Article Text

Abstract
Background Using the promoter methylation assay, we have shown that MDGA2 (MAM domain containing glycosylphosphatidylinositol anchor 2) is preferentially methylated in gastric cancer. We analysed its biological effects and prognostic significance in gastric cancer.
Methods MDGA2 methylation status was evaluated by combined bisulfite restriction analysis and bisulfite genomic sequencing. The effects of MDGA2 re-expression or knockdown on cell proliferation, apoptosis and the cell cycle were determined. MDGA2 interacting protein was identified by mass spectrometry and MDGA2-related cancer pathways by reporter activity and PCR array analyses. The clinical impact of MDGA2 was assessed in 218 patients with gastric cancer.
Results MDGA2 was commonly silenced in gastric cancer cells (10/11) and primary gastric cancers due to promoter hypermethylation. MDGA2 significantly inhibited cell proliferation by causing G1–S cell cycle arrest and inducing cell apoptosis in vitro, and suppressed xenograft tumour growth in both subcutaneous and orthotopic xenograft mouse models (both p<0.001). The anti-tumorigenic effect of MDGA2 was mediated through direct stabilising of DNA methyltransferase 1 associated protein 1 (DMAP1), which played a tumour suppressive role in gastric cancer. This interaction activated their downstream key elements of p53/p21 signalling cascades. Moreover, promoter methylation of MDGA2 was detected in 62.4% (136/218) of gastric cancers. Multivariate analysis showed that patients with MDGA2 hypermethylation had a significantly decreased survival (p=0.005). Kaplan–Meier survival curves showed that MDGA2 hypermethylation was significantly associated with shortened survival in patients with early gastric cancer.
Conclusions MDGA2 is a critical tumour suppressor in gastric carcinogenesis; its hypermethylation is an independent prognostic factor in patients with gastric cancer.
- GASTRIC CANCER
- TUMOUR MARKERS
- GENE REGULATION
- METHYLATION
- MOLECULAR ONCOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
MDGA2 belongs to the brain-derived immunoglobulin superfamily and has been reported to regulate the growth of axons and the development of inhibitory synapse.
Expression of human MDGA2 is detected in normal human stomach.
What are the new findings?
MDGA2 is frequently silenced or downregulated in gastric cancer by promoter hypermethylation.
MDGA2 acts as a novel tumour suppressor in gastric cancer through inhibiting cell proliferation, suppressing G1–S cell cycle transition and inducing cell apoptosis.
The tumour suppressive effect of MDGA2 is mediated by directly cooperating with DMAP1 and consequently inducing p53/p21 signalling cascade.
MDGA2 methylation is an independent prognostic biomarker in patients with gastric cancer.
How might it impact on clinical practice in the foreseeable future?
Detection of methylated MDGA2 may serve as a new biomarker for the prognosis of patients with gastric cancer.
Introduction
Gastric cancer is the fifth leading cause of cancer and the third leading cause of cancer-related mortality globally. The prognosis of patients with gastric cancer continues to be poor, despite improving surgical and adjuvant treatment approaches, with a 5-year overall survival of less than 25%.1 Inactivation of tumour suppressor genes by promoter hypermethylation contributes to the development of gastric cancer.2 Identification of novel tumour suppressive genes targeted by promoter hypermethylation in gastric cancer may provide insights into the epigenetic mechanisms for the inactivation of tumour suppressive pathways. This is also important for the identification of new markers and therapeutic targets for diagnosis and treatment of this disease. In a previous study using MeDIP-chip for a genome-wide screen for hypermethylated candidates in gastric cancer, we found the gene MDGA2 (MAM domain containing glycosylphosphatidylinositol anchor 2), the function of which remains largely uncharacterised, was regulated by promoter methylation in gastric cancer cells.3
MDGA2, also known as MAMDC1, is located on chromosome 14q21.3 and belongs to the brain-derived immunoglobulin superfamily (MDGA1 and MDGA2) in rat.4 MDGAs are classified as cell adhesion molecules and have been reported to regulate the growth of axons and the development of inhibitory synapses.5 ,6 However, the function of MDGAs has not yet been investigated in other organs or diseases. Data in the Human Protein Altas show high or medium protein expression of MDGA2 in some normal human tissues including the stomach (http://www.proteinatlas.org/ENSG00000139915/tissue), but it was not detected in all 12 tested stomach cancer tissues according to data in the Human Protein Altas (http://www.proteinatlas.org/ENSG00000139915/cancer). These findings collectively suggest that inactivation of MDGA2 may play a role during gastric carcinogenesis.
We conducted the first study on MDGA2 in gastric cancer with the aim of elucidating the functional significance and molecular mechanism of MDGA2, as well as the clinical implications of its promoter methylation in this malignancy.
Materials and methods
Subjects and sample collection
Two hundred and eighteen primary gastric cancer samples were collected during surgical resection at the First Affiliated Hospital of Sun Yat-sen University in Guangzhou, China. In addition, 22 paired biopsy specimens of gastric tumour and adjacent non-tumour sites from patients with gastric cancer and 20 biopsy specimens of normal gastric mucosa from healthy controls were obtained during endoscopy at the Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong. None of these patients received preoperative chemotherapy or radiotherapy. The samples were all confirmed histologically and all subjects provided informed consent for obtaining the study specimens. Human normal stomach cDNA was purchased commercially (Stratagene, La Jolla, California, USA).
In vivo subcutaneous and orthotopic xenograft models
BGC823 cells (1×107 cells in 0.1 mL phosphate-buffered saline) stably transfected with MDGA2 expression vector or empty vector were injected subcutaneously into the dorsal right flank of 4-week-old male Balb/c nude mice (n=10 per group). Tumour diameter was measured every 2 days for 2–3 weeks. Tumour volume (mm3) was estimated by measuring the longest and shortest diameters of the tumour and calculating as previously described.7 Orthotopic gastric cancer mouse models were also established to determine the intragastric tumorigenicity. Subcutaneous tumours were harvested 1 week after subcutaneous injection, cut into 1.0 mm3 pieces and then implanted into the stomach mucosa of nude mice (11/group). The mice were sacrificed after 10 days and the tumour size and tumour weight were measured. All experimental procedures were approved by the Animal Ethics Committee of the Chinese University of Hong Kong.
