Article Text

Abstract
Objective Helper T (Th) cell responses are critical for the pathogenesis of Helicobacter pylori-induced gastritis. Th22 cells represent a newly discovered Th cell subset, but their relevance to H. pylori-induced gastritis is unknown.
Design Flow cytometry, real-time PCR and ELISA analyses were performed to examine cell, protein and transcript levels in gastric samples from patients and mice infected with H. pylori. Gastric tissues from interleukin (IL)-22-deficient and wild-type (control) mice were also examined. Tissue inflammation was determined for pro-inflammatory cell infiltration and pro-inflammatory protein production. Gastric epithelial cells and myeloid-derived suppressor cells (MDSC) were isolated, stimulated and/or cultured for Th22 cell function assays.
Results Th22 cells accumulated in gastric mucosa of both patients and mice infected with H. pylori. Th22 cell polarisation was promoted via the production of IL-23 by dendritic cells (DC) during H. pylori infection, and resulted in increased inflammation within the gastric mucosa. This inflammation was characterised by the CXCR2-dependent influx of MDSCs, whose migration was induced via the IL-22-dependent production of CXCL2 by gastric epithelial cells. Under the influence of IL-22, MDSCs, in turn, produced pro-inflammatory proteins, such as S100A8 and S100A9, and suppressed Th1 cell responses, thereby contributing to the development of H. pylori-associated gastritis.
Conclusions This study, therefore, identifies a novel regulatory network involving H. pylori, DCs, Th22 cells, gastric epithelial cells and MDSCs, which collectively exert a pro-inflammatory effect within the gastric microenvironment. Efforts to inhibit this Th22-dependent pathway may therefore prove a valuable strategy in the therapy of H. pylori-associated gastritis.
- HELICOBACTER PYLORI
- GASTRITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Th22 cells represent a newly discovered Th cell subset, exerting either pathogenic or protective properties depending on the context of inflammation.
Th22 cells and IL-22 have been confirmed to play critical roles in chronic inflammatory conditions.
IL-22 is expressed in the mucosa of various species, including humans.
Inflammatory cell infiltration is the most likely result in pathophysiology for gastritis and is considered a clinical hallmark of the disease.
What are the new findings?
Th22 cells accumulated in gastric mucosa of both patients and mice infected with Helicobacter pylori.
Gastric IL-22 expression correlated with H. pylori colonisation and the severity of gastritis and contributed to a proinflammatory role.
H. pylori infection-induced inflammation was characterised by the CXCR2-dependent influx of myeloid-derived suppressor cells (MDSCs), whose migration was induced via the IL-22-dependent production of CXCL2 by gastric epithelial cells
How might it impact on clinical practice in the foreseeable future?
Our in vitro and in vivo data together provide a multistep model of inflammation during H. pylori infection involving interactions between H. pylori, Th22 cells, dendritic cells, gastric epithelial cells and MDSCs within the gastric mucosa. In this regard, our findings suggest several possible therapeutic targets, including IL-22, S100A8 and S100A9. Given the apparent relationship between IL-22 levels and the severity of gastric inflammation observed in H. pylori-infected patients, thought should be given to the use of IL-22 and/or Th22 cells as novel diagnostic biomarkers for H. pylori infection.
Introduction
Helicobacter pylori is a human pathogen that infects nearly half the world's population. Infection with H. pylori is frequently associated with chronic inflammation of the gastric mucosa (gastritis) and can lead to peptic ulceration and gastric cancer.1 Although the development of H. pylori-associated gastritis remains poorly understood, it is believed that the nature of the CD4+ helper T (Th) cell response is a key contributing factor. For example, a mouse model of H. pylori-induced gastritis, Th cells were found to be both ‘necessary and sufficient’ for the development of H. pylori-associated gastritis.2
Cells of the Th22 lineage secrete IL-22 (Th22 cells) represent a newly discovered Th cell subset and can contribute to both protective and pathological immune responses. In mice, it has been reported that IL-22 has protective effects in IBD3 and hepatitis.4 By contrast, others have found that IL-22 has a pathological role in mouse models of Toxoplasma gondii infection5 and psoriasis,6 while in human IBD, IL-22 appeared to be pro-inflammatory.7 To date, virtually nothing is known about Th22 cells during H. pylori infection in either humans or mice and we were therefore interested to explore a possible relationship.
In the current study, we have for the first time demonstrated that H. pylori-infected patients have an over-abundance of Th22 cells and that this result is paralleled in mice infected with H. pylori. The differentiation of these Th22 cells is induced by IL-23 derived from H. pylori-activated dendritic cells (DC) and its overall effect is to promote inflammation. In this regard, Th22 polarisation stimulates gastric epithelial cells to secrete CXCL2, which, in turn, recruits myeloid-derived suppressor cells (MDSC) that produce the pro-inflammatory proteins S100A8 and S100A9 and inhibit Th1 cell responses. Collectively, these data highlight a pathological role for Th22 cells in H. pylori-induced gastritis.
