Article Text

Abstract
Pancreatic ductal adenocarcinoma (PDA) exhibits one of the poorest prognosis of all solid tumours and poses an unsolved problem in cancer medicine. Despite the recent success of two combination chemotherapies for palliative patients, the modest survival benefits are often traded against significant side effects and a compromised quality of life. Although the molecular events underlying the initiation and progression of PDA have been intensively studied and are increasingly understood, the reasons for the poor therapeutic response are hardly apprehended. One leading hypothesis over the last few years has been that the pronounced tumour microenvironment in PDA not only promotes carcinogenesis and tumour progression but also mediates therapeutic resistance. To this end, targeting of various stromal components and pathways was considered a promising strategy to biochemically and biophysically enhance therapeutic response. However, none of the efforts have yet led to efficacious and approved therapies in patients. Additionally, recent data have shown that tumour-associated fibroblasts may restrain rather than promote tumour growth, reinforcing the need to critically revisit the complexity and complicity of the tumour–stroma with translational implications for future therapy and clinical trial design.
- PANCREATIC DISEASE
- PANCREATIC FIBROSIS
- PANCREATIC PATHOLOGY
- PANCREATIC TUMOURS
- PANCREATITIS
Statistics from Altmetric.com
Key messages
Basic science
A pronounced desmoplastic and hypovascular microenvironment is a histological hallmark feature in pancreatic cancer.
The tumour microenvironment consists of activated fibroblasts, pancreatic stellate cells, infiltrating immune cells, macrophages and soluble growth factors and cytokines that are secreted in the stiff extracellular matrix scaffold.
Activated fibroblast govern local immune surveillance through CxCl12 secretion and additional unknown mechanisms.
Pharmacological and genetic depletion of activated fibroblasts result in accelerated pancreatic tumour growth and immune-modulation, whereas reprogramming of activated fibroblasts promotes improved response to cytotoxic drugs.
Clinical practice
Despite the massive desmoplastic reaction characteristic for pancreatic ductal adenocarcinoma no antistromal therapies are currently approved for clinical use.
Therapies aimed at depleting or modifying cellular and acellular components of the stroma may open new avenues for immunological checkpoint antagonists or other targeted therapies (eg, antivascular endothelial growth factor) that otherwise fail in patients with pancreatic ductal adenocarcinoma.
Stromal depletion or modification therapies should be investigated carefully since tumour-suppressive and promoting functions of the stroma have been described.
The prognostic role of the activated tumour stroma in patients should be revisited as results are highly controversial to date.
Introduction
With a median survival of 6 months, pancreatic ductal adenocarcinoma (PDA) is a notoriously aggressive disease with a dismal prognosis and steadily increasing incidence rates in the industrialised world.1 In the UK, about 9000 new cases are diagnosed each year, and in the USA the number rises to almost 44 000 cases per year.1 Due to the lack of effective means for an early diagnosis, most patients diagnosed with PDA have locally advanced or metastasised disease, and are therefore not candidates for surgical resection. Therefore, >80% of patients qualify only for palliative treatment and gemcitabine has been the standard of care chemotherapy for many years. The use of gemcitabine is associated with a significant, though marginal, survival extension of approximately one month.2 The only targeted agent that has been successfully combined with gemcitabine is erlotinib, an inhibitor of tyrosine kinase domain of the epithelial growth factor receptor, however, achieving an only marginal survival benefit of a median of 2 weeks in unselected patients.3 Notably, the development of skin rash (grade ≥2) is considered a predictive clinical marker for this therapy and results in a median survival of almost 12 months.4 Combinations of gemcitabine with a second cytotoxic drug such as 5-fluorouracil, capecitabine, cisplatin, irinotecan or oxaliplatin all failed to achieve meaningful improvement in overall survival, and thus did not enter clinical routine.5–11 However, more recently, two novel combination chemotherapies have considerably changed the clinical oncological landscape for patients with PDA. In 2011, the gemcitabine-free FOLFIRINOX regimen (folinic acid, fluorouracil, irinotecan and oxaliplatin) was shown to significantly improve overall median survival in metastasised PDA compared with gemcitabine (11.1 vs 6.8 months).12 Two years later, in 2013, results from the international phase III Metastatic Pancreatic Adenocarcinoma Trial revealed a significant survival benefit for nab-paclitaxel and gemcitabine compared with gemcitabine alone (8.5 vs 6.7 months).13 Even though these chemotherapy protocols are a major improvement in the pancreatic cancer space, increased efficacy can only be achieved on the cost of more severe side effects. Much hope has been put into approaches targeting the tumour stroma in PDA. Indeed, PDA is an extremely stroma-rich, hard and scirrhous mass of which almost 90% are made up of extracellular matrix (ECM) consisting of a highly complex assembly of activated fibroblasts, immune cells, blood vessels, neural cells and a variety of matricellular proteins (figure 1A). Furthermore, fibrillar collagen, fibronectin, hyaluronic acid as well as soluble growth factors such as transforming growth factor-β (TGF-β), fibroblast growth factor, vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF/CCN2) are secreted and stored in this highly dynamic location, which is termed the tumour microenvironment.14 For many years, the pronounced stroma reaction (also termed desmoplastic reaction or desmoplasia) was considered to be a passive bystander during carcinogenesis and tumour progression.15 However, during the last 10 years it has dawned that the tumour microenvironment may in fact act as ‘partner in crime’ with tumour cells, promoting tumour initiation and progression.16–18 Many efforts were undertaken to elucidate the complex stroma–tumour interactions and provided interesting and fascinating insights into the stromal biology of pancreatic tumours.19 These investigations were predominantly executed in vitro or in transplanted immunodeficient in vivo models that only partly recapitulate the complex composition of the microenvironment.20 Still, the concept of antistromal therapies emerged soon thereafter and fuelled great enthusiasm in the pancreas cancer community.21 However, all of the promising experimental and early clinical findings failed when rigorous clinical phase II or III studies were conducted and no approved antistromal therapy has actually entered the clinical routine.22–24 Recent experimental evidence has shown that stromal depletion approaches may favour tumour aggressiveness and spread, and thus have reignited the discussion whether the tumour stroma in PDA is ‘friend or foe’.25 In fact, the mechanisms and functional consequences of the tumour–stroma crosstalk may be by far more complex than previously anticipated and should therefore be reassessed in an unbiased manner. This review revisits the translational aspects of stromal biology in pancreatic cancer with potential implications for future therapy and clinical trial design.