Co-immunoprecipitation of MDGA2 and DMAP1 in gastric cancer cell lines and mass spectrometry
The total protein of AGS and BGC823 cells stably transfected with MDGA2 (His-tagged) expression vector or empty vector was extracted in radioimmunoprecipitation assay (RIPA) buffer supplemented with proteinase inhibitor (Novagen, Darmstadt, Germany). Lysate (100 μg protein) was incubated with anti-His-tag (Abcam, Cambridge, Massachusetts, USA) or IgG (as a negative control) (Santa Cruz, Dallas, Texas, USA) (see online supplementary table S1). The immune complexes were precipitated by centrifugation and separated by SDS-PAGE. Candidate targets were excised from gels and subjected to protein identification by undertaking the in-gel digestion approach and using matrix-assisted laser desorption/ionization time-of-flight/time-of-flight (MALDI-TOF/TOF) mass spectrometry.8
Whole cell lysate (150 μg protein) and co-immunoprecipitation (Co-IP) precipitant by anti-His-tag, anti-DNA methyltransferase 1 associated protein 1 (DMAP1) antibody or IgG were immunoblotted with either anti-DMAP1 or anti-MDGA2 antibody to confirm the interaction of MDGA2 and DMAP1 that was identified by mass spectrometry. The lysate (6% input, 10 μg protein) was also used as a control.
Glutathione S-transferase protein pull-down assay
Purified recombinant human glutathione S-transferase (GST)-DMAP1 protein (Abnova, Taipei City, Taiwan) and His-tagged recombinant human MDGA2 (MDGA2-His; Creative Biomart, Shirley, New York, USA) were used for pull-down assay. GST protein was expressed in E coli strain BL21 (DE3), and the GST protein was purified using a GSTrap column (GE Healthcare, Buckinghamshire, UK). GST-DMAP1 and GST proteins (0.2 μg each) were captured by 30 μL glutathione sepharose beads (GE Healthcare) at 4°C for 2 h; 0.2 μg of MDGA2-His was then incubated with the beads loaded with GST-DMAP1 or GST for 2 h at 4°C. The beads with protein complexes were then washed and separated by SDS-PAGE and visualised by Coomassie Brilliant Blue staining.
Ubiquitination assay
AGS and BGC823 cells stably transfected with MDGA2 expression vector or empty vector were incubated in the presence or absence of 30 μM MG132 (Cell Signaling Technology, Danvers, Massachusetts, USA) for 24 h. Total proteins were extracted using RIPA buffer supplemented with proteinase inhibitor. Immunoprecipitation was then performed using anti-DMAP1 or anti-IgG, respectively. The immunoprecipitated proteins were subjected to western blotting using anti-ubiquitin to evaluate the ubiquitination level. The inputs were subjected to western blot analysis with antibodies against MDGA2, DMAP1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), respectively. Other details and experimental procedures are provided in the online supplementary materials and methods.
Results
MDGA2 is downregulated by promoter hypermethylation in gastric cancer cells
We first determined the expression level of MDGA2 by reverse transcription PCR (RT-PCR). MDGA2 was silenced in 10 of the 11 gastric cancer cell lines (90.9%) but was readily expressed in normal human stomach mucosa. Using methylation-specific PCR, almost full methylation was detected in all the 10 silenced gastric cancer cell lines (AGS, BGC823, MGC803, MKN28, MKN45, NCI-N87, SNU1, SNU16, SNU638 and SNU719), whereas methylation was not detected in MKN1 which expressed MDGA2 or normal stomach mucosa (figure 1A). The promoter methylation status of MDGA2 was further evaluated by bisulfite genomic sequencing (BGS). As shown in figure 1B, CpG sites of MDGA2 promoter region were densely methylated in the 10 silenced cell lines (81.5–99.1%), but only 17.8% methylated in MKN1 cells and no methylation was found in normal stomach mucosa. To further validate this, four randomly selected MDGA2-silenced cells (AGS, BGC823, N87 and SNU719) were treated with demethylation agent 5-Aza. MDGA2 expression was subsequently restored in all treated cells (figure 1C). In concordance with the restored MDGA2 expression, the promoter methylation level of MDGA2 was significantly reduced in these cells by 5-Aza treatment (from 94.2±4.7% to 36.0±3.2%; p=0.0002, paired t test). These results show that transcriptional silence of MDGA2 is mediated by promoter hypermethylation in gastric cancer cells.

MDGA2 is inactivated by promoter methylation in gastric cancer cell lines. (A) MDGA2 was silenced or downregulated in 10 out of 11 gastric cancer cell lines but readily expression in normal gastric tissue. The methylation status of MDGA2 was determined by methylation-specific PCR (MSP). M, methylated; U, unmethylated. (B) A typical CpG island is present at the promoter region of MDGA2. Each vertical bar represents a single CpG site. The transcription start site (TSS) is indicated by a curved arrow. A region for bisulfite genomic sequencing (BGS) and combined bisulfite restriction analysis (COBRA) is denoted. BGS analysis confirmed high levels of promoter methylation in MDGA2-silenced cells and no/low methylation in MDGA2-expressing samples. (C) MDGA2 mRNA expression was restored after treatment with the demethylation reagent 5-Aza. Decreased methylation was revealed by BGS after 5-Aza treatment.
MDGA2 is downregulated by promoter hypermethylation in primary gastric tumours
We then examined the expression and methylation status of MDGA2 in primary gastric cancers. Both mRNA and protein expression of MDGA2 were downregulated in primary gastric tumours compared with adjacent normal tissues of 20 gastric cancers (p<0.001; figure 2A, B), while the promoter methylation level of MDGA2 was significantly higher in tumours than in adjacent non-tumour tissues (63.0±8.3% vs 22.3±9.1%; p<0.0001; figure 2C).

Epigenetic inactivation of MDGA2 in primary gastric cancers. (A) Transcriptional downregulation of MDGA2 in gastric tumours compared with adjacent normal tissues was shown by reverse transcription PCR (RT-PCR). (B) Protein levels of MDGA2 were quantitated as the percentage of cells with positive staining by immunohistochemistry in 20 pairs of gastric tumour and adjacent non-tumour samples. (C) Methylation-specific PCR (MSP) showed high methylation of MDGA2 promoter in 10 gastric tumours and low methylation in their adjacent non-tumour tissues, which was confirmed by bisulfite genomic sequencing (BGS). M, methylated; U, unmethylated. (D) Kaplan–Meier curves showed that MDGA2 methylation is significantly associated with shortened survival in overall patients with gastric cancer and (E) in patients at an early stage but not patients at late stages (log rank test).