Materials and methods
Patients and specimens
The gastric biopsy specimens and blood were collected from 78 H. pylori-infected and 59 uninfected patients who underwent upper oesophagogastroduodenoscopy for dyspeptic symptoms at XinQiao Hospital (see online supplementary table S1). H. pylori infection was determined by [14C] urea breath test and rapid urease test of biopsy specimens taken from the antrum and subsequently conformed by real-time PCR for 16S rDNA and serology test for specific anti-H. pylori antibodies (Abs). For isolation of human primary gastric epithelial cells, fresh non-tumour gastric tissues (at least 5 cm distant from the tumour site) were obtained from patients with gastric cancer who underwent surgical resection and were determined as H. pylori-negative individuals as above at the Southwest Hospital. None of these patients had received chemotherapy or radiotherapy before sampling. Individuals with atrophic gastritis, hypochlorhydria, antibiotics treatment, autoimmune disease, infectious diseases and multiprimary cancer were excluded. The study was approved by the ethics committee of XinQiao Hospital and Southwest Hospital of Third Military Medical University. Written informed consent was obtained from each patient.
Antibodies and other reagents
See online supplementary methods.
Mice
All breeding and experiments were undertaken with review and approval from the Animal Ethical and Experimental Committee of Third Military Medical University. Specific pathogen free (SPF) female BALB/c and C57BL/6 wild-type (WT) mice were purchased from the Experimental Animal Centre of the Third Military Medicine University. Through material transfer agreements, C57BL/6 IL-23p19 knockout (KO) (IL-23 KO) and BALB/c IL-22 KO mice were obtained from Dr Wenjun Ouyang (Genentech). All mice were viral Ab free for pathogenic murine viruses and negative for pathogenic bacteria including Helicobacter spp and parasites (see online supplementary table S2), and were maintained under SPF conditions in a barrier-sustained facility and provided with sterile food and water.
Bacteria culture and infection of mice with bacteria
H. pylori NCTC 11637 (cagA positive) (WT H. pylori) and cagA-KO mutant H. pylori NCTC 11637 (ΔcagA) were grown in brain-heart infusion plates containing 10% rabbit blood at 37°C under microaerophilic conditions. For infecting mouse, bacteria were amplified in Brucella broth with 5% fetal bovine serum (FBS) with gentle shaking at 37°C under microaerobic conditions. After culture for 1 day, live bacteria were collected and adjusted to 109 colony forming units (CFU)/mL. The mice were fasted overnight and orogastrically inoculated twice at a 1-day interval with 3×108 CFU bacteria. H. pylori infection status and H. pylori-induced gastritis in murine experiments were confirmed (data not shown).
Generation of bone marrow chimaera mice
See online supplementary methods.
Cytokine/antibodies/CXCR2 antagonist administration
One day after infection, mice were injected intraperitoneally with 25 μg of recombinant murine IL-22 or IL-23, or anti-IL-22, anti-IL-17A, anti-IL-17F, anti-interferon (IFN)-γ or isotype control Abs (100 μg), or anti-CXCR2 and/or anti-CXCL2 Abs or rat immunoglobin (Ig)G2a and/or IgG2b (100 μg), or 4 mg/kg SB225002 or dimethylsulfoxide (DMSO) control, and repeated every week until the mice were sacrificed.
Evaluation of inflammation
The mice were sacrificed at the indicated times. The greater curvature of the stomach was cut to perform H&E staining and immunofluorescence. The intensity of inflammation was evaluated independently by two pathologists according to previously established criteria.8
Isolation of single cells from tissues and DCs preparation
See online supplementary methods.
Cell/tissue culture and stimulation
Human primary gastric epithelial cells were purified from gastric tissue single-cell suspensions in a Magnetic-activated cell sorting (MACS) column purification system using anti-CD326 magnetic beads (Miltenyi Biotec). Human gastric epithelial cell line AGS cells, primary gastric epithelial cells or gastric tissues were stimulated with WT H. pylori and/or ΔcagA at different multiplicity of infection (MOI). AGS cells and primary gastric epithelial cells were also stimulated with IL-22 (100 ng/mL) for 1, 3, 6, 12 and/or 24 h. For signal pathway inhibition experiments, AGS cells were pretreated with FLLL32 (10 μM) for 2 h, or STAT3 siRNA or control siRNA (100 nM) for 24 h. DCs were stimulated with WT H. pylori and/or ΔcagA at different MOI for 6 h. Then the gentamycin was added to kill the bacteria for 2 h and then cells were washed three times. MDSCs were sorted with FACSAria II (BD Biosciences) from blood of H. pylori-infected patients and stimulated with IL-22 (100 ng/mL) for 1, 3, 6, 12 and/or 24 h. After coculture, cells were collected for microarray, real-time PCR and western blot, and the culture supernatants were harvested for ELISA.
In vitro T cell culture system
In a 5-day incubation, purified human peripheral or mouse spleen CD4+ T cells (StemCell Technologies) were cocultured (2×105 cells/well) with WT H. pylori or ΔcagA stimulated-DCs from autologous blood; or WT H. pylori or ΔcagA stimulated-bone marrow–derived dendritic cells (BMDCs) from WT or IL-23 KO mice at 2:1 ratio. Alternatively, CD4+ T cells were cocultured with autologous ΔcagA-stimulated DCs at 2:1 ratio supplemented with IL-23 (10 ng/mL) or media alone, or with autologous WT H. pylori-stimulated DCs at 2:1 ratio supplemented with IL-23 Ab (10 μg/mL) or control IgG (10 μg/mL). CD4+ T cells were also first labelled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and cocultured (1×105 cells/well) with MDSCs at different ratios. After such 5-d incubation, cells were collected and analysed by intracellular cytokine staining.