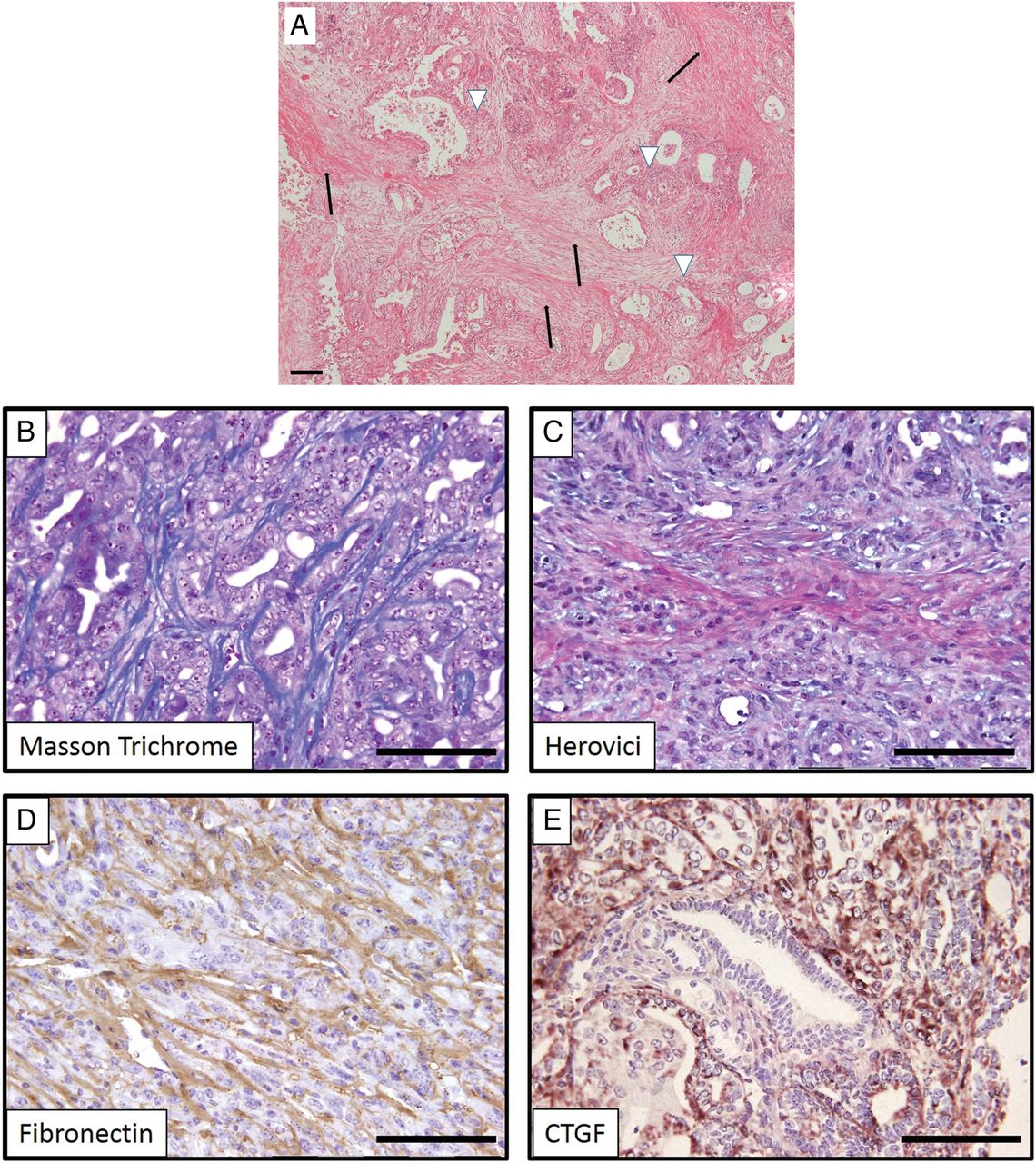
(A) H&E stain of human pancreatic ductal adenocarcinoma displaying a prominent desmoplastic reaction (black arrows) in which neoplastic ductal cells are embedded (white arrowheads). (B–E) Genetically engineered mice (here: KrasG12D/+;p53172H/+;Pdx-Cre) faithfully recapitulate the dense collagen-rich stroma as evidenced by several chemical and immunohistochemical staining methods. CTGF, connective tissue growth factor.