Promoter methylation of MDGA2 is an independent predictor of poor outcome in patients with gastric cancer
To evaluate the clinical significance of MDGA2 in gastric cancer, MDGA2 methylation status was examined in 218 primary gastric tumour tissues. Dense MDGA2 promoter methylation was detected in 62.4% (136/218) of gastric cancers. There was no correlation between MDGA2 methylation status and clinicopathological features such as age, gender, Helicobacter pylori infection, histological type or pathological stage (see online supplementary table S2). However, MDGA2 methylation was associated with an increased risk of cancer-related death by univariate Cox regression (relative risk (RR) 2.00 (95% CI 1.31 to 3.06), p=0.001; table 1) after a mean follow-up of 22.1±18.6 months. As expected, the Tumor-Node-Metastasis (TNM) stage was also a significant prognostic factor (p<0.0001). In particular, after adjustment for potential confounding factors including age, gender and TNM stage, MDGA2 methylation was found to be an independent risk factor for shortened survival in patients with gastric cancer by multivariate Cox regression analysis (RR 1.85 (95% CI 1.21 to 2.84), p=0.005; table 1). As shown in the Kaplan–Meier survival curves, in all the patients with gastric cancer examined, those with MDGA2 hypermethylation had significantly shorter survival than those without MDGA2 methylation (median 1.59 vs 4.87 years; p=0.001, log-rank test; figure 2D). After stratification by TNM stage, MDGA2 methylated patients had significantly shorter survival in stage I/II (p<0.05) but not in stages III or IV (p>0.05; figure 2E). These findings indicate that MDGA2 hypermethylation predicts a poor prognosis in patients with gastric cancer, especially in the early stages.
Univariate and multivariate Cox regression analyses of potential poor prognostic factors in gastric cancer
Ectopic expression of MDGA2 suppresses gastric cancer cell growth
To elucidate the tumour suppressive function of MDGA2 in gastric cancer, MDGA2 expression vector was stably transfected into the MDGA2-silenced AGS and BGC823 cells, with empty vector transfection as control. Re-expression of MDGA2 mRNA and protein in these cells was evidenced by RT-PCR and western blot analysis (figure 3A). Ectopic expression of MDGA2 significantly inhibited cell growth, as evidenced by cell viability and colony formation assays in AGS and BGC823 (figure 3B).

In vitro gain- and loss-of-function assays on MDGA2. (A) Ectopic expression of MDGA2 in AGS and BGC823 cells at mRNA and protein levels was confirmed by reverse transcription PCR (RT-PCR) and western blot analysis. (B) MDGA2 significantly inhibited cell viability and colony formation ability. (C) MDGA2 caused cell cycle arrest at G1–S transition, as indicated by flow cytometry. Reduced cell proliferation by MDGA2 was further shown by Ki-67 staining (magnification ×400) and altered cell cycle-related protein expression by western blot analysis. (D) Cell apoptosis by flow cytometry analysis after annexin V-APC and 7-aminoactinomycin (7-AAD) double staining. Q2 shows the late apoptotic cells and Q4 shows the early apoptotic cells. Cell apoptosis was confirmed by terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) staining (magnification ×400) and upregulation of apoptosis-related proteins in MDGA2-expressing cells was confirmed by western blot analysis. (E) Growth inhibitory effect of cisplatin on MDGA2-overexpressing and control vector-transfected AGS and BGC823 cells. Cell viability was measured by MTT assay after 48 h treatment with cisplatin. Data are mean±SD from three independent experiments. (F) Knockdown of MDGA2 in MKN1 cells by short interference RNA (siRNA) transfection was confirmed by RT-PCR, qRT-PCR and western blot analysis. (G) Knockdown of MDGA2 significantly increased cell viability of MKN1 cells, promoted colony formation and (H) promoted cell cycle progression, but reduced cell apoptosis as indicated by flow cytometry. *p<0.05, **p<0.01, ***p<0.001.
MDGA2 inhibits G1–S cell cycle transition and induces cell apoptosis
To determine the molecular mechanism by which MDGA2 suppresses cell growth, we investigated the effect of MDGA2 on cell cycle distribution and apoptosis by flow cytometry. Ectopic expression of MDGA2 led to a significant increase in cells of the G1 phase and a concomitantly significant decrease in cells of the S phase in AGS and BGC823 cells (figure 3C). Consistently, significantly fewer proliferating cells were detected in AGS and BGC823 with MDGA2 overexpression compared with control cells by Ki-67 staining (p<0.005; figure 3C). G1 cell cycle arrest by MDGA2 was confirmed by reduced protein expression of key G1 cell cycle regulators (cyclin D1 and CDK4) and elevated G1 cell cycle inhibitors (p21 and p27) by western blot analysis. The decrease in S phase was confirmed by the reduced proliferation marker proliferating cell nuclear antigen (PCNA) in MDGA2-overexpressing cells (figure 3C), suggesting that MDGA2 inhibits G1–S transition in gastric cancer cells.
Apoptosis analysis by flow cytometry showed that MDGA2 expression induced a significant increase in both early and late apoptotic cells compared with empty vector transfection in AGS and BGC823 cells (figure 3D). Consistent with this finding by flow cytometry, there were significantly more terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL)-positive cells in MDGA2-expressing AGS and BGC823 cells than in control cells (p<0.0005; figure 3D). The cell apoptosis induced by MDGA2 was found to be mediated by activation of caspase-8, caspase-9, caspase-7, caspase-3 and poly (ADP-ribose) polymerase (PARP) by western blot (figure 3D).
MDGA2 re-sensitises gastric cancer cells to cisplatin
To determine the effect of MDGA2 expression on the sensitivity of gastric cancer cells to chemotherapeutic agents, AGS and BGC823 cells stably transfected with MDGA2 expression vector or empty vector were treated with cisplatin at different concentrations for 48 h. As shown in figure 3E, MDGA2 significantly re-sensitised AGS and BGC823 cells to cisplatin.
MDGA2 suppresses invasion and migration of gastric cancer cells
Ectopic expression of MDGA2 markedly suppressed cell migration ability in both AGS and BGC823 cells (see online supplementary figure S1A). Quantitative analyses at 48 h confirmed a significant reduction in wound closure in MDGA2-expressing cells compared with control cells (39% in AGS and 35% in BGC823, respectively). Matrigel invasion assay also showed that MDGA2 significantly impaired the invasiveness of both AGS and BGC823 cells (see online supplementary figure S1B). Moreover, protein expression of epithelial–mesenchymal transition (EMT) markers including E-cadherin, N-cadherin, Vimentin, Snail and Slug in MDGA2-overexpressing cells were examined by western blot analysis. As shown in online supplementary figure S1C, ectopic expression of MDGA2 enhanced protein levels of E-cadherin while reducing N-cadherin, Vimentin, Snail and Slug in both AGS and BGC823 cells. Thus, MDGA2 suppresses the migration and invasive abilities of gastric cancer cells by modulating key EMT regulating factors.
Knockdown of MDGA2 increases cell growth, promotes cell cycle progression and reduces cell apoptosis
To further confirm the tumour suppressive role of MDGA2 in gastric cancer, MDGA2 was knocked down by RNA interference in MKN1 cells which showed endogenous MDGA2 expression. Knockdown of MDGA2 (figure 3F) markedly enhanced cell viability (p=0.01) and clonogenicity (p<0.0005; figure 3G). Moreover, MDGA2 knockdown decreased cells in the G1 phase while increasing cells in the S phase, and reduced cell apoptosis (figure 3H), further supporting the tumour suppressive role of MDGA2 by causing cell cycle arrest and inducing apoptosis in gastric cancer.