Chemotaxis assay
Th cell-polarising culture supernatants derived from WT H. pylori or ΔcagA-stimulated DCs were collected to stimulate primary gastric epithelial cells for 48 h. Then, the secondary primary gastric epithelial cell culture supernatants were again collected as chemoattractant liquids. Sorted MDSCs (1×105) were transferred into the upper chambers of transwells (Corning). CXCL2 (10 ng/mL) and chemoattractant liquids under various conditions were placed in the lower chambers. After 24 h culture, migration was quantified by counting cells in the lower chamber and cells adhering to the bottom of the membrane. In some cases, blocking Ab for CXCR2 (10 μg/mL) were added into MDSC suspensions and incubated for 2 h before chemotaxis assay.
Immunofluorescence, real-time PCR, flow cytometry, ELISA, western blot analysis and microarray experiments are described in online supplementary methods.
Statistical analysis
Results are expressed as mean±SEM. Student t test was generally used to analyse the differences between two groups, but when the variances differed, the Mann–Whitney U test was used. Inflammation score data were analysed by the Mann–Whitney U test. For multigroup data analysis, an analysis of variance was used. Correlations between parameters were assessed using Pearson correlation analysis and linear regression analysis, as appropriate. SPSS statistical software (V.13.0) was used for all statistical analysis. All data were analysed using two-tailed tests, and p<0.05 was considered statistically significant. Microarray data analysis was performed with the assistance of Genminix Informatics. Clustering was performed using Cluster V.3.0 and patterns were created and viewed using Java TreeView V.1.0.13 software. Raw data from each array were analysed using TwoClassDif.
Results
Th22 cells are enriched in gastric mucosa of H. pylori-infected patients and mice
To evaluate the potential role of Th22 cells in H. pylori-associated pathology, we compared the Th22 cell levels in gastric tissues. Notably, the gastric mucosa of H. pylori-infected patients showed a higher frequency of Th22 cells (figure 1A). Also, the overall levels of IL-22 mRNA (figure 1B) and protein (figure 1C) were higher, respectively, in the gastric mucosa of H. pylori-infected patients. Next, IL-22 expression was positively correlated with H. pylori colonisation (figure 1D), suggesting induction and/or maintenance of Th22 cells by H. pylori.

Th22 cells accumulated in gastric mucosa of Helicobacter pylori-infected patients and mice. (A) The percentage of T helper type 22 (Th22) cells in CD3+ cells in gastric mucosa of H. pylori-infected (n=22) and uninfected donors (n=12) was compared. Results are expressed as percentage of Th22 cells in CD3+ T cells. (B and C) Interleukin (IL)-22 mRNA expression (B) and IL-22 protein concentrations (C) in gastric mucosa of H. pylori-infected (n=46) and uninfected donors (n=27) were compared. (D) The correlation between IL-22 expression and H. pylori colonisation was analysed. (E) IL-22 mRNA expression in gastric mucosa of cagA+ H. pylori-infected (n=35), cagA− H. pylori-infected (n=11) and uninfected donors (n=29) were compared. (F) Dynamic changes of Th22 cell response and IL-22 mRNA expression in wild-type (WT) H. pylori-infected, ΔcagA-infected and uninfected BALB/c mice. n=5 per group per time point in F. *p<0.05, **p<0.01 n.s. p>0.05 for groups connected by horizontal lines compared, or compared with uninfected mice. n.s. not significant.
The presence of cagA is strongly associated with the development of gastritis.9 Notably, we found that IL-22 expression in cagA-positive patients was significantly higher than that in cagA-negative individuals (figure 1E). Consistent with our findings in humans, Th22 cells were only detected in WT H. pylori-infected mice, reaching a peak 35 days postinfection (p.i.) (figure 1F). Similar observations were made in C57BL/6 mice (see online supplementary figure S1A, B), indicating a role for cagA across multiple host genetic backgrounds.
It has previously been reported that—apart from Th cells—IL-22 can also be produced by natural killer cells, lymphoid tissue inducer-like cells and innate lymphoid cells.10 Using our mouse model of H. pylori infection, we found no evidence for IL-22 expression in these cells (see online supplementary figure S1E), suggesting that Th cells are the only immune cells that produce IL-22 in gastric mucosa during H. pylori infection. Finally, we also assessed whether we could detect Th22 cells outside the gastric mucosa during H. pylori infection in mice, but found minimal numbers of Th22 cells in bone marrow (BM), blood, spleen, mesenteric lymph node and Peyer's patches (see online supplementary figure S2).
DCs stimulated by H. pylori induce Th22 cells via IL-23
DCs are known to be critically important in both priming and maintaining Th22 cells.11 We, therefore, sought to determine whether DCs were responsible for the development of Th22 cells during H. pylori infection. Interestingly, H. pylori-stimulated DCs were able to potently induce CD4+ T cells to differentiate into Th22 cells (figure 2A), and this was most noticeable when using a WT H. pylori strain. Similarly in mice, BMDCs can effectively induce Th22 cell differentiation following WT H. pylori exposure (figure 2B).