Cellular and molecular hallmarks in PDA: from men to mice
The cellular and molecular evolution of pancreatic carcinogenesis has been intensively studied over the last few decades. Human and murine data suggest that PDAs develop through an increasing number of dysregulated cell-autonomous and non-tumour cell-autonomous pathways paralleled by typical morphological changes that are termed pancreatic intraepithelial neoplasia (PanIN 1–3) and originally arise from acinar to ductal metaplasia (ADM).26 Activating mutations in the K-ras-oncogene represents a critical event and can be found in >90% of PDA cases.27 Subsequent somatic mutations of tumour suppressor genes such as p16, p53 and DPC4/SMAD4 lead to epithelial transformation and ultimately pave the way for progression to frank adenocarcinoma that commonly metastasise to the liver and lymph nodes at early stages.28–30 Histologically, PDA is characterised by a marked fibrotic tissue reorganisation. The activation of fibroblasts, myofibroblasts and pancreatic stellate cells (PSCs) can already be observed at early pre-neoplastic stages, leading to the production of large amounts of ECM components (eg, hyluronan, collagen, fibronectin) as well as a large number of growth factors and matricellular proteins (eg, SPARC) that determine the biophysical properties and the biochemical complexity around epithelial cells.14 ,31
The molecular and histopathological evolution of human PDA has also been recapitulated in genetically engineered mouse models (GEMMs).32 ,33 Mice with conditionally engineered activating mutations (eg, by conventional Cre-loxP-based techniques) of K-ras progressively develop murine PanIN lesions and ultimately murine PDA (mPDA) with long latency (12–15 months).34 Additional genetic ablation of p16, TGF-β or p53 dramatically accelerates tumorigenesis, and locally invasive and metastasised mPDA can be observed starting at 3–4 months of age.35–38 Importantly, GEMMs not only recapitulate the clinical symptoms such as biliary obstruction, anaemia, ascites, cachexia and chemoresistance to gemcitabine, but also faithfully mimic the histological features of the tumour microenvironment within the pancreas (figure 1B–E). The introduction of GEMMs has marked a milestone in pancreatic cancer research and hugely contributed to a more detailed understanding of the complex biology of the disease.20 Moreover, numerous preclinical drug trials have been conducted in GEMMs in so-called ‘mouse-hospitals’ to foster insights into the poor therapeutic responsiveness of PDA.39–41 However, simultaneous embryonic activation of oncogenes and inactivation of tumour suppressor genes do not reflect the genuine sequential multistep development of human cancer and represent a profound limitation of traditional GEMMs. More recently, several modifications of GEMMs have been developed. Interestingly, the next-generation dual-recombinase system (DRS) allows spatiotemporal genetic engineering that resembles the natural course of cancer more closely. To this end, the Flp-FRT recombinase system is combined with the Cre-loxP system for secondary genetic manipulation. This sophisticated approach opens new experimental avenues to rigorously investigate genetic alterations during multistep carcinogenesis, scrutinise tumour cell subpopulations (eg, stem cells), functionally dissect different cellular compartments within the tumour microenvironment (ie, immune cells, fibroblasts, nerves) and ultimately assess therapeutic targets and resistance mechanisms.42 Using this next-generation dual recombinase system, Schonhuber et al provided first evidence that mast cells that had traditionally been regarded as oncogenic players are surprisingly dispensable for tumour formation. Besides the Cre-loxP, Flp-FRT DRS, tetracycline-inducible expression systems,43 ,44 novel embryonic stem cell-based PDA models45 and recombination systems such as Dre-Rox system are valuable additions to the genomic toolbox that will spur the field. Notably, Kras alone is insufficient to induce PDA, and several transposon-based insertional mutagenesis screens in mice have led to the discovery of genetic alterations that cooperate with oncogenic K-ras to promote tumorigenesis and have not previously been implicated in pancreatic carcinogenesis.46–48
However, the generation, imaging, treatment and maintenance of GEMMs require resources and staffing that are often not feasible for single laboratories. Notably, in order to draw robust conclusions from preclinical and genetic experiments, cohort sizes need to consist of at least 5–8 animals per treatment cohort, and large-scale drug screening experiments cannot be performed.49 ,50 Therefore, three-dimensional and organotypic culture systems are currently developed to investigate the complex interplay between stromal cells, tumour cells and immune cells within a reconstituted ECM gel.51–54 Recent work showed that pancreatic organoids can be rapidly derived from resected tumours and biopsies, survive cryopreservation and exhibit biological and cellular hallmark features of PDA.55 Orthotopically transplanted neoplastic organoids recapitulate preneoplastic stages and progress to locally invasive and metastatic carcinomas. Whether results from drug screens using organoid models are predictive remains to be seen, but genetic and pharmacological manipulation provide a platform to probe these issues. Therefore, organoids will certainly expand the experimental armamentarium, thus possibly bridging the gap between GEMMs and cell culture-based or xenotransplanted models.
Chronic inflammation and immune cells
General concept of inflammatory-driven pancreatic carcinogenesis
The transition of chronic pancreatitis to pancreatic carcinoma represents an exemplary clinical observation of the paradigm of chronic inflammation evolving into malignancy. The risk correlates with the duration of recurrent pancreatitis and chronic inflammation. The lag of time between pancreatitis diagnosis and PDA onset is usually about 10–20 years. Despite these positive correlations, chronic pancreatitis as risk for PDA is still uncommon since only about 4–6% of patients with chronic pancreatitis develop frank carcinoma within 20 years of diagnosis.56 Chronic inflammation is thought to be the mechanism by which chronic pancreatitis develops to PDA.57 Damaged acinar cells are responsible for releasing the first inflammatory signals (interleukin (IL)-1b, IL-6, tumour necrosis factor (TNF)) in response to pancreatic injury, leading to the activation of the immune system. Activated immune cells eliminate genetically altered cells. However, over time, the anticancer response is insufficient to eliminate these cells.58 Such altered cells activate proinflammatory transcriptional programmes such as nuclear factor of activated T cells and release further inflammatory cytokines promoting not only inflammation but also growth of pre-neoplastic/neoplastic cells.59 ,60 During the process of chronic inflammation, quiescent PSC are converted into an ‘activated or myofibroblastic’ state. PSCs do not only retain their ability to produce large amounts of ECM proteins in the inflamed pancreas transisting to PDA, they establish a network of interactions between inflammatory cells, acinar cells and cancer cells.61–64 Thus, PSCs might assume a coupling role in inflammation-associated carcinogenesis (figure 2). Once the tumour is established, the tumour microenvironment has a highly immunosuppressive composition comprising increased numbers of tumour-associated macrophages (TAMs) with an M2 phenotype, neutrophils with an N2 phenotype, Treg cells as well as a Th1/Th2 cell shift.65 All these cells further contribute to immune evasion.

