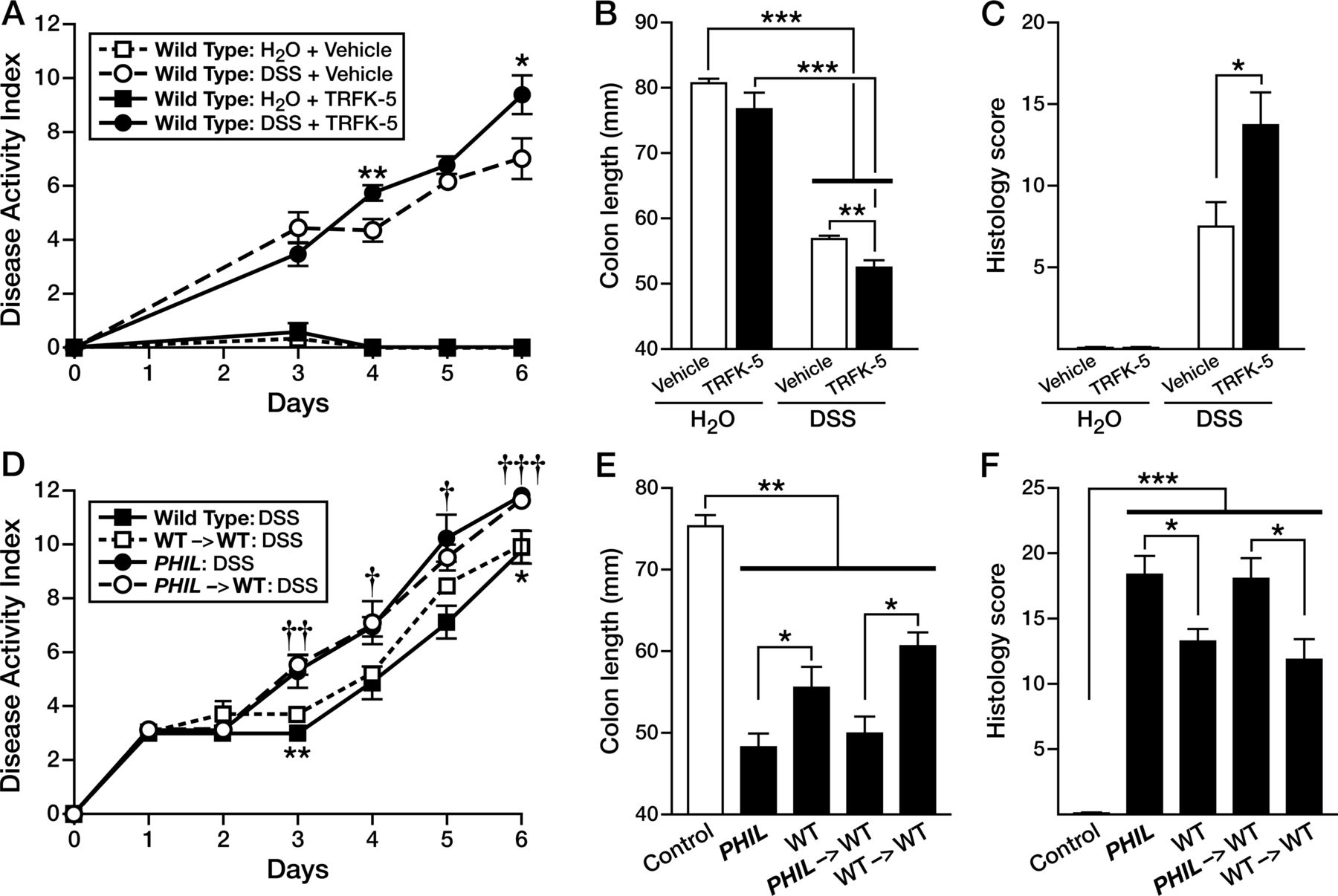
We used two other methods, antibody targeting and bone marrow depletion, to address eosinophils’ role in colitis as described below. The first model used the IL-5-depleting monoclonal antibody (TRFK-5) to reduce the eosinophil colonic population by 50% in WT-mice (see online supplementary figure S2). Induction of DSS-colitis in TRFK-5-treated WT-mice (WTTRFK-5), led to similarly increased colonic inflammation as observed in PHIL-mice. WTTRFK-5 mice displayed increased DAI relative to WT-controls (1.4 points greater on day 4, p<0.01 and 2.4 points greater on day 6, p<0.05; figure 3A) and shorter colon lengths (52.4 vs 56.7 mm; p<0.01; WTTRFK-5-DSS vs WTVeh-DSS; figure 3B). Histological scores revealed a significantly greater degree of tissue injury in WTTRFK-5 eosinophil-depleted mice compared with WT-control animals (13.8 vs 7.5, p<0.05; figure 3C). The second model of eosinophil depletion used was bone marrow engraftment of WT recipient mice with donor marrow from PHIL-mice (see online supplementary figure S3). In this series of experiments, we induced DSS-colitis in bone marrow chimeric mice. WT-recipient animals receiving PHIL marrow (PHIL->WT) with DSS-induced colitis uniquely displayed an eosinophil deficiency that did not occur in DSS-induced WT-recipients of WT-marrow (WT->WT) or DSS-induced WT-mice (see online supplementary figure S3A, B). Similar to both PHIL and TRFK-5-treated mice, DSS-induced colitis in eosinophil-less PHIL->WT chimera-mice was significantly greater than that observed in eosinophil-sufficient WT->WT chimeras (figure 3D), including significant changes in DAI (1.8 points greater day 3, p<0.01; 1.9 points greater day 4, p<0.05; 1 point greater day 5, p<0.05; 1.6 points greater day 6, p<0.001 and greater colon shortening (50.0 mm vs 60.5 mm; PHIL->WT vs WT->WT, respectively, p<0.05) (figure 3E). Comparisons of histology scores from colons of DSS-treated mice also confirmed that PHIL and PHIL->WT chimeras displayed the same increased inflammatory phenotype relative to WT and WT->WT chimeras (figure 3F).