Helicobacter pylori-stimulated DCs induce Th22 cell polarisation via IL-23. (A and B) Th22 cell polarisation was assessed by flow cytometry, as described in the Methods section, and statistically analysed. Results are expressed as percentage of Th22 cells in CD4+ T cells. (C) Concentrations of IL-22 protein in unstimulated, WT H. pylori-stimulated or ΔcagA-stimulated DCs derived from blood monocytes and in the DC supernatants were compared (n=3). (D) Th22 cell polarisation was assessed by flow cytometry, as described in the Methods section, and statistically analysed. Results are expressed as percentage of Th22 cells in CD4+ T cells. Results are representative of three independent experiments. (E and F) Th22 cell response in gastric mucosa of WT H. pylori-infected WT C57BL/6 and IL-23 KO (E) or WT H. pylori-infected BM chimaera mice (F) on day 35 postinfection were compared. *p<0.05, **p<0.01, n.s. p>0.05 for groups connected by horizontal lines compared. DC, dendritic cells; Th; helper T cells; WT; wild-type; KO, knockout; BM, bone marrow; MOI, multiplicity of infection; BMDC, bone marrow–derived dendritic cells; IL, interleukin; n.s., not significant.
It has previously been shown that Th22 cells are induced by IL-2312 and that DCs are potent producers of IL-23 at sites of bacterial infection.13 To see whether similar mechanisms might operate in H. pylori infection, we first found that IL-23 protein were significantly upregulated in WT H. pylori-stimulated DCs compared with those stimulated with ΔcagA or no bacteria (figure 2C). Next, we found that blocking IL-23 with neutralising Ab effectively inhibited the generation of Th22 cells (figure 2D). Consistent with this, BMDCs from IL-23 KO mice failed to induce Th22 cell polarisation (figure 2B). Conversely, provision of exogenous IL-23 significantly increased Th22 cell polarisation (figure 2D). Collectively, these findings indicate that H. pylori-stimulated DCs express IL-23 which, in turn, promotes Th22 cell differentiation in vitro.
To assess if a similar phenomenon might occur in vivo, we infected WT and IL-23 KO mice with H. pylori and found that, compared with WT mice, IL-23 KO mice developed significantly fewer Th22 cells in gastric mucosa (figure 2E), indicating that IL-23 does indeed have a permissive role in inducing Th22 cell development in vivo. By generation of BM chimaera mice, we found that IL-23-producing BM-derived cells are largely responsible for Th22 cell development during H. pylori infection in this model (figure 2F). Taken together, our data demonstrate that IL-23 plays an essential role in Th22 cell induction by DCs in vitro and are consistent with the operation of similar mechanisms in vivo.
IL-22 has proinflammatory effects during H. pylori infection
To understand the possible biological effects of Th22 cell induction during H. pylori infection, we compared IL-22 expression within the gastric mucosa with the severity of gastritis observed in patients infected with H. pylori. Notably, higher IL-22 expression was strongly associated with more severe gastritis (figure 3A). This led us to hypothesise that IL-22 might exert proinflammatory effects during H. pylori infection and, thus, contribute to gastritis.
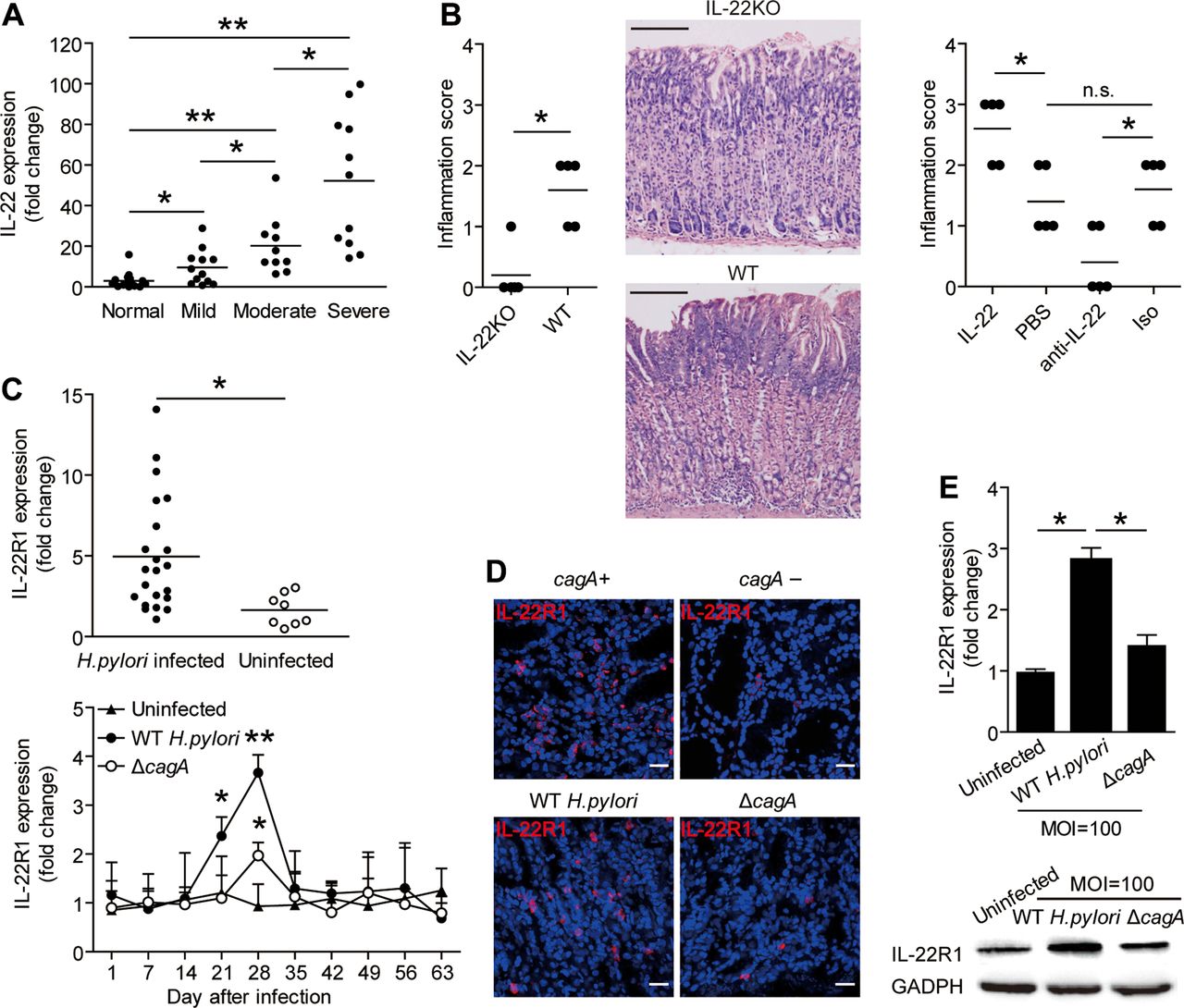