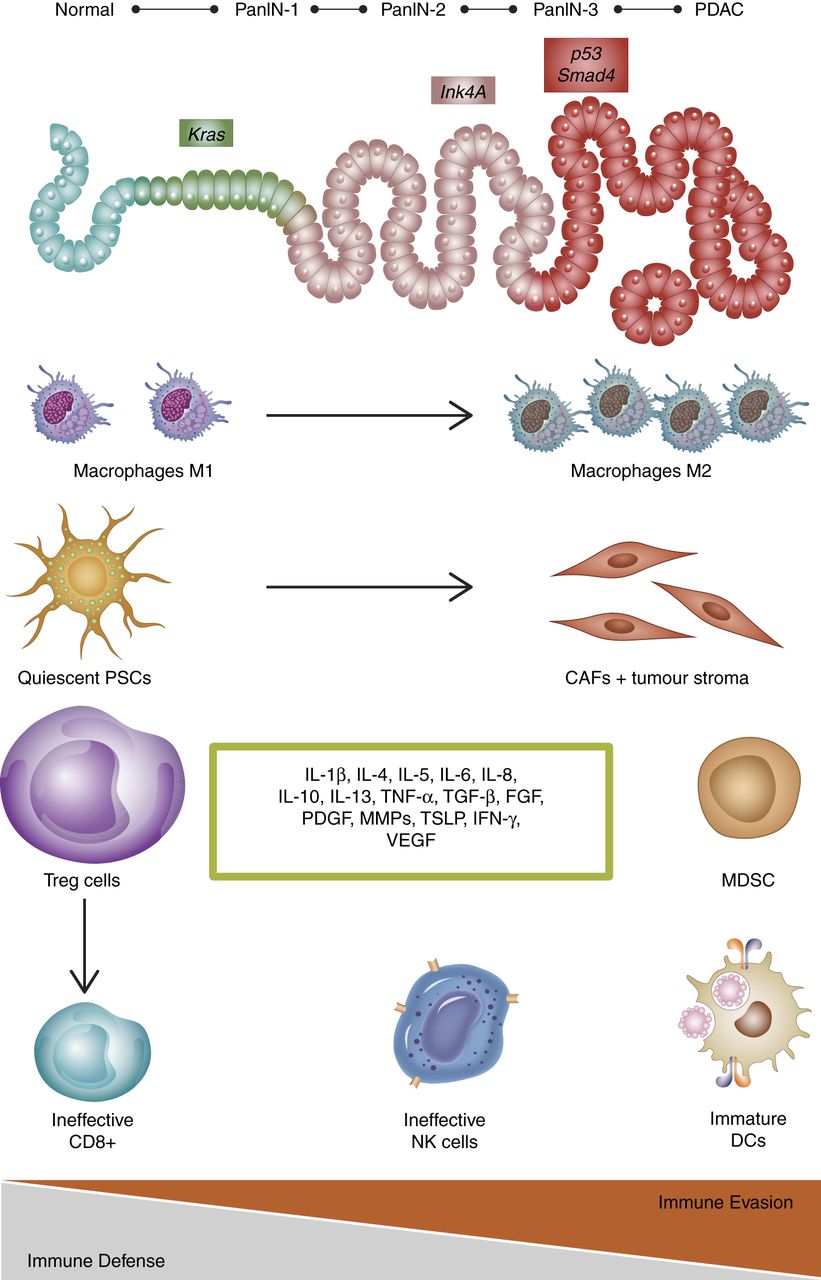
Following expression of oncogenic Kras, ductal cells induce a cascade of cell-autonomous and non-cell-autonomous alterations. Accumulating genetic mutations govern pancreatic ductal adenocarcinoma (PDA) progression from low-grade pancreatic intraepithelial neoplasia (PanIN) to high-grade PanIN and ultimately frank carcinoma (top line). Mechanisms of innate and adaptive immune responses with accumulation of cytotoxic CD8 T cells, mature dendritic cells (DCs), activated proinflammatory macrophages (M1), natural killer (NK) cells and pancreatic stellate cells (PSCs) are sequentially inhibited by premalignant and malignant cells that culminate in both local and systemic immune dysfunction, which hampers the detection and clearance of transformed cells (immune evasion). Thus, a tumour-induced immunosuppressive environment is established in which macrophages switch to the immunosuppressive M2 state, while T regulatory cells (Tregs) and myeloid derived suppressor cells (MDSC) inhibit effector immune response. PSCs are activated to cancer associated fibroblasts (CAFs) that produce abundant extracellular matrix components. Furthermore, various cell types within the tumour microenvironment support the immunosuppressive phenotype by secreting a multitude of soluble growth factors and cytokines (green box) that further support the immunosuppressive phenotype in pancreatic cancer. IFN, interferon; IL, interleukin; MMP, matrix metalloproteinase; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β; TNF, tumour necrosis factor-α; TSLP, thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor.