Antibody depletion or myeloablation of eosinophils exacerbates acute colitis. Mice were depleted of eosinophils by antibody mediated interleukin-5 diminution (anti-TRFK-5), induced with dextran sodium sulfate (DSS)-colitis and (A) disease activity, (B) colonic length and (C) histology scores of H&E-stained colon tissues were reported. Bone marrow chimera was performed between PHIL-donors and wild type (WT)-recipients (PHIL->WT) and between WT-donors and WT-recipients (WT->WT) as controls. Age and sex matched PHIL-mice and WT-mice were maintained as controls. Mice were induced with colitis and (D) disease activity, (E) colonic length and (F) histology scores of H&E-stained colon tissues were reported. Data are expressed as means±SEM of 3-7 individual mice per group and represent 2-independent experimental repeats following 6 days of colitis. *†p≤0.05, ††p≤0.01, †††p≤0.001 PHIL->WT-DSS-versus-WT->WT-DSS.
Taken together, induction of colitis in three models of eosinophil reduction demonstrates that disease-ameliorating effects dynamically track with the presence of eosinophils.
Neutrophils are the predominant infiltrate in eosinophil-deficient DSS-treated mice
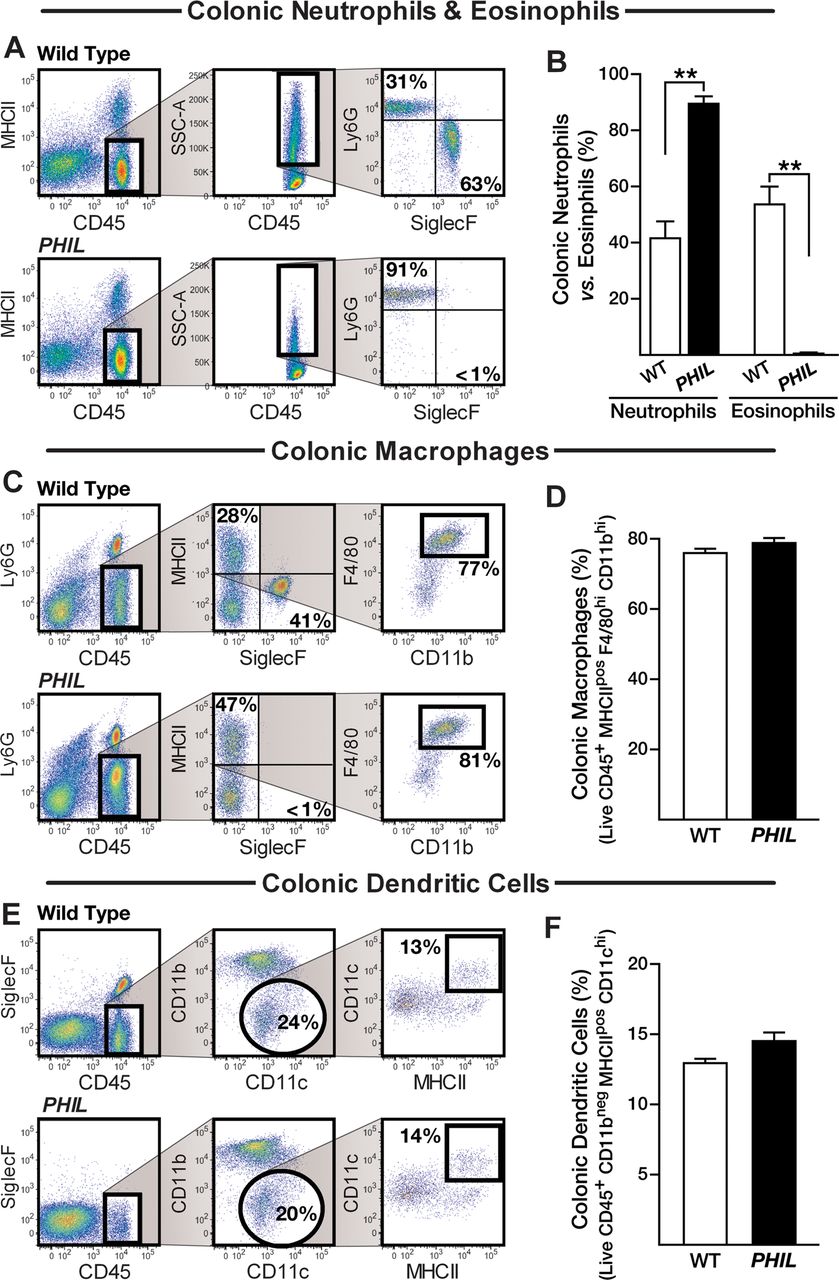
We assessed the inflammatory cell infiltrate of colonic lamina propria in DSS-treated mice to define changes in the tissue milieu that occurred in the absence of eosinophils. While PHIL colonic mucosae were virtually devoid of eosinophils compared with 50% eosinophils in DSS-treated WT-mice (WT-control vs WT-DSS, p<0.05) (figure 4A, B), there were no differences observed in the frequencies of macrophages and dendritic cells (figure 4C–F). Quantitated immunohistochemistry for F4/80 positive cells per high-powered field also showed no difference when comparing PHIL-DSS with WT-DSS mouse colons (55.99±6.4 vs 49.95±2.4; p=0.74). By contrast, as shown in figure 4, the higher inflammatory responses occurring in the colons of DSS-treated PHIL-mice (relative to DSS-treated WT-mice) resulted in a further differential increase in colonic neutrophils ((90%) 13.5×103 vs (42%) 7.2×103, PHIL-DSS vs WT-DSS; p<0.05). There were very few neutrophils observed in normal PHIL and WT colons (0.4×103 vs 0.3×103, PHIL vs WT; n.s.). These data demonstrated that a lack of tissue eosinophils appears to promote the accumulation of colonic neutrophils during inflammation (figure 4A, B). Immunohistochemistry for MPO on colonic tissue sections was used as an additional method of validation. Quantification determined a significant increase in the number of MPO-positive cells per high-powered field in PHIL-DSS versus WT-DSS colons (124 vs 97, p<0.05) (see online supplementary figure S4A). Congruent with this observation, a significant increase in colonic neutrophils during DSS-colitis was also observed following TRFK-5 or bone marrow chimeric eosinophil depletion (see online supplementary figures S1 and S2) and in PHIL mice following TNBS colitis (see online supplementary figure S4B).

