Article Text

Abstract
Objective Hepatitis E virus (HEV) infection can take chronic courses in immunocompromised patients potentially leading to liver cirrhosis and liver failure. Ribavirin (RBV) is currently the only treatment option for many patients, but treatment failure can occur which has been associated with the appearance of a distinct HEV polymerase mutant (G1634R). Here, we performed a detailed analysis of HEV viral intrahost evolution during chronic hepatitis E infections.
Design Illumina deep sequencing was performed for the detection of intrahost variation in the HEV genome of chronically infected patients. Novel polymerase mutants were investigated in vitro using state-of-the-art HEV cell culture models.
Results Together, these data revealed that (1) viral diversity differed markedly between patients but did not show major intraindividual short-term variations in untreated patients with chronic