Tumour-associated macrophages
TAMs have been identified as a major population of inflammatory cells in solid tumours and identified as potential therapeutic targets.66–68 For instance, the recently approved cytotoxic agent trabectedin selectively targets mononuclear phagocytes through caspase-8-dependent apoptosis; and selectivity is achieved due to differential expression of signalling and decoy TRAIL receptors in macrophages.69 Trabectedin has been used in a clinical phase II trial in metastasised PDA (NCT01339754), but results have not been published so far. Interestingly, recent data revealed that TAMs induced upregulation of cytidine deaminase (CDA), the main enzyme responsible for gemcitabine inactivation, and CDA inhibition sensitised murine tumours to gemcitabine.70 ,71 TAMs have also been implicated significantly in tumour progression via secretion of IL-6 and activation of Stat3.72 ,73 Recruited and activated macrophages release IL-6 to activate the Stat3/Socs3 pathway in the pancreas. This IL-6-dependent cascade accelerates pancreatic carcinogenesis.72 The complexity of the local immune system during pancreatic cancer progression is depicted in figure 2.
Myeloid cell infiltrations
However, even in the absence of chronic inflammation, pancreatic cancer contains infiltrates of diverse leucocyte subsets. Although these infiltrates, composed of both myeloid-lineage and lymphoid-lineage cells, do not meet the classical definition of an inflammatory immune response, they seem to be instrumental in mediating tumour progression. This form of immune cell infiltration that is detectable in many solid tumours has been described as infiltrating immune cells (IICs) to encompass both classic inflammation and a more subtle involvement of immune cells.74 IICs are found very early in the tissue surrounding early and late mPanIN lesions and are still present in full-blown pancreatic cancer.75 It is becoming increasingly evident that immune cells in the tumour microenvironment fail to mount an effective antitumour immune response as the majority of the IICs are immunosuppressive. The granulocyte macrophage colony-stimulating factor (GM-CSF) seems to play a central role not only in establishing the inflammatory tumour microenvironment but also in mediating immunosuppression. Oncogenic, activated Kras expressed in pancreatic ductal epithelial cells seems to reprogram the tumour microenvironment by directing transcription of the inflammatory cytokine GM-CSF.76 ,77 Tumour-derived GM-CSF promotes recruitment of myeloid progenitor cells to the surrounding stroma and subsequent differentiation into myeloid-derived suppressor cells (MDSCs). MDSCs, in turn, suppress the immune surveillance function of CD8+ killer T cells, preventing them from recognising and clearing transformed pancreatic ductal cells. Data from the Hingorani group show that targeted depletion of granulocytic MDSC (Gr-MDSC), a single myeloid subset, triggers an endogenous antitumour T cell response, suggesting that therapeutic targeting of Gr-MDSC might be potential strategy for patients with PDA.78
T cell infiltration
Among infiltrating T lymphocytes, CD8+ T cells are rare whereas CD4+ T cells are abundant.75 Recent studies addressed the functional role of CD4+ T cells within the pancreatic cancer microenvironment.79 ,80 These studies show that CD4+ T cells promote PanIN formation by blocking the antitumour immune responses mediated by CD8+ T cells. A predominant infiltration of pancreatic cancer stroma by Th2 (GATA3+) in contrast to Th1 (Tbet+) cells correlated with disease progression.81 The ratio of GATA3+/Tbet+ tumour-infiltrating lymphocytes was proposed as an independent predictive marker for survival after surgery in patients with (stage IB/III) pancreatic cancer. Activation of the TNF receptor superfamily member CD40 has been shown to be a key regulatory step in the development of T-cell-dependent antitumour immunity. CD40 activation can reverse immune suppression and drive antitumour T cell responses. Systemic CD40 activation with an agonist CD40 monoclonal antibody was sufficient to circumvent tumour-induced immune suppression and invoke productive macrophage and T-cell-dependent antitumour immunity in pancreatic cancer in mice and humans.82 These studies argue for an important role of the immune system in established pancreatic cancer. CD4+ T cells are a heterogeneous population. High numbers of regulatory T cells and T helper 17 (TH17) cells were observed in the pancreatic microenvironment in a Kras-dependent manner. Levels of TH17 cells were found to be elevated in the pancreatic immune-infiltrates in humans as well as in mice. CD4+ T lymphocytes and IL-17 signalling are required for oncogenic Kras-driven pancreatic carcinogenesis. Intrapancreatic CD4+ T cells suppress the antitumour activity of CD8+ T cells during Kras-driven PanIN formation. TH17 cells (as well as γδ T cells) secrete IL-17A that signals through IL-17RA in acinar–ductal metaplasia and PanINs, thereby inducing tumour initiation and progression. CD4+ T cell ablation enables effector CD8+ T cell function and induces apoptosis in PanIN cells, thus blocking the onset of pancreatic cancer initiation.80 Moreover, pattern recognition receptors were found to be expressed in pancreatic cancer. Studies have shown that Toll-like receptors (TLRs) and the adaptor protein MyD88 impact pancreatic cancer development through the regulation of immune response. Ligation of TLR7, for example, also involved various signalling pathways such as Stat3.83 ,84 This information is now culminating in various clinical trials. A phase III trial addressing the additional role of Jak1/2/Stat3 inhibitors in patients with metastatic pancreatic cancer are under way (NCT02117479; NCT02119663).