Inflammatory infiltrate in eosinophil null mucosa is predominated by neutrophils. Whole colon flow cytometry analysis for infiltrating myeloid leucocytes in wild type (WT)-colons and PHIL-colons in dextran sodium sulfate (DSS)-colitis. Derivation and quantification of relative proportions of (A and B) neutrophils and eosinophils, (C and D) macrophage and (E and F) dendritic cell infiltrates comparing WT-DSS with PHIL-DSS-colitis. Data are expressed as means±SEM of seven individual mice per group and represent >3-independent experimental repeats following 6 days of colitis. **p≤0.01, Wild Type versus PHIL.
Consistent with this finding is the molecular characterisation of the colonic inflammatory microenvironment. While the proinflammatory cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 were all elevated in the colons of DSS-treated WT-mice, the level of each proinflammatory cytokine was further augmented in the colons of DSS-treated PHIL-mice (figure 5A–C). By contrast, type-2 cytokines typical of Th2 responses (ie, IL-4, IL-5 and IL-13) were not increased in PHIL or WT-mice (figure 5D–F). Enhanced neutrophilic inflammation occurring in DSS-treated PHIL colons compared with WT-mice was accompanied by a differential increase in neutrophil-associated chemokines CXCL1 (mKC) and CXCL2 (macrophage inflammatory protein (MIP)-2β), likely a finding secondary to the increased state of mucosal inflammation, and not directly related to eosinophil-specific effects on production (figure 6A, B). In situ hybridisation analysis for CXCL1 found the cellular sources for this chemokine in mouse colon sections from PHIL-DSS and WT-DSS mice to be very similar. Here we found that both epithelial cells as well as lamina propria infiltrating leucocytes produce this chemokine in equal abundance (see online supplementary figure S5). We also measured NOS2 (iNOS) expression in the colonic mucosa as a confirmatory metric of the enhanced neutrophilic inflammation occurring in DSS-treated PHIL-versus-WT-mice. Similar to what was observed with other neutrophil-associated events occurring in DSS-treated PHIL-versus-WT-mouse colons, the level of iNOS expression was significantly higher in PHIL-mouse colons relative to the increase observed in DSS-treated WT-mice (figure 6C). Previously published work has provided a link between the myeloid immune system, particularly neutrophils, and lipid mediator synthesis. We thus measured the lipid oxidation enzymes prostaglandin-endoperoxide synthase 1 (PTGS1), PTGS2, arachidonate 5-lipoxygenase (ALOX5), ALOX12 and ALOX15 in the colons of eosinophil null, neutrophil-predominant DSS-treated PHIL-versus-WT-mice (figure 6D–I). A significant inhibition of ALOX15 induction was observed in PHIL-DSS versus WT-DSS colons (figure 6H).

Proinflammatory and type-2 cytokine expression profile in PHIL-mice during acute colitis. mRNA was isolated from whole colons and assessed by Taqman technologies. (A–C) Major acute proinflammatory cytokines tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 as well as (D–F) Type-2 cytokines IL-4, IL-5 and IL-13 were measured. Data are expressed as means±SEM of 7–10 individual mice per group and represent 2-independent experimental repeats following 6 days of colitis. *p≤0.05, **p≤0.01, ***p≤0.001 versus genotype matched water controls. †p≤0.05, ††p≤0.01, †††p≤0.001 versus wild type-dextran sodium sulfate (DSS).