IL-22 has pro-inflammatory effects during Helicobacter pylori infection and H. pylori induce gastric epithelial cells to upregulate IL-22R1. (A) IL-22 mRNA expression in gastric mucosa of H. pylori-infected patients with mild (n=12), moderate (n=10), severe inflammation (n=11) and uninfected donors with normal gastric histopathology (n=15) was compared. (B) Histological scores of inflammation in gastric antra of the WT H. pylori-infected WT BALB/c or IL-22 KO mice or WT H. pylori-infected WT BALB/c mice injected with IL-22 or Abs against IL-22 on day 49 postinfection were compared. H&E staining, scale bars: 100 μ. (C) Expression of IL-22 mRNA in gastric mucosa of H. pylori-infected patients (n=22) and uninfected donors (n=8) was compared. Dynamic change of IL-22R1 mRNA expression in WT H. pylori-infected, ΔcagA-infected and uninfected BALB/c mice. (D) Representative immunofluorescence staining images showed IL-22R1 expression in gastric mucosa of WT H. pylori-infected and ΔcagA-infected mice or cagA+ H. pylori-infected and cagA− H. pylori-infected patients. Scale bars: 20 μ. (E) IL-22R1 mRNA expression or IL-22R1 protein in WT H. pylori-infected, ΔcagA-infected, and uninfected primary gastric epithelial cells from uninfected donors were compared (n=3) or analysed by western blot. *p<0.05, **p<0.01, n.s. p>0.05 for groups connected by horizontal lines compared. WT; wild-type; KO, knockout; IL, interleukin; MOI, multiplicity of infection; PBS, phosphate-buffered saline; GADPH, glyceraldehyde 3-phosphate dehydrogenase; Abs, antibodies; n.s., not significant.
To test this hypothesis in vivo, we conducted a series of loss-of-function and gain-of-function experiments involving IL-22 and evaluated the inflammatory response in gastric mucosa on day 49 p.i. Compared with WT mice, IL-22 KO mice showed significantly less inflammation in gastric mucosa (figure 3B). Neutralisation of IL-22 significantly reduced gastric inflammation (figure 3B). Conversely, injection of IL-22 significantly increased gastric inflammation (figure 3B). Finally, consistent IL-22 being derived from Th cells, the effect of IL-22 appears to be mediated by BM-derived cells (see online supplementary figure S3A). Collectively, these results suggest that IL-22 has proinflammatory effects during H. pylori infection in vivo.
Gastric epithelial cells are induced by H. pylori to upregulate IL-22R1
Given the critical importance for IL-22R1 in IL-22 signalling, we sought to evaluate IL-22R1 expression during H. pylori infection. First, IL-22R1 expression was increased in gastric mucosa of H. pylori-infected patients (figure 3C), and was also higher in individuals carrying cagA-positive strains (figure 3D). Similarly, in mice, IL-22R1 expression was significantly increased in gastric mucosa of mice infected with WT H. pylori compared with those either uninfected or infected with ΔcagA (figure 3C, D). Since IL-22 typically acts on epithelial cells, we next examined expression of IL-22R1 on human primary gastric epithelial cells. Infection with WT H. pylori infection, upregulated IL-22R1 gene expression compared with either no infection or infection with ΔcagA (figure 3E). Similar results were obtained with AGS cells, an immortalised human gastric epithelial cell line (see online supplementary figure S3D). Collectively, these results indicate that H. pylori infection induces IL-22R1 expression on gastric epithelial cells, implying that these cells are a major target of IL-22 action within the inflamed gastric mucosa.
IL-22 promotes CXCL2 production and attracts MDSCs into the gastric mucosa during H. pylori infection via CXCR2
IL-22 is known to induce the production of various chemokines within the brain.14 We were, therefore, interested to know if IL-22 similarly induces chemokine production in gastric mucosa. To begin, we found that IL-22 induced AGS cells to produce CXCL2 in a dose-dependent and STAT3-dependent manner (figure 4A). Similarly, IL-22 (but not IL-23) induced CXCL2 production by primary gastric epithelial cells (figure 4B). IL-22 KO mice or neutralisation of IL-22 significantly reduced CXCL2 production in gastric mucosa (figure 4C). Conversely, injection of IL-22 significantly increased CXCL2 production (figure 4C).