Stromal depletion in PDA: therapeutic and biological implications
The first preclinical trial in GEMMs that introduced the stromal depletion hypothesis was conducted by Olive et al. The authors demonstrated that pharmacological inhibition of the pro-stromal sonic hedgehog signalling cascade by the Smoothened inhibitor (IPI-926) led to a significant reduction of tumour stroma and increased perfusion and mean vessel density. Paralleled by these alterations, intratumoural gemcitabine delivery was elevated and therapeutic response and median survival increased significantly.21 This preclinical work prompted clinicians and scientists likewise to postulate that the biophysical rigidity of the ECM may considerably contribute to therapeutic resistance in PDA by compressing blood vessels, leading to reduced perfusion that ultimately impedes the delivery of drugs to neoplastic cells.16 ,85 ,86
Indeed, subsequent preclinical investigations introduced alternative approaches to successfully relieve vessel compression and improve drug delivery. For instance, hyaluronan is highly overexpressed by tumour and stromal cells and accumulates in PDA.87 ,88 As megadalton glycosaminoglycan, HA retains water due to its high colloid osmotic pressure and provides elasticity to connective tissue in healthy organs. If excessive accumulation of HA occurs, interstitial fluid pressure and solid stress result in blood vessel compression. In line with previous data from a prostate cancer xenograft model,89 we and others have conducted preclinical trials in which HA degradation was achieved by hyaluronidase PEGPH20 in pancreatic GEMM tumours.90 ,91 Although the preclinical trial design was slightly different, increased vessel patency, drug delivery and survival was reported in both publications. Whether this concept holds true in human pancreatic cancer remains to be seen when the results of an ongoing phase I/II trial of hyaluronidase (PEGPH20) plus gemcitabine will be released (NCT01453153).
Other investigators have shown that the angiotensin inhibitor losartan decreases stromal collagen and hyaluronan secretion in PDA.92 Moreover, losartan treatment was accompanied by reduction of profibrotic signals such as TGF-β1, CCN2/CTGF.92 The authors concluded that angiotensin inhibitors reduce solid stress in tumours, resulting in increased vascular perfusion, oxygen and drug delivery. A currently ongoing clinical trial with FOLFIRINOX and losartan in patients with pancreatic cancer will show whether this class of drugs may serve as inexpensive anticancer therapeutics to sensitise PDA to standard chemotherapy (NCT01821729).
However, the possibility to increase drug delivery to stroma-rich pancreatic tumours does not necessarily imply that this strategy can be successfully translated to patient care. First, disruption of the dense tumour stroma leads to both biophysical and biochemical modifications that may independently or collectively contribute to a favourable therapeutic response, and such information may guide the optimal translation of these preclinical findings to patients. To this end, our group attempted to uncouple stromal depletion from drug delivery by using a chemical inhibitor for CDA, the main inactivating enzyme within the gemcitabine metabolism. Although pharmacological inhibition of CDA raised the levels of activated intratumoural gemcitabine to a comparable level to PEGPH20 or SHH inhibition, the apoptotic rate in neoplastic cells and overall tumour growth was surprisingly unaffected.93 These data and prior work from our group suggest that disrupting the stromal barrier to increase drug delivery is not the only factor that increases antitumour responses.40 Rather, increased drug delivery may be most effective when various intratumoural survival cues are concomitantly targeted. However, elegant studies in patients with PDA using CT-derived transport properties have revealed significant interpatient and intratumoural heterogeneity of gemcitabine DNA incorporation despite similar intravascular pharmacokinetics. Notably, stromal content was inversely correlated with gemcitabine incorporation, and pretherapy CT-derived properties correlated with clinically relevant endpoints such as survival.94 ,95 Furthermore, clinical data have shown that high levels of the gemcitabine transporter hENT1 and activating enzyme deoxycytidine kinase are associated with improved survival in patients treated with adjuvant gemcitabine,96 ,97 suggesting that gemcitabine metabolism rather than biophysical delivery matters most.
Very recently, first results were published regarding the biological effects of prolonged myofibroblast depletion in GEMM by pharmacological and genetic methods. To this end, Pdx1-Cre;KrasLSL-G12D/+;p53flox/+;Rosa26LSL-YFP/+ were intercrossed with Shhfl/fl mice to conditionally ablate the Shh pathway in murine pancreata. As expected, depletion of the pro-stromal Shh pathway resulted in a marked reduction of myofibroblasts and the corresponding tumour stroma. Surprisingly, however, Shh-ablated tumours exhibited more frequent ADM and PanIN lesions during early carcinogenesis, while established tumours were undifferentiated, more metastatic and highly proliferative compared with control littermates98 (figure 3). Furthermore, epithelial to mesenchymal transition (EMT) markers such as Zeb1 and Slug and reduced CD45+-positive myeloid cells were observed following Shh depletion98 (figure 3). In line with earlier observations,21 Shh-deficient mice revealed a more prominent tumour vasculature as evidenced by increased mean vessel density. Although VEGF expression was not increased, Shh-depleted tumours responded to VEGF inhibition compared with wildtype controls, suggesting that stromal depletion may open a narrow therapeutic window for targeted therapies.98 The mechanism by which permanent stromal depletion via Shh inhibition led to such aggressive pancreatic carcinomas has not been answered yet. However, increased tumour vasculature and reduced autophagy might be pro-tumorigenic, whereas fibroblasts seem to mediate some tumour-suppressive functions. As the data could also be recapitulated by chronically administering a Smoothed inhibitor (Smo) IPI-926 as monotherapy, it may explain the poor clinical performance of Smo inhibitors in patients with pancreatic cancer that resulted in early termination of several clinical trials and caused scepticism regarding the concept of stromal depletion as relevant therapeutic approach. However, Shh and Gli1 protein abundance in resected PDA specimen were recently reported to be associated with a poor prognosis (overall survival and disease-free survival),99 further complicating the understanding of the current data.