Chemokine, iNOS and lipid oxygenation enzyme expression profile in PHIL-mice during acute colitis. mRNA was isolated from whole colons and assessed by Taqman technologies. (A–C) Neutrophil-associated transcripts CXCL1/KC, CXCL2 and NOS2 were assessed. (D–H) Lipid oxygenation enzymes critical for lipid mediator biosynthesis PTGS1, PTGS2, ALOX5, ALOX12 and ALOX15 were also measured. Data are expressed as means±SEM of 7–10 individual mice per group and represent 2-independent experimental repeats following 6 days of colitis. *p≤0.05, **p≤0.01, ***p≤0.001 versus genotype-matched water controls. †p≤0.05, ††p≤0.01, †††p≤0.001 versus wild type-dextran sodium sulfate (DSS).
Eosinophil depletion is associated with decreased 12/15-LOX derived lipid metabolites
Since our finding that lipid oxidation enzyme production was altered, most specifically ALOX15 (figure 6H), and lipid mediators generated at local tissue sites are influenced by and in turn influence accumulation of select leucocyte populations in inflammation,36 we reasoned that eosinophils might influence the lipid mediator profile of DSS-colitis. LC-MS/MS-based lipidomic analyses performed on colonic tissue from PHIL and WT-mice revealed that loss of eosinophils in colons of PHIL-mice was associated with a significant decline in lipid molecules derived from 12/15-LOX dependent pathways, congruent with our observation of inhibited ALOX15 induction. In particular, colon levels of eosinophil-associated anti-inflammatory PD124 ,37 were virtually absent in healthy-PHIL-mice (0.6 ng/g-colon) and DSS-treated PHIL-mice (1 ng/g-colon) relative to eosinophil sufficient healthy (3 ng/g-colon) and DSS-treated (11 ng/g-colon) WT-mice (PHIL-DSS vs WT-DSS; p<0.05) (figure 7-inset). No difference in levels of the PD1 precursor DHA was observed in either PHIL or WT colons prior to, or following, the induction of colitis. However, 17-hydroxy-DHA, a marker of DHA utilisation, was significantly decreased in PHIL-colons prior to and following induction of colitis (p<0.001). Levels of lipid mediators derived from the other major metabolic networks linked with cyclo-oxygenase (COX) and/or 5-LOX were not different in PHIL and WT healthy or DSS-treated colons. Congruent with our finding of increased neutrophils in inflamed eosinophil-deficient colons, a significant increase in the neutrophil-associated chemokine LTB4 was observed (figure 7).
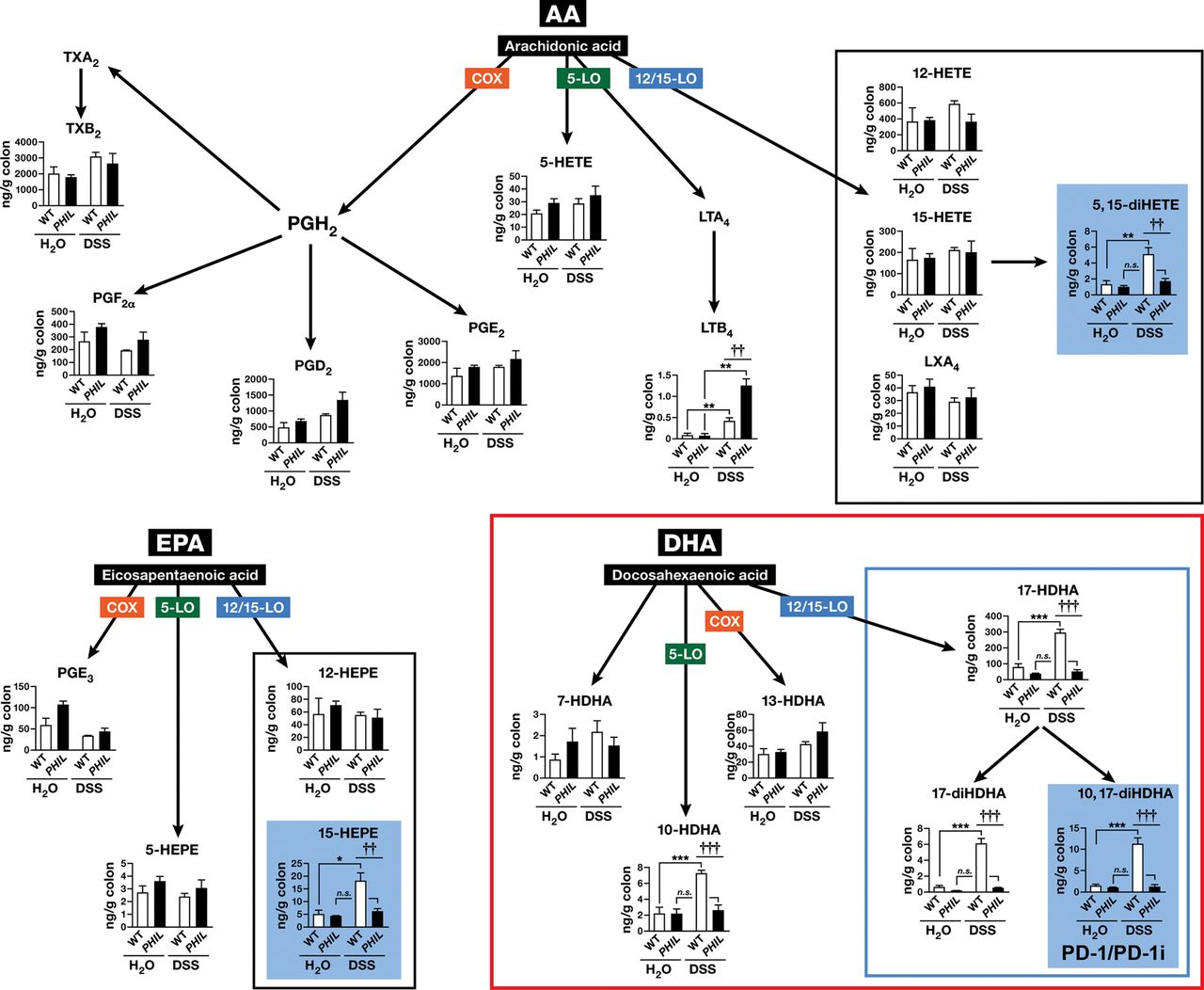