MDGA2 attenuates the growth of both subcutaneous and orthotopic xenograft tumours in nude mice
To examine the effect of MDGA2 on gastric tumour growth in vivo, we first set up subcutaneous xenograft tumour models by subcutaneously injecting MDGA2-transfected and empty vector-transfected BGC823 cells in nude mice. Subcutaneous tumour growth was then monitored and compared between the two groups. As shown in figure 4A, the growth of tumour volume by MDGA2-transfected cells was significantly suppressed compared with control cells (p<0.0001). The net weight of tumours formed by MDGA2-transfected cells was also significantly reduced compared with controls at termination of the experiment (p<0.01; figure 4B). Protein expression of MDGA2 was detected in xenograft tumours of the MDGA2-transfected group but not in the control group by immunohistochemistry (figure 4B), indicating that the tumour suppressive effect was exerted by MDGA2 expression. In concordance with the in vitro findings, significantly fewer proliferating cells and more apoptotic cells were detected in MDGA2-expressed xenografts, as indicated by Ki-67 and TUNEL assays, respectively (both p<0.001; figure 4C).
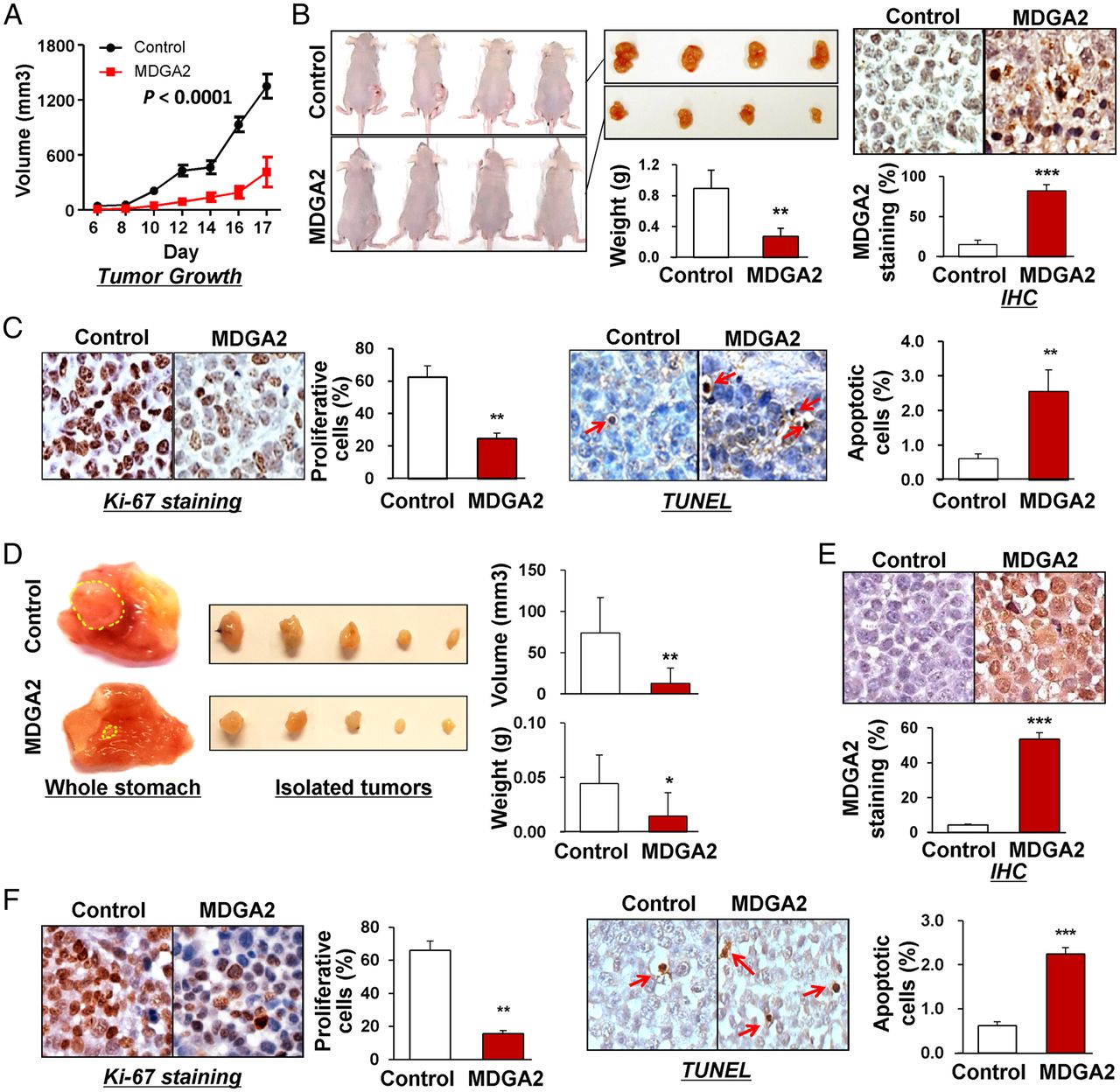
In vivo subcutaneous and orthotopic xenograft models confirmed the suppressive effect of MDGA2 on tumorigenicity. (A) Subcutaneous tumour growth curve of MDGA2-expressing BGC823 cells in nude mice was compared with control vector (pcDNA3.1) transfected cells. The data are mean±SD (n=5/group) of three separate experiments. (B) A representative image of tumour growth in nude mice subcutaneously inoculated with MDGA2- or control vector-transfected cells. Tumour weight was compared at the end of the experiment. MDGA2 expression in subcutaneous xenografts was confirmed by immunohistochemistry (IHC). (C) Cell proliferative activity was evaluated by Ki-67 staining and apoptotic activity by terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) staining in subcutaneous xenografts. (D) Representative images of orthotopic xenograft tumours. Both volume and weight of the orthotopic xenograft tumours were significantly smaller in the MDGA2 group than in the control group. (E) Confirmation of MDGA2 expression in orthotopic xenograft tumours by IHC. (F) Ki-67 staining in orthotopic xenografts and TUNEL staining in orthotopic xenografts. *p<0.05, **p<0.001, ***p<0.0001.
Orthotopic xenograft tumour models were then established using subcutaneous xenograft tumours with and without MDGA2 expression. Orthotopic tumours in the stomach were significantly smaller (p<0.001) and of less net weight (p<0.01) in the MDGA2 group than in the control group (figure 4D). MDGA2 expression was confirmed in orthotopic tumours from the MDGA2 group only, indicating that the tumour suppressive effect was exerted by MDGA2 (figure 4E). Similar to the findings in in vitro experiments and subcutaneous xenografts, significantly fewer proliferating cells (p<0.001) and more apoptotic cells (p<0.0001) were detected in MDGA2-expressing orthotopic tumours than in controls, as indicated by Ki-67 and TUNEL assays, respectively (figure 4F).