Stromal depletion approaches such as sonic hedgehog depletion or genetic myofibroblast ablation can tip the balance towards tumour-promoting effects, resulting in highly proliferative and invasive tumours with increased epithelial to mesenchymal transition and stemness features. Dependent on the targeted stromal component and the length of treatment, tumour-suppressive effects such as increased oxygen delivery (sensitisation for radiation) and drug delivery as well as biochemical and immunological sensitisation (eg, activation of pro-apoptotic pathways) can also be observed. EMT, epithelial to mesenchymal transition.
Approaches that sensitise tumour cells to chemotherapy by biochemical and/or biophysical alterations may be promising new strategies. We have attempted to inhibit stromal-derived CTGF/CNN2 in a GEMM by using a monoclonal antibody FG-3019. Inhibition of CTGF by FG-3019 resulted in increased rates of tumour cell apoptosis when combined with standard chemotherapy gemcitabine. Although the gross appearance of the cellular and acellular tumour stroma was unchanged, the biochemical pattern of the murine tumours changed profoundly, and antiapoptotic and pro-survival proteins such as X-linked inhibitor of apoptosis (XIAP) Birc6, Psen1, Ubqln2 and Hif1α were all downregulated.93 The identified mechanism of chemosensitisation with FG-3019 and gemcitabine is distinct from increased drug delivery that was unaffected in these experiments. To this end, Straussman et al100 reported that primary human diploid fibroblasts suppressed the response of PDA cell lines to chemotherapy when co-cultured in vitro, but did not identify the molecular pathways involved. Only recently, Sherman et al have provided further evidence that regulation of activated PSCs via vitamin D receptor (VDR) is an important determinant for the reactive pancreatic cancer stroma. This study showed that VDR activation reduces PSCs activation and fibrosis, thus reprogramming the tumour microenvironment to a more quiescent, non-inflammatory and physiological state. Treatment with standard chemotherapies such as gemcitabine resulted in improved delivery and antitumour response in GEMMs, suggesting that reprogramming the tumour stroma instead of ablating it might evolve as a new paradigm and promising perspective for patients with PDA.101
Immune cells and fibroblasts
The fibroblast compartment of the tumour microenvironment has been mainly the research interest of cancer biologist rather than cancer immunologists.102 However, the close proximity of inflammatory and immune cell infiltrations to cancer-associated fibroblasts (CAFs) may imply certain regulatory functions and interactions between those compartments. And indeed, increasing evidence suggests that CAFs and myofibroblasts not only shape the soluble and solid stromal environment in PDA, but also control and mediate local immune suppression, thus promoting tumour progression, invasion and distant metastasis.103–106 For instance, fibroblast activation protein-α (FAP) expressing fibroblasts govern immune suppression by CXCL12, the chemokine that signals via the CXCL12 receptor CXCR4. CXCL12 binds to tumour cells and excludes T cells, and conditional depletion of the FAP-positive CAFs permits immune control in various transplanted and autochthonous models of PDA.107 Pharmacological inhibition of CXCR4 thus leads to killing of tumour cells through the rapid, intratumoural accumulation of PDA-specific CD8-positive T cells, and shows the therapeutic efficacy of the T cell checkpoint antagonist anti-PD-L1 that otherwise fails in PDA (figure 4).107 These data elegantly demonstrate that the immune-suppressive environment is controlled by CAFs and accounts for the failure of T cell checkpoint antagonists. Moreover, compelling data have provided insights into additional functions of FAP expressing CAFs being involved in cachexia and anaemia, symptoms that are most relevant for patients with cancer.108 Very