12/15-lipoxygenase (12/15-LOX) lipid derivative deficiency in colitis in PHIL-mice. Whole mouse colons from dextran sodium sulfate (DSS)-colitis and water control wild type (WT)-mice and PHIL-mice were assessed by LC-MS/MS-based mediator lipidomics following 6 days of colitis. Arachidonic acid (AA)-derived, eicosapentaenoic acid-derived and docosahexaenoic acid (DHA)-derived products were quantified by LC-MS/MS, and multiple reaction monitoring. Levels of 12/15-LOX products are boxed in black or blue and 12/15-LOX biosynthetic end products with statistically significant differences are in blue shaded boxes. Data are expressed as means±SEM of 3-H2O and 6-DSS individual mice. *p≤0.05, **p≤0.01, ***p≤0.001 DSS versus genotype-matched water controls. ††p≤0.01, †††p≤0.001 PHIL-DSS versus WT-DSS.
Exogenous administration of PD1-isomer rescues PHIL-mice from colitis and diminishes neutrophil chemotaxis and inflammation
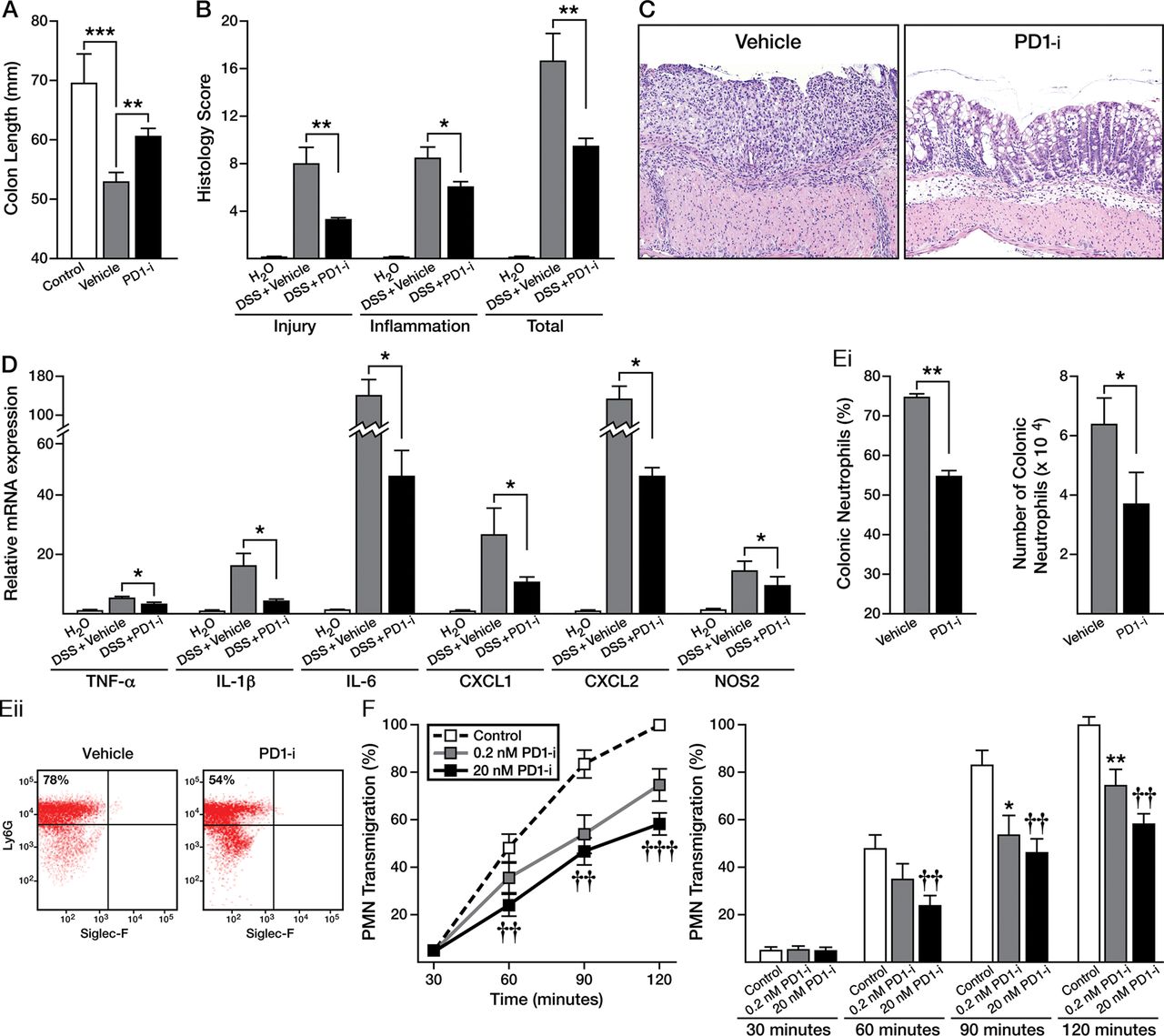
Administration of PD1-isomer to DSS-treated PHIL-mice resulted in significant improvement in colonic inflammation (figure 8). Specifically, PD1-isomer treatment of PHIL-mice resulted in significant improvement in colon shortening (69.5 mm vs 60.6 mm vs 52.8 mm, Control PHIL vs. PD1-isomer-treated PHIL vs vehicle-treated PHIL, p<0.01) and histological scores (9.4 vs 16.5, PD1-isomer-treated PHIL vs vehicle-treated PHIL, p<0.01) (figure 8A, B). Representative photomicrographs of colonic mucosa from PD1-isomer-treated mice revealed improvements in epithelial cell loss, inflammatory infiltrates, and architectural disruption (figure 8C). Consistent with these findings, proinflammatory molecules, including TNF-α, IL-1β and IL-6 decreased significantly (figure 8D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Protectin D1 (PD1)-isomer (PD1-i) treatment attenuates colitis in PHIL-mice via attenuation of neutrophil migration. PHIL- dextran sodium sulfate (DSS)-colitis mice were treated with PD1-isomer or vehicle via intraperitoneal injection 0.05 mg/kg daily throughout the course of colitis which lasted 6 days. Upon sacrifice, colons were excised and (A) colon length measured. (B) Injury, inflammatory and total histology scores were assessed and (C) representative H&E-stained sections from PHIL-DSS-mice treated with PD1-isomer or vehicle (100× magnification). (D) Proinflammatory cytokine and neutrophil-associated chemokine mRNA production was examined in PD1-isomer versus vehicle-treated PHIL-DSS-mice. (E and F) Flow cytometric analysis of lamina propria infiltrates for neutrophil percent and absolute number comparing PD1-isomer with vehicle-treated PHIL-DSS-controls. Data are expressed as means±SEM of 3–7 individual mice per group, and represent 2 independent experimental repeats. (G) In vitro examination of the effects of PD1-isomer on Formyl–Methionyl–Leucyl–Phenylalanine-induced neutrophil-colon epithelial monolayer (Caco-2) transmigration; dose-dependence and time-dependence. Data are expressed as means±SEM of three individual donors and three independent experimental repeats. *p≤0.05, **p≤0.01 0.2 nM PD1 versus controls. ††p≤0.01, †††p≤0.001 20 nM PD1-isomer versus controls.