IL-22 promotes CXCL2 production in vivo and in vivo and CXCR2-expressing MDSCs accumulated in gastric mucosa during Helicobacter pylori infection. (A and B) AGS cells (A) and primary gastric epithelial cells (B) were pretreated and stimulated as described in Methods. CXC12 production was detected in cell supernatants by ELISA (n=3). STAT3 and p-STAT3 proteins were analysed by western blot. (C) Concentrations of CXCL2 protein in gastric mucosa of WT H. pylori-infected WT BALB/c mice injected with IL-22 or PBS control, or Abs against IL-22 or corresponding isotype control Ab, or of WT H. pylori-infected WT BALB/c and IL-22 KO mice on day 42 postinfection (p.i) were compared. (D) Dynamic change of MDSCs in WT H. pylori-infected, ΔcagA-infected and uninfected BALB/c mice. (E) Representative dot plots of MDSCs by gating on CD45+ cells and expression of Ly6C, Ly6G and CXCR2 on MDSCs in gastric mucosa of WT H. pylori-infected mice on day 49 p.i., and representative dot plots of MDSCs by gating on CD45+ cells and expression of CXCR2 on MDSCs in peripheral blood of H. pylori-infected and uninfected donors. Numbers indicate relative percentages in CD45+ cells. (F) MDSC level in peripheral blood of H. pylori-infected patients (n=22) and uninfected donors (n=21) was compared; n=5 per group per time point in D. *p<0.05, **p<0.01, n.s. p>0.05 for groups connected by horizontal lines compared, or compared with uninfected mice. WT; wild-type; KO, knockout; IL, interleukin; PBS, phosphate-buffered saline; GADPH, glyceraldehyde 3-phosphate dehydrogenase; Abs, antibodies; MDSC, myeloid-derived suppressor cell; DMSO, dimethylsulfoxide; n.s., not significant.
CXCL2 promotes cell migration by binding to the chemokine receptor CXCR2. We first found that mice infected with WT H. pylori showed a higher frequency of MDSCs with abundant expression of CXCR2 in gastric mucosa than those infected with ΔcagA or uninfected (figure 4D,E and see online supplementary figure S4C). This accumulation of MDSCs peaked on day 49 p.i. Notably, MDSCs expressed high levels of Ly6C and minimal Ly6G, and should thus be regarded as monocytic MDSC (M-MDSCs)15 (figure 4E). Consistent with this, we found a higher frequency of MDSCs with a CD14+HLA-DRlow/− M-MDSC phenotype in peripheral blood of H. pylori-infected patients compared with uninfected donors. Notably, these MDSCs showed abundant expression of CXCR2 (figure 4E, F).
Next, neutralisation of IL-22 significantly reduced MDSC accumulation, whereas, neutralisation of IL-17A, IL-17F and IFN-γ all had no effect (see online supplementary figure S5A). Similarly, both IL-22 KO and IL-23 KO mice showed significantly fewer MDSCs (figure 5B). However, injection of IL-22 significantly increased MDSC accumulation in IL-23 KO mice, while injection of IL-23 had no significant effect on MDSC numbers in IL-22 KO mice (figure 5B), suggesting that IL-22 and not IL-23 is a more proximal influence on MDSC accumulation.

IL-22 promotes MDSC accumulation in gastric mucosa in vivo and migration in vitro during Helicobacter pylori infection by CXCL2-CXCR2 axis. (A) MDSC responses in gastric mucosa of WT H. pylori-infected WT BALB/c mice injected with IL-22 or PBS control, Abs against IL-22 (IgG2a), CXCR2 (IgG2a) and/or CXCL2 (IgG2b) or corresponding isotype control Abs, or SB225002 or DMSO control on day 49 p.i. were compared. (B) MDSC responses in gastric mucosa of WT H. pylori-infected WT BALB/c and IL-22 KO mice, or WT C57BL/6 and IL-23 KO mice, or WT H. pylori-infected IL-22 KO mice injected with IL-23 and IL-23 KO mice injected with IL-22 (B) on day 49 p.i. were compared. (C) MDSC migration was assessed by transwell assay, as described in Methods, and statistically analysed (n=3). *p<0.05, **p<0.01, n.s. p>0.05 for groups connected by horizontal lines compared. WT; wild-type; KO, knockout; IL, interleukin; PBS, phosphate-buffered saline; Abs, antibodies; MDSC, myeloid-derived suppressor cell; DMSO, dimethylsulfoxide; DC, dendritic cells; p.i., postinfection; n.s., not significant.
To evaluate the contribution of an IL-22-CXCL2-CXCR2 axis to the accumulation of MDSCs, MDSC chemotaxis assay was performed and demonstrated that culture supernatants from primary gastric epithelial cells treated with WT H. pylori-stimulated DC-derived Th22 cell-polarising culture supernatants induced significantly more MDSC migration than those supernatants from gastric epithelial cells treated with ΔcagA-stimulated DC-derived culture supernatants and this effect was lost upon pretreatment with neutralising Abs against IL-22 and CXCR2 (figure 5C). Neutralisation of IL-22 or CXCL2 with Ab, inhibition of CXCR2 with SB225002 or simultaneous blocking of both CXCL2 and CXCR2, all significantly reduced H. pylori-induced MDSC accumulation (figure 5A and see online supplementary figure S5B). Conversely, injection of IL-22 significantly increased H. pylori-induced MDSC accumulation (figure 5A and see online supplementary figure S5B). Finally, the effect of IL-22 again appears to be mediated by BM-derived cells (see online supplementary figure S5C). Collectively, these results, therefore, suggest that an IL-22-CXCL2-CXCR2 axis contributes to MDSC accumulation within the gastric mucosa of H. pylori-infected mice.
IL-22 stimulates production of proinflammatory proteins S100A8 and S100A9 by MDSCs
It has previously been shown that proinflammatory S100A8 and S100A9 proteins are highly expressed in the inflamed gastric mucosa of H. pylori-infected individuals,16 and our own recent data provide a similar result (data not shown). The underlying basis for this induction of S100 proteins has remained unclear. We were, therefore, interested to observe a positive correlation between IL-22 and S100A8/S100A9 (see online supplementary figure S6A), prompting us to speculate whether IL-22 might regulate S100A8/S100A9. Since S100A8/S100A9 are known to be secreted by MDSCs,17 we isolated MDSCs and stimulated them with IL-22. This potently induced MDSCs to synthesise and express S100A8/S100A9 in vitro (figure 6A). Further,IL-22 KO mice or neutralisation of IL-22 produced significantly less S100A8/S100A9 in gastric mucosa (figure 6B, C). Conversely, injection of IL-22 significantly increased S100A8/S100A9 production (figure 6C). Finally, the effect of IL-22 once again appears to be mediated by BM-derived cells (see online supplementary figure S6F). Collectively, our data demonstrate that IL-22 plays an essential role in inducing S100A8/S100A9 expression in gastric mucosa during H. pylori infection.

IL-22 induces proinflammatory proteins S100A8 and S100A9 production from MDSCs and regulates S100A8 and S100A9 in vivo, and IL-22-induced MDSCs suppress Th1 cell response in Helicobacter pylori infection. (A) S100A8 and S100A9 proteins in IL-22-stimulated human CD45+CD14+HLA-DRlow/− MDSCs for different time points or 24 h were analysed. (B and C) S100A8 and S100A9 protein in gastric mucosa of WT H pylori-infected WT BALB/c and IL-22 KO mice (B), or WT H. pylori-infected WT BALB/c mice injected with IL-22 or Abs against IL-22 (C) on day 49 p.i. were compared. (D and E) Th1 cell responses in gastric mucosa of H. pylori-infected WT BALB/c mice injected with IL-22 or PBS control, Abs against IL-22 (IgG2a), CXCR2 (IgG2a), and/or CXCL2 (IgG2b) or corresponding isotype control Abs, or SB225002 or DMSO control (D), or H pylori-infected WT BALB/c and IL-22 KO mice (E) on day 49 p.i. were compared. (F) T cell-MDSC coculture was assessed by flow cytometry as described in Methods and statistically analysed (n=3). Results are expressed as percentage of proliferated Th1 cells in CD4+ T cells. Results are representative of three independent experiments. *p<0.05, **p<0.01, n.s p>0.05 for groups connected by horizontal lines compared, or compared with uninfected mice. WT; wild-type; KO, knockout; IL, interleukin; PBS, phosphate-buffered saline; Abs, antibodies; MDSC, myeloid-derived suppressor cell; DMSO, dimethylsulfoxide; DC, dendritic cells; p.i., postinfection; GADPH, glyceraldehyde 3-phosphate dehydrogenase; IFN, interferon; CFSE, carboxyfluorescein diacetate succinimidyl ester; n.s., not significant.
IL-22-induced MDSCs suppress Th1 cell responses in H. pylori infection
Th1 cells have previously been implicated in H. pylori gastritis.2 ,18 Since Th1 cell responses can be inhibited by MDSCs in other contexts,19 we were interested to learn whether IL-22-induced MDSCs would affect Th1 cell responses during H. pylori infection. Neutralisation of IL-22 or CXCL2 with Ab, inhibition of CXCR2 with SB225002 or simultaneous blocking of both CXCL2 and CXCR2 all significantly increased H. pylori-induced Th1 cell responses (figure 6D and see online supplementary figure S7A). Similarly, IL-22 KO mice showed significantly greater Th1 cell responses (figure 6E). Conversely, injection of IL-22 significantly reduced H. pylori-induced Th1 cell response (figure 6D and see online supplementary figure S7A). Finally, the effect of IL-22 again appears to be mediated by BM-derived cells (see online supplementary figure S7B). Collectively, these data demonstrated that IL-22 plays an essential role in the inhibition of Th1 cells in gastric mucosa during H. pylori infection in vivo.
Next, we wanted to test whether MDSCs might directly suppress Th1 cell development. We, therefore, cocultured CFSE-labelled peripheral CD4+ T cells of healthy donors with peripheral CD14+HLA-DRlow/− MDSCs from H. pylori-infected patients for 5 days, and found that—in comparison with CD14+HLA-DRhigh monocytes—CD14+HLA-DRlow/− MDSCs suppressed Th1 cell development (figure 6F and see online supplementary figure S7C).
Discussion
Th22 cells and IL-22 appear to have different roles depending upon the nature of the infection. For example, in infections with Trichuris trichiura roundworm20 and Influenza A virus,21 Th22 cells and IL-22 provide the host with protection. By contrast, in Toxoplasma gondii,5 West Nile virus,14 Hepatitis B virus22 and persistent fungal23 infections, Th22 cells and IL-22 contribute to pathology by promoting inflammation. Our data are in keeping with the latter studies, since they clearly indicate that, during H pylori infection, Th22 cells and IL-22 are proinflammatory and contribute to the pathogenesis of gastritis. What remains unclear is why Th22 cell responses are protective in some infections, but pathogenic in others. To this end, our findings that the H. pylori-associated virulence factor cagA was necessary to induce maximal IL-22R1 expression, CXCL2 production, MDSC accumulation and S100A8/9 expression suggest that intrinsic factors encoded by the infection itself are likely to be important in influencing the outcome of the Th22 cell response. Previous studies detailed that mice were preferentially infected with cagPAI-negative H. pylori clinic isolates24 and H. pylori B128 strains25 that induced less inflammation, which resembles our data on ΔcagA compared with WT H. pylori (data not shown).
The proinflammatory nature of IL-22 has been suggested to involve various mechanisms in the past. For example, enhanced expression of IL-22 was accompanied by increased CXCL2 levels in a mouse model of psoriasis.26 Meanwhile, IL-22 was observed to promote myeloid cell infiltration into the central nervous system.27 And IL-22 was also reported to induce S100 family members in acanthosis.6 Our findings are consistent with these studies and demonstrate for the first time how each of these individual findings fit together in the context of a single experimental model: in this case, H. pylori gastritis. Specifically, our in vitro and in vivo data together provide a multistep model of inflammation during H. pylori infection involving interactions between H. pylori, Th22 cells, DCs, gastric epithelial cells and MDSCs within the gastric mucosa (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A proposed model of cross-talk among Helicobacter pylori, dendritic cells (DC), gastric epithelial cells and myeloid-derived suppressor cells (MDSC) leading to Th22 cell differentiation and MDSC-mediated proinflammation in gastric mucosa during H. pylori infection. H. pylori stimulate DCs to secrete interleukin (IL)-23 and H. pylori induce gastric epithelial cells to express IL-22R1. Release of IL-23 induces the polarisation of Th22 cells. Polarised Th22 cells expand in gastric mucosa where they release cytokine IL-22 that stimulates gastric epithelial cells to secrete CXCL2. Responding to the CXCL2 chemokine gradient, myeloid progenitor cell-derived, CXCR2-expressing MDSCs migrate into gastric mucosa where they exert a proinflammatory effect by producing inflammatory proteins, S100A8 and S100A9, and suppressing T helper type 1 (Th1) cell response.
MDSCs are a heterogeneous population of immature myeloid cells with the capacity to potently suppress T cell immunity.15 MDSCs have been most intensively studied in the context of cancer, but their role in the pathogenesis of viral,28 parasitic20 and bacterial29 diseases is now starting to be appreciated. Our data suggest that MDSCs may be a key cellular player in H. pylori-induced gastritis via the inhibition of Th1 responses that differed in C57BL/6 and BALB/c mice30 and were associated with cagPAI+ H. pylori-induced and IFN-γ-inducible chemokine responses in gastric epithelial cells31—which is consistent with their canonical role in T cell suppression—and also via the production of key inflammatory mediators, such as S100A8 and S100A9.
The clinical outcome for patients with H. pylori-associated gastritis remains diverse, with sequelae ranging from an asymptomatic illness at one of the spectrum to life-threatening peptic ulceration and gastric carcinoma at the other end. Given the apparent relationship between IL-22 levels and the severity of gastric inflammation observed in H. pylori-infected patients (figure 3A), thought should be given to the use of IL-22 and/or Th22 cells as novel diagnostic biomarkers for H. pylori infection.
Although H. pylori remains reasonably straightforward to eradicate in most patients using oral antibiotics, it is noteworthy that chronic gastritis commonly persists even after successful eradication therapy.32 Treatments that can address the underlying inflammatory process may therefore be of clinical value in such cases. In this regard, our findings suggest several possible therapeutic targets, including IL-22, S100A8 and S100A9. At the same time, it will be interesting to test whether the same proinflammatory cellular networks and molecular pathways described here for H. pylori gastritis operate in other chronic infections where eradication is more difficult (eg, hepatitis B). If this is true, then targeting these same molecular pathways may also prove to be of clinical benefit.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
- Data supplement 5 - Online table 3
Footnotes
YZ and X-fL contributed equally.
Correction Notice This article has been corrected since it was published online first. The funding number 81201265 has now been added.
Contributors Conception and design, data analysis, drafting the manuscript: YZ and Q-mZ. Manuscript revision: YZ, WC and KCP. Statistical analysis: YZ, PC, X-fL, L-sP, B-sL, T-tW, NC, W-hL and YS. Obtained funding: YZ and Q-mZ. Technical support: YZ, PC, MZ, X-hM, S-mY, HG, GG, TL, Q-fZ, H-jY, L-yY, F-yM and Y-pL. Final approval of submitted version: YZ and Q-mZ.
Funding This work was supported by a grant of Medical Science Youth Training Project of Chinese People's Liberation Army (13QNP108), National Natural Science Foundation of China (31270174) and 81201265 and National Basic Research Program of China (973 Program, No. 2009CB522606).
Competing interests None.
Patient consent Obtained.
Ethics approval The biopsy specimens were obtained under protocols approved by the ethics committees of XinQiao Hospital and Southwest Hospital of Third Military Medical University. All animal experiments were undertaken with approval from the Animal Ethical and Experimental Committee of Third Military Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.