DMAP1 is a direct interactor with MDGA2
To gain insight into the molecular mechanistic basis of the tumour suppressive effect of MDGA2 in gastric cancer, we sought to identify its interacting partners by Co-IP followed by identification of associated proteins using mass spectrometry (see online supplementary table S3). DMAP1 was found to interact with MDGA2 by comparing the anti-His IP product of AGS cells overexpressing MDGA2-His with AGS cells transfected with control vector (pcDNA3.1/His) or with anti-IgG IP product (figure 5A). To validate the interaction between MDGA2 and DMAP1, Co-IP using DMAP1 antibody was also performed in AGS and BGC823 cells. MDGA2 could also be co-precipitated by DMAP1 antibody in both cell lines, as shown by western blot analysis (figure 5B), confirming the physical interaction between MDGA2 and DMAP1. We then performed a GST pull-down assay using purified recombinant GST-DMAP1 and MDGA2-His proteins to verify the specificity of the interaction between DMAP1 and MDGA2 under cell-free conditions. Coomassie blots showed that MDGA2 existed in a complex with GST-DMAP1 but not with GST (figure 5C), demonstrating specific binding between MDGA2 and DMAP1.
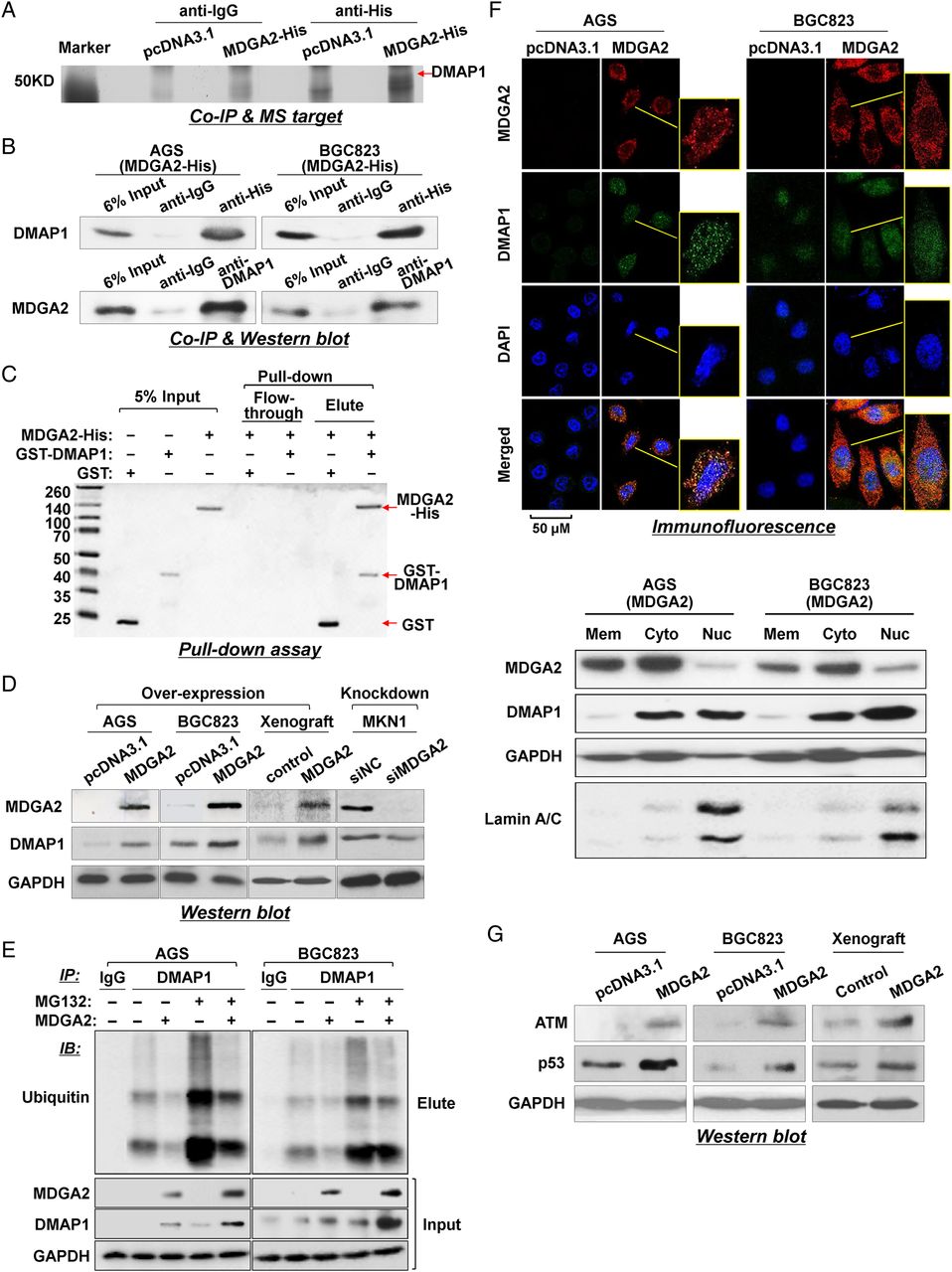
MDGA2 interacts with DNA methyltransferase 1 associated protein 1 (DMAP1) in gastric cancer. (A) Co-immunoprecipitation (Co-IP) of MDGA2 binding proteins followed by mass spectrometry identified DMAP1 to be a MDGA2-binding protein. (B) Co-IP of MDGA2 and DMAP1 with each other from proteins of MDGA2-transfected AGS and BGC823 cells. The presence of MDGA2 and DMAP1 in the Co-IP products was confirmed by western blot analysis using specific antibodies. (C) Direct interaction between MDGA2 and DMAP1 was shown by glutathione S-transferase (GST) pull-down assay. (D) Western blot analysis for DMAP1 expression. (E) MDGA2 increased the DMAP1 level by inhibiting its ubiquitin-mediated degradation. (F1) Confocal immunofluorescence analysis of MDGA2 and DMAP1 expression in MDGA2- and empty vector-transfected cells. The scale bar of 50 M is for unframed images of both cell groups. The yellow-frame fields were enlarged arbitrarily. (F2) Western blot detection of MDGA2 and DMAP1 in the membrane, cytoplasmic and nuclear fractions of AGS and BGC823 cells transfected with MDGA2 expression vector. (G) Western blot analysis showed increased levels of ataxia telangiectasia mutated (ATM) and p53 by MDGA2 over-expression in AGS and BGC823 cells and xenograft tumours.