PD1-isomer add-back also led to a significant decrease in neutrophilic inflammation in DSS-treated PHIL-mice (figure 8E), as well as a decrease in neutrophil-specific chemokines CXCL-1/KC and CXCL-2/MIP2-α and proinflammatory NOS2 (figure 8D). Ex vivo chemotaxis studies further substantiated the anti-inflammatory effects of PD1-isomer leading to a concentration-dependent decrease in PMN chemotaxis across intestinal epithelial monolayers compared with control PMNs (figure 8G). Such findings identify PD1/PD1-isomers as a prominent molecular signature that attenuates neutrophil accumulation during ongoing colonic inflammation.
Discussion
Although eosinophil accumulation in tissues of patients with IBD has been described, the specific and unique contributions of eosinophils to IBD has yet to be clearly delineated.2 ,6 ,8 ,38 Here, we demonstrate a protective role for eosinophils during mouse colitis. We show that the physical, histological and molecular features of acute colonic inflammation are significantly increased without these granulocytes. Our data demonstrates acute colitis in eosinophil-competent mice correlates with biosynthesis of anti-inflammatory derivatives of omega-3 fatty acids, including PD1, a molecule known to dampen neutrophil influx and proinflammatory cytokine production.23 The enzyme necessary for PD1 synthesis, 15-LOX was not induced in PHIL-mice and, thus, PD1 was barely detectable in PHIL-mice at baseline or during colitis suggesting an important role for these leucocytes in PD1 synthesis in this myeloid predominant acute experimental colitis model. Bypassing the deficiency in the 15-LOX enzyme, exogenous administration of PD1-isomer to eosinophil-deficient mice following colonic injury markedly attenuated weight loss, colon shortening, histological indices of injury and inflammation, PMN infiltration and proinflammatory cytokine production including TNF-α, IL-1β, IL-6 and inducible nitric oxide. These findings suggest a role for acute-phase-responding eosinophils, either directly or indirectly, in the resolution of early inflammatory responses, possibly through alterations in gene expression and leucocyte recruitment, such as that mediated by the 12/15-LOX-derived lipid mediator PD1/PD1-isomers.