recently, Oezdemir et al provided further evidence of the complex interaction of myofibroblasts and immune cells in PDA. The authors genetically depleted α-smooth muscle actin (α-SMA)-positive fibroblasts in KrasG12D/+;Tgfbr2fl/fl mice by intercrossing them with α-SMA-tk transgenic mice. In line with Rhim et al, depletion of myofibroblasts resulted in highly undifferentiated, invasive and necrotic murine carcinomas that exhibited EMT and stemness features (figure 3).109 Moreover, stromal depletion was also paralleled by a profound alteration in IICs that was characterised by decreased CD4+ effector cells and cytotoxic CD8+ cells while regulatory T cells CD3+/Foxp3+ (Tregs) were increased. Upon myofibroblast depletion, cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) expression was elevated and CTLA-4-blocking antibodies attenuated tumour progression and prolonged survival in mice (figure 4).109
{kind=link}
{kind=link}
{kind=link}
{kind=link}
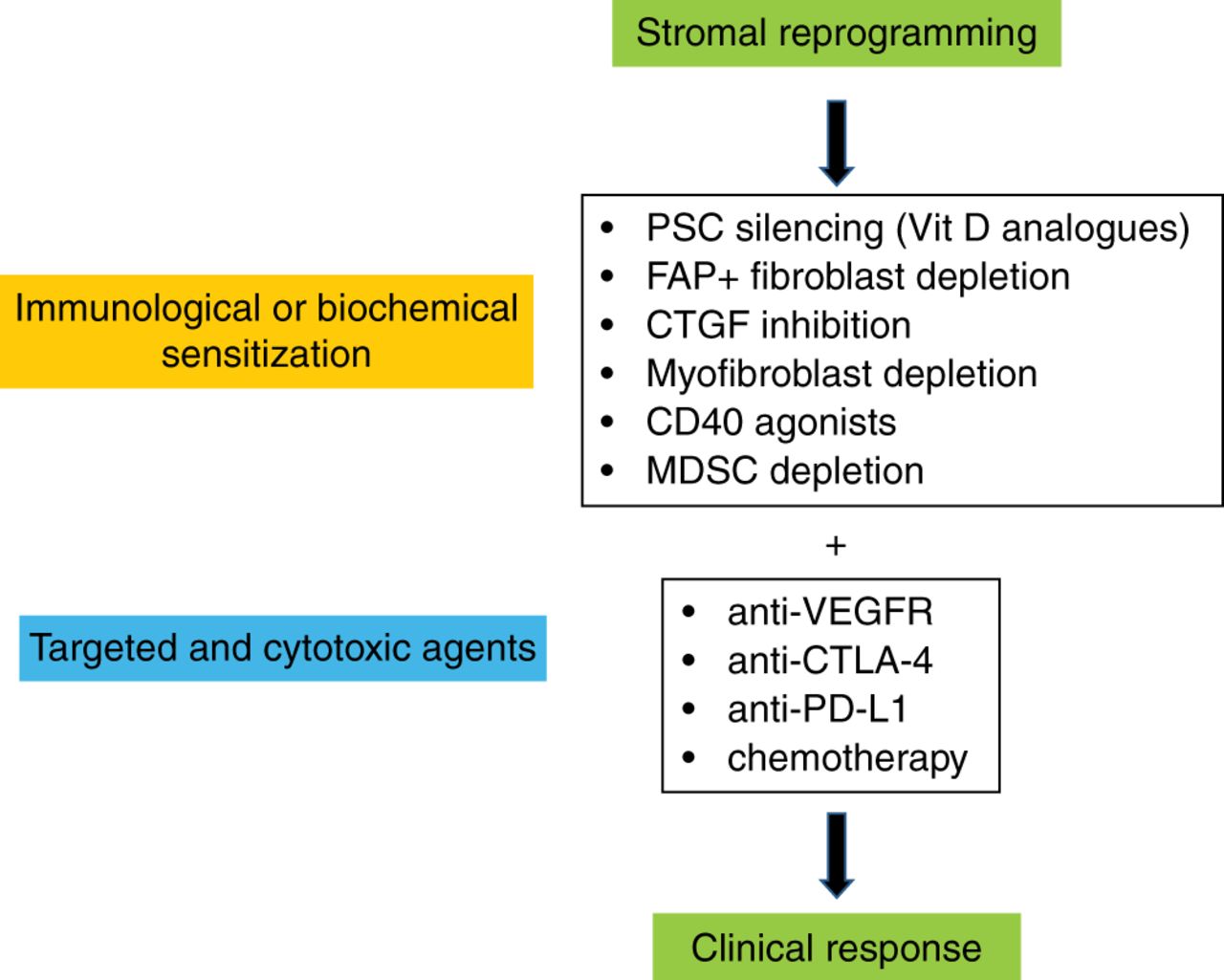
Stromal reprogramming: potential future two-tiered therapeutic strategy for patients with pancreatic ductal adenocarcinoma. Initial biochemical and/or immunological sensitisation of tumours via various targets. Subsequent treatment with immunological checkpoint antagonists, targeted agents or chemotherapy may achieve robust clinical responses. CTGF, connective tissue growth factor; CTLA-4, cytotoxic T-lymphocyte-associated protein-4; FAP, fibroblast activation protein-α; MDSC, myeloid-derived suppressor cell; PSC, pancreatic stellate cell; VEGFR, vascular endothelial growth factor receptor.
Conclusions
In conclusion, recent data have provided novel and fascinating insights into the role of the tumour microenvironment in pancreatic cancer development, progression and therapeutic resistance. (1) In contrast to the previous notion that tumour stroma is solely tumour promoting, certain components such as myofibroblasts may operate as tumour suppressor in preneoplastic and neoplastic pancreas tissue. However, this function may be highly dependent on the tumour stage, the specific tissue context and also variable for certain components of the microenvironment. For instance, while fibroblast depletion may promote tumour progression and invasiveness, blockade of stroma-derived soluble growth factors may result in opposite effects. Therefore, a detailed understanding of the different components involved is crucial for the development of future therapies. (2) Bearing the potential risks in mind, stromal depletion and/or reprogramming might still be a very promising therapeutic strategy in PDA as this approach may open small therapeutic windows to sensitise for targeted therapeutic interventions such as immunological checkpoint blockade or antiangiogenic therapy that otherwise fail in patients with PDA. Accumulating knowledge suggests intensive fibroblast-immune cell crosstalk that could provide a novel and much needed vantage point of attack. (3) The prognostic role of the activated stroma in human PDA tissue is conflicting. Unbiased and joint efforts need to be undertaken to resolve the current discrepancies. While past investigations mainly described a poor prognostic role for stroma-rich PDA,110 recent data contradict these findings and propose that high α-SMA content correlates with increased survival in patients with PDA.98 ,109 ,111 One might even imagine a scenario where chemotherapy that targets proliferating cells might be deleterious for tumours with a high mitotic rate in fibroblasts. Thus, stromal density and proliferation should be carefully considered for both prognostic and therapeutic classifications as the biological behaviour, differentiation and tumour vasculature might be profoundly different.
References
Footnotes
Contributors All authors wrote and critically revised the manuscript.
Funding This work was supported by the Deutsche Krebshilfe, Max Eder Research Group to AN (110972).
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.