We evaluated the interplay between MDGA2 and DMAP1 and found that DMAP1 was upregulated with the ectopic expression of MDGA2 in cultured cells (AGS, BGC823) and xenograft tumours, and decreased after knockdown of the endogenous MDGA2 expression in MKN1 cells (figure 5D). To investigate whether the MDGA2–DMAP1 interaction increased the level of DMAP1 by affecting ubiquitination/degradation of DMAP1, we examined the ubiquitination and protein level of DMAP1 in the presence or absence of MDGA2 and/or the proteasome inhibitor MG132. As shown in figure 5E, MDGA2 decreased the ubiquitination level of DMAP1 (anti-DMAP1 IP product) while both MDGA2 expression and MG132 treatment increased the level of DMAP1, suggesting that the MDGA2–DMAP1 interaction stabilises DMAP1 by inhibiting its ubiquitin-mediated degradation.
We next examined the expression and intracellular distribution of MDGA2 and DMAP1 by immunofluorescence. Confocal microscopy images showed that neither MDGA2 nor DMAP1 were observed in AGS and BGC823 cells transfected with control vector. Following stable transfection of MDGA2 in these two cell lines, MDGA2 was found mainly in the membrane and cytoplasm of AGS and BGC823 cells; DMAP1 was found in both the cytoplasm and nucleus and co-localised with MDGA2 in the cytoplasm (figure 5F1). The localisation of the two proteins was further confirmed by western blotting of membrane, cytoplasmic and nuclear fractions, which showed that MDGA2 was mainly localised in the membrane and cytoplasm and DMAP1 was mainly localized in the cytoplasm and nucleus (figure 5F2).
It is known that ataxia telangiectasia mutated (ATM) is a key downstream effector of DMAP1.9 To further confirm the interaction between MDGA2 and DMAP1, we investigated the effect of MDGA2 on the expression of ATM, a DNA damage repair gene. Ectopic expression of MDGA2 in AGS and BGC823 cells induced ATM protein expression (figure 5G), further confirming the interplay between MDGA2 and DMAP1. The upregulation of p53, a downstream factor of ATM, was also detected in parallel with induction of ATM in MDGA2 overexpressed cells and xenograft tumours (figure 5G).
The tumour suppressive function of MDGA2 is dependent on DMAP1
We examined the importance of DMAP1 in MDGA2-mediated tumour suppressive function. Unlike the changes in DMAP1 following upregulation or downregulation of MDGA2, inversely, MDGA2 expression was not changed following overexpression or knockdown of DMAP1 in AGS and BGC823 cells (figure 6A), suggesting that DMAP1 is a downstream effector of MDGA2. In addition, ectopic expression of DMAP1 significantly suppressed cell growth (figure 6B) and colony formation ability (figure 6C) in AGS and BGC823 cells. Moreover, the inhibitory effect on cell growth and colony formation ability by MDGA2 overexpression in AGS and BGC823 cells was significantly attenuated by DMAP1 knockdown (figure 6D–F). These results collectively indicate that MDGA2 plays a tumour suppressive function, at least in part, depending on DMAP1 in gastric cancer.

Suppression of gastric cancer growth by MDGA2 is partly dependent on DNA methyltransferase 1 associated protein 1 (DMAP1). (A) Expression of MDGA2 was not affected by DMAP1. (B) Cell viability of AGS and BGC823 cells by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. (C) Colony formation in AGS and BGC823 cells. (D) DMAP1 was knocked down in cells with stable MDGA2 over-expression by short interference RNA (siRNA) transfection. (E) Effect on gastric cancer cell growth by different levels of MDGA2 and DMAP1. Cell growth was monitored by the xCelligence system. Data shown are mean±SD. (F) Effect of different levels of MDGA2 and DMAP1 on colony formation ability of gastric cancer cells. *p<0.05, **p<0.001, ***p<0.0001.
MDGA2 induces p53/p21 signalling cascade
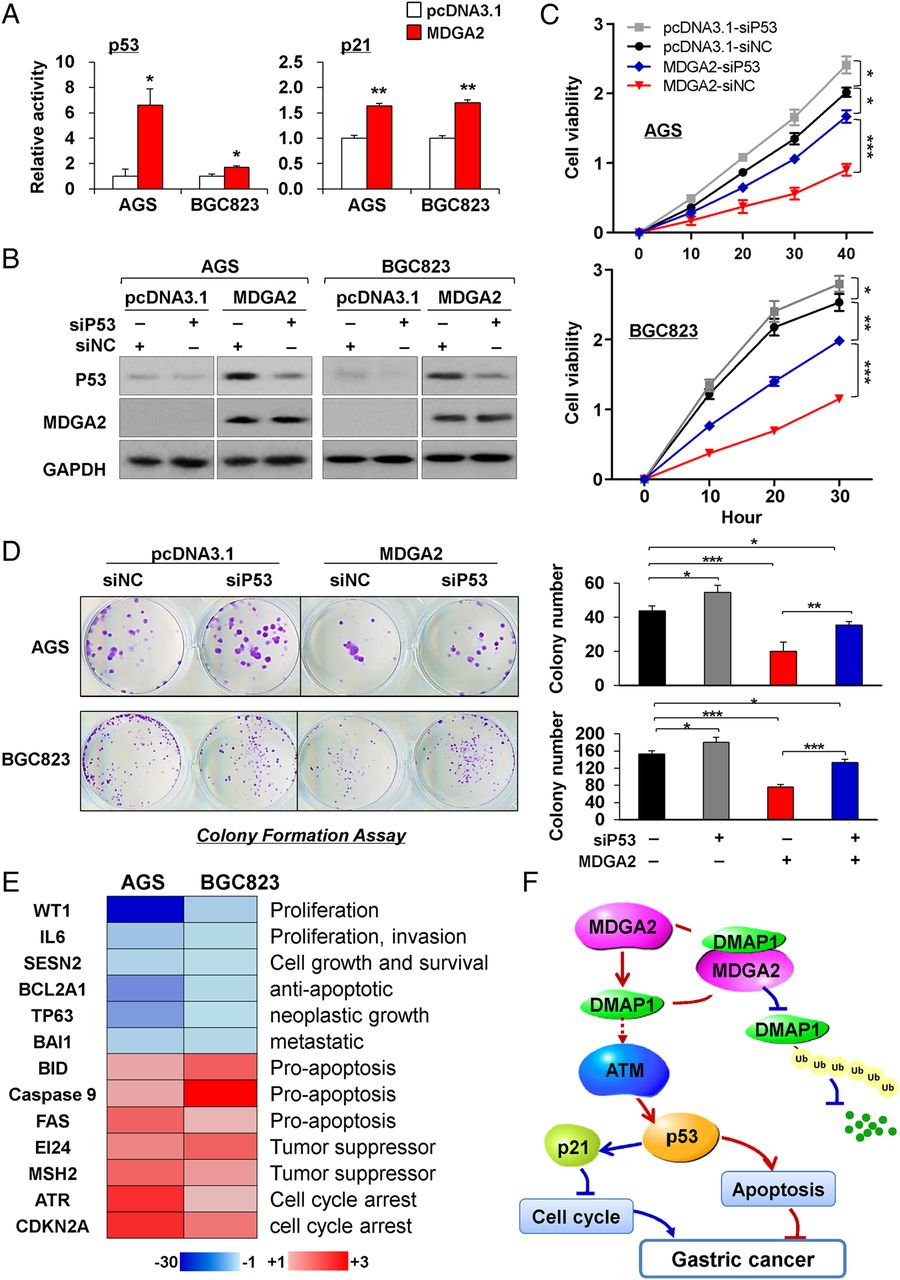
To understand the molecular basis of the tumour suppressive property of MDGA2, we performed luciferase reporter assays to assess the effect of MDGA2 on the activities of p53, p21, NF-κB, MAPK and Wnt signalling pathways. MDGA2 increased p53 and p21 luciferase reporter activities in both AGS and BGC823 cells, but not other pathways (figure 7A). We thus tested whether p53 knockdown could blunt the effect of MDGA2 overexpression. Our results show that p53 knockdown in AGS and BGC823 cells transfected with MDGA2 could partially block the effects of MDGA2 on cell growth, as evidenced by cell viability and colony formation assays (figure 7B–D). To further determine the downstream mediators of p53 signalling derived by MDGA2, gene expression profiles in MDGA2 stably transfected AGS and BGC823 cells were analysed by p53 signalling pathway PCR array. When compared with empty vector-transfected cells, MDGA2 modulated the expression of p53 signalling-related genes involved in apoptosis, proliferation, cell cycle and tumour suppressive function (figure 7E). In addition, knockdown DMAP1 by siDMAP1 transfection in AGS and BGC823 cells with stable MDGA2 overexpression partially decreased the protein expression of p53 and p21 as determined by western blot analysis (see online supplementary figure S2), suggesting that MDGA2 induces p53/p21 signalling cascade partly depending on DMAP1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MDGA2 induces p53/p21 signalling pathway. (A) Luciferase reporter assays showed that p53 and p21 were significantly activated by MDGA2 expression. Date are mean±SD. *p<0.05, **p<0.001. (B) p53 knockdown by short interference RNA (siRNA) in cells with stable MDGA2 transfection was examined by western