To date, the role of eosinophils during IBD has remained elusive. A number of studies suggest an end-stage destructive role(s) for eosinophils in the pathogenesis of gut inflammation in addition to other mucosal organs.10 ,39 For instance, eosinophil granule protein null mice were relatively protected from colitis indicating a pathological role for these proteins.10 ,39–41 The emerging dual role for eosinophils in contributing to host health and to inflammation is a fascinating one. Eosinophils have been reported as important for the maintenance of host health in the retention of memory plasma cells to the bone marrow.42 They are also important for the maintenance of microbial homeostasis of commensal organisms in the intestines during health by retaining IgA-producing plasma cells in peyer's patches.43 It is important to note that eosinophil-deficient mice are not devoid of IgA, however, as other mechanisms of IgA production were maintained. Both these studies implicate eosinophils as a primary source for the critical plasma cell chemokine, APRIL. In the setting of acute peritoneal inflammation, eosinophils have also been demonstrated as protective sentinels, producing anti-inflammatory lipids including PD1, promoting the rate of resolution.24 Experimental models of colonic inflammation, such as the IL-10−/− mouse present with significant colonic eosinophilia prior to and during disease.44 In this model, the absence of IL-4 and, thus, eosinophils resulted in diminished disease and led the authors to suggest important role(s) for eosinophils in the initiation of colitis. However, IL-4 mediates significant influences on a number of other potential pathogenic effector cells. In light of our findings, one could speculate eosinophils’ increase prior to the onset of inflammation, as observed in the IL-10−/− model, may be an early attempt to dampen inflammation, potentially via mechanisms including the production of anti-inflammatory lipid mediators, among others, for example, PD1. It is important to keep in context that eosinophils may play a role, but are not the sole mediators in the maintenance of intestinal health and homeostasis. It appears that eosinophils may react to acute insults in attempt to resolve and restore intestinal health.
Clinical studies have identified increased numbers of colonic eosinophils in disease states and a reduction following treatment providing circumstantial evidence for a pathogenic role for eosinophils in patients.45 Contrasting with this, others have found an increase in eosinophils during disease remission.46 However, studies have not addressed the impact of eosinophils in the acute/early phase of colitis induction, including whether the baseline population of these granulocytes may offer host benefits as a part of innate host defence. Recent review speculates that eosinophils may mediate host health by antimicrobial and tissue repair functions.2 When these cells are chronically present in excess however, they may indeed participate in a deleterious manner towards organ dysfunction.
Two previous studies have addressed the impact of eosinophils on the development of colitis.9 ,47 Comparison of those results with findings shown here, yielded different conclusions, thus demonstrating the complexity of studies in mouse models of disease. For example, results from two models of eosinophil deficiency (PHIL and ΔdblGATA) at two locations (USA and Brazil) suggested that the loss of eosinophils leads to the amelioration of colitis.9 ,47 Potential factors that explain these conflicting results include technical aspects of experimental colitis induction, institutional differences including dietary source and composition, age of mice and variability in eosinophil-deficient strains. For example, Ahrens et al studied PHIL-mice at younger ages than our study and measured histology and colon length as the primary readouts. Vieira et al examined the ΔdblGATA mouse, deficient in basophils, in addition to eosinophils,48 and used a higher concentration of DSS than our studies (4% vs 2.5%). Additionally, the dietary availability and ratio of the generally considered proinflammatory omega-6 and anti-inflammatory omega-3 precursors of potent lipid mediators in different mouse chow will play a critical role in the balance between inflammation and its resolution. We suggest that these experimental differences are significant and contribute to the disparity in our findings. More importantly, we propose that this likely reflects the multifaceted role of eosinophils in disease initially contributing anti-inflammatory activities following acute provocation and then proinflammatory effects in chronic settings. Our ongoing studies of eosinophil deficiency in a variety of environmental settings (eg, barrier facilities vs conventional housing venues)49 provide an additional confounding perspective; the rapidly responsive microbiome and/or exposure to environmental toxicants (eg, endotoxins) present in the resident animal facilities are likely to be key issues modulating the immune/inflammatory responses linked with eosinophils in mouse models of disease.50 We suggest that the confounding interinstitutional issues are a consequence of these extrinsic modulatory events, altering the proinflammatory versus anti-inflammatory roles of eosinophils in a given setting. It may be possible that each, or any combination, of these factors may be necessary to stimulate the eosinophil population to act in a regulatory manner. Thus, both conclusions may be correct, and the specific circumstances surrounding a given model and protocol will need to be noted in future studies with the multiple eosinophil-deficient mouse models now currently available.