blot analysis. (C) p53 knockdown in AGS and BGC823 cells transfected with MDGA2 partially blunts the effects of MDGA2 overexpression on cell growth, as evidenced by cell viability and (D) colony formation assays. Data shown are mean±SD. *p<0.05, **p<0.01, ***p<0.005. (E) Downstream targets of MDGA2 identified by p53 signalling pathway PCR array. (F) Schematic illustration of the molecular mechanism of MDGA2 in gastric cancer. MDGA2 binds to DNA methyltransferase 1 associated protein 1 (DMAP1) to stabilise DMAP1 by inhibiting its ubiquitin-mediated degradation, which subsequently activates the ataxia telangiectasia mutated (ATM)/p53/p21 signalling pathway to mediate the tumour suppressive function by inhibiting cell cycle progression and promoting apoptosis. Besides activation of p21 and p53, upregulation of p21 (figure 3C), ATM and p53 (figure 5G) by MDGA2 was well confirmed at the protein level by western blot analysis.
Discussion
In this study we have shown that MDGA2 is commonly silenced or downregulated in gastric cancer cell lines and primary gastric cancers due to promoter hypermethylation. We further investigated the clinical importance of MDGA2 methylation in 218 patients with gastric cancer and found that promoter hypermethylation of MDGA2 was detected in 62.4% of patients with primary gastric cancer. MDGA2 methylation was an independent risk factor of poor survival in patients with gastric cancer by multivariate Cox regression analysis (RR 1.85, p=0.005). The disease-free survival of patients with MDGA2 methylation was significantly shorter than that of other patients with gastric cancer (median survival time 1.59 years vs 4.87 years, p=0.001; figure 2D) by Kaplan–Meier survival curve analysis. In particular, methylation was significantly associated with shorter survival for patients with stage I/II gastric cancer (p=0.024, figure 2E). The clinical outcome of gastric cancer varies greatly depending on the aggressiveness of individual tumours. The most important clinical prognostic indicator of disease outcome is TNM staging. Nevertheless, many patients experience disease recurrence following radical surgery. Although adjuvant chemotherapy may benefit patients with TNM stage I/II gastric cancer, its role remains controversial due to the lack of data showing a definite benefit in this group of patients. Thus, additional prognostic biomarkers may provide better risk assessment that can guide personalised chemotherapy. Promoter methylation has been reported as a promising predictive biomarker in gastric cancers.7 ,10–12 Our results suggest that MDGA2 hypermethylation may serve as a valuable new prognostic marker for patients with early gastric cancer. MDGA2 was found to be commonly downregulated in patients with gastric cancer. This also implies the importance of the functional loss of MDGA2 by promoter methylation during gastric carcinogenesis.
In this connection, we investigated the function of MDGA2 in gastric cancer both in vitro and in vivo. Ectopic expression of MDGA2 in AGS and BGC823 cells which had silenced MDGA2 due to full promoter methylation significantly suppressed cell viability and colony formation ability compared with empty vector transfection. Conversely, knockdown of MDGA2 in MKN1 cells, which showed high endogenous expression of MDGA2, significantly promoted cell growth. Furthermore, both subcutaneous xenograft and orthotopic stomach implantation models confirmed that MDGA2 significantly attenuated tumorigenicity of gastric cancer cells in nude mice. In keeping with this, significantly fewer proliferative cells and more apoptotic cells were detected in MDGA2-expressing subcutaneous and orthotopic xenograft tumours compared with corresponding controls (figure 4). The mechanism by which MDGA2 suppressed gastric cancer cell growth was mediated by inhibiting G1–S cell cycle transition and inducing cell apoptosis (figure 3). G1 arrest by MDGA2 was associated with the suppression of cyclin D1 and CDK4 as well as the induction of p27 and p21. The cyclin D1/CDK4 complex is a key regulator of the transition through the G1 phase of the cell cycle which governs cell cycle progression at the restriction and late transition points of G1, respectively. p27 is a potent inhibitor of cyclin D/CDK4 activity. The role of p27 as a major player in G1 arrest has been well accepted, and p27 is generally expressed at high levels in quiescent cells.13 It is well known that p53 and its downstream target p21 also play an important role in suppressing G1–S cell cycle transition.14 ,15 The significantly reduced cell proliferation by MDGA2 was confirmed by decreased S phase cells, downregulated proliferation marker PCNA and reduced Ki-67 index. In addition to inhibition of cell proliferation, the growth inhibitory effect of MDGA2 was also related to induction of apoptosis. The mechanisms by which MDGA2 may induce apoptosis in gastric cancer cells include activation of both the intrinsic and extrinsic apoptotic cascades, including cleavage of caspases-8, -9, -7, -3 and PARP. The activated caspase-8 and caspase-9 triggered downstream caspase effectors (caspase-3 and caspase-7), which further stimulated the proteolytic cleavage of PARP to facilitate cellular disassembly during apoptosis.16 ,17 Moreover, we revealed that MDGA2 increased the sensitivity of gastric cancer cells to the chemotherapeutic agent cisplatin. These findings support the notion that MDGA2 is a novel tumour suppressor by suppressing G1–S transition and inducing apoptosis, and may serve as a therapeutic target in gastric cancer.
The significance of MDGA2 on cell migration and invasion was also evaluated. Re-expression of MDGA2 in AGS and BGC823 cells significantly inhibited their migration and invasion abilities. The molecular mechanism by which MDGA2 exerts its anti-invasive function was shown to be mediated via enhancing E-cadherin expression and inhibiting N-cadherin, Vimentin, Snail and Slug expression. E-cadherin functions as an invasion suppressor while N-cadherin, Vimentin, Snail and Slug promote cell motility and invasion in cancers.18–22 Taken together, MDGA2 inhibited cell migration and invasion through modulating key elements of EMT such as E-cadherin, N-cadherin, Vimentin, Snail and Slug which contribute to its tumour suppressive effect in gastric cancer.
To understand the molecular basis of the role of MDGA2, we performed Co-IP of MDGA2 followed by protein sequencing for the identification of the MDGA2 interacting partner. DMAP1 was identified as a potential functional partner interacting with MDGA2 (figure 5). The direct interaction between MDGA2 and DMAP1 was confirmed by western blot analysis of Co-IP products as well as by GST pull-down assay using purified MDGA2 and DMAP1 recombinant proteins in cell-free conditions. It is known that DMAP1 is an essential regulator of the activity and function of ATM9; the interaction of MDGA2 and DMAP1 was further confirmed by the upregulation of ATM following ectopic expression of MDGA2 in gastric cancer cells and tumour xenografts. DMAP1 was found to be upregulated following overexpression of MDGA2 and downregulated following MDGA2 knockdown. Upregulation of DMAP1 by MDGA2 was mediated through reducing the ubiquitination/degradation of DMAP1 in gastric cancer cells. Moreover, MDGA2 was co-localised in the cytoplasm with DMAP1 by confocal immunofluorescence assay. Conversely, MDGA2 expression could not be altered by DMAP1 overexpression or knockdown in AGS and BGC823 cells (figure 6). These results collectively suggest that DMAP1 is a direct downstream interacting partner of MDGA2.
We then examined the functional role of DMAP1 in gastric cancer and revealed that DMAP1 significantly suppressed gastric cell growth. In keeping with our findings, DMAP1 was reported to suppress tumour growth in neuroblastoma,23 signifying the tumour suppressive role of DMAP1 in human cancer. We further evaluated whether the tumour suppressive function of MDGA2 is dependent on DMAP1, and observed that the tumour suppressive effect of MDGA2 was significantly blunted due to DMAP1 knockdown in gastric cancer cells, although the effect is still significant compared with the cells without MDGA2/DMAP1 expression. The tumour suppressive property of MDGA2 is therefore at least in part dependent on DMAP1 in gastric cancer.
Moreover, we elucidated the cancer pathways involved by MDGA2 using luciferase reporter assays and found that MDGA2 activated the p53/p21 signalling pathway. Enhanced protein expression of p53 and p21 by MDGA2 was confirmed in MDGA2 overexpressed cells and xenograft tumours (figures 3C, 5G and 7A). It is known that p53 is a tumour suppressor and the downstream target of ATM,24 which is regulated by DMAP1.9 We revealed that the tumour suppressive role of MDGA2 was partially dependent on p53 (figure 7B–D). To better define the tumour suppressive effect of MDGA2 through activation of p53 in gastric cancer, we examined the downstream consequences on p53 signalling by overexpression of MDGA2 using p53 signalling pathway PCR array in MDGA2-tranfected AGS and BGC823 cells (figure 7E). p53 target genes modulating the apoptosis, cell growth and DNA repair pathways were characterised. We observed that MDGA2-mediated tumour suppressive properties occur through the p53 pathway by downregulation of oncogenic genes (WT1, IL6, SESN2)25–27 and genes associated with neoplastic phenotype (TP63, BAI1),28 ,29 upregulation of tumour suppressors (EI24, MSH2),30 ,31 upregulation of the cell apoptosis regulator BH3 interacting domain death agonist (BID) induced by caspase-8, intrinsic apoptosis initiator caspase-9 and Fas cell surface death receptor16 ,17 and downregulation of the anti-apoptotic gene BCL2A1.32 ,33 These further support our finding that MDGA2 induced apoptosis by activating both the intrinsic and extrinsic apoptotic cascades (figure 3D) and upregulation of ataxia telangiectasia and Rad3 related (ATR) and CDKN2A, which contribute to cell cycle arrest.34 ,35 The regulation of p53/p21 signalling and p53-mediated anti-oncogenic and pro-apoptotic cascade induced by MDGA2 therefore adds further weight to the tumour suppressive effect exerted by MDGA2 in gastric cancer (figure 7F). Moreover, MDGA2 induces the p53/p21 signalling cascade partially dependent on DMAP1 (see online supplementary figure S2).
In summary, we have identified a novel tumour suppressor gene, MDGA2, which is frequently inactivated by promoter methylation in gastric cancer. MDGA2 exerts a tumour suppressive function by cooperating with DMAP1 to activate the p53/p21 signalling cascade. Promoter hypermethylation of MDGA2 represents a prognostic biomarker in patients with early stage gastric cancer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
KW and QL are joint first authors.
Contributors KW, XL, HT, JZ, HW and MYYG performed the experiments. KW, QL and JY analysed the data and wrote the paper. PWYC and EKWN provided human samples. JJYS commented on the study. JY supervised the research.
Funding This project was supported by research funds from RGC GRF Hong Kong (766613), Shenzhen Municipal Science and Technology R&D fund (JCYJ20130401151108652), the China 863 Program (2012AA02A504), the National Natural Science Foundation of China (81272304) and Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics approval was obtained from the clinical research ethics committee of the Sun Yat-sen University of Medical Sciences and the ethics committee of the Chinese University of Hong Kong.
Provenance and peer review Not commissioned; externally peer reviewed.