Our study contributes to a growing body of basic and clinical studies that support a role for omega-3 PUFAs and their derivatives in IBD treatment. 17–20 ,51–53 While we do not rule out the potential contributions of anti-inflammatory lipids derived from COX or 5-LO biosynthesis pathways,54 we specifically see a global deficiency in 12/15-LOX and its derived lipid mediators in the absence of eosinophils. Moreover, we do not discount the potential anti-inflammatory utility of AA or eicosapentaenoic acid-derived 12/15-LOX mediated lipids, including 5,15-diHETE and 15-HEPE, however 5,15-diHETE may potentiate PMN degranulation and thus exacerbate inflammation in this model.55 Our studies have focused on the DHA-derived, previously established eosinophil-associated, PD1 molecule.24 ,37 The data reported here suggest a link between eosinophil activities and the levels of the DHA-derived PD1 family of lipid mediators present in the initial stages of colonic inflammation, an attempt by the host to limit colonic inflammation. However, in addition to eosinophils, 12/15-LOX-derived biosynthesis of PD1 and its isomers may occur through other cells present during DSS-colitis, such as tissue-resident macrophages, dendritic cells, mast cells as well as epithelial cells.21 Our experiments do not rule out these other cell types as potential contributors of PD1/PD1-isomers. Nonetheless, it is noteworthy that no differences were observed in the frequencies of these other cells between colitic PHIL and control mice, suggesting the critical role, specifically for the increased eosinophil infiltrate in either directly or indirectly producing or inducing 15-LOX, and the proresolving lipid mediator PD1/PD1-isomers. Thus, these findings are consistent with observations that recruitment of eosinophils and production of PD1/PD1-isomers are associated with reduced numbers of neutrophils in inflamed tissue and the resolution of inflammation.22 ,24 The importance of eosinophil-derived PD1/PD1-isomer expression awaits future studies using eosinophil-specific 12/15-LOX knockout mice generated by using 12/15-LOXflox/flox animals crossed with eosinophil-specific Cre-expressing mice.
Reconstitution studies, and in vitro modelling identify the impact of PD1-isomer on reducing PMN inflammation in colitis as has been demonstrated in other organ systems.23 ,36 ,56 Our data extends these findings by showing that proinflammatory gene expression (TNF-α, IL-1β, IL-6, nitric oxide, CXCL1 and CXCL2) associated with PMN recruitment in colonic tissues decreases following PD1-isomer treatment. This reduction in proinflammatory cytokines and PMN infiltration, mediated, in part, by pharmacological reconstitution with PD1-isomers, may be an effective approach in the prevention and maintenance of remission in patients with IBD.
In summary, the present study highlights a critical role for eosinophils in regulating inflammatory tone during acute experimental mouse colitis. Mechanistically, we identify that the omega-3 PUFA-derived lipid mediator, PD1/PD1-isomer specifically limits colon PMN infiltration. Interestingly, complete ablation of neutrophil recruitment in animal models of colitis is itself deleterious, and may suggest that a balance between eosinophils and neutrophils may be necessary for resolution of inflammation.57 ,58 These findings support a potentially protective role for resident eosinophils in mouse colitis and, thus, challenge the paradigm for a solely proinflammatory role in human disease. We speculate that in certain circumstances, the increased presence of eosinophils in early stages of mucosal inflammation may not necessarily be an indicator of disease, but rather a host response, fostering mucosal healing and a pathway to return to homoeostasis (figure 1). Thus, future translational studies linking numbers of eosinophils in a given patient with observed histopathology and clinical manifestation of disease are necessary to test our hypotheses.
Acknowledgments
The authors wish to thank the members of all the participating laboratories for insightful discussions and critical comments. We also wish to acknowledge the invaluable assistance in animal husbandry and care at University of Colorado School of Medicine (Kristann Magee), the Mayo Clinic Arizona medical graphic artist (Marv Ruona), and the administrative support provided by Linda Mardel and Shirley (‘Charlie’) Kern. We gratefully thank Ms Michiko Kamio for technical assistance on LC-MS/MS analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Contributors Study concept and design (JCM, ENMcN, SPC, MA, JJL, GTF); acquisition of data (JCM, ENMcN, SAF, LH, RH, SDF, PJ, RI, GTF); analysis and interpretation of data (JCM, ENMcN, SDF, PJ, RI, SPC, MA, JJL, GTF); drafting of the manuscript (JCM, JJL, GTF); critical revision of the manuscript for important intellectual content (JCM, ENMcN, SPC, MA, JJL, GTF); statistical analysis (JCM); obtained funding (JCM, HKE, MA, JJL, GTF); technical or material support (EJ, CP); study supervision (JCM, SPC, MA, JJL, GTF).
Funding Grants from the USA National Institutes of Health (R01-DK62245 and K24-DK 100303) (GTF), (R01-HL065228 and K26-RR0109709) (JJL), and (DK097075, R01-HL0921, R01-DK083385, R01-HL098294 and POIHL114457-01) (HKE), Crohn's and Colitis Foundation of America (JCM, HKE), the North American Society for Pediatric Gastroenterology Hepatology and Nutrition (JCM, GTF), the Mayo Foundation for Medical Education and Research (JJL), a Grant-in-aid from the Japan Science and Technology Agency Precursory Research for Embryonic Science and Technology (MA) and a Grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MA) were the sources of funding used in the performance of studies including data analysis and manuscript preparation.
Competing interests None.
Ethics approval Colorado Multiple Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Available data in this paper will be shared upon request to the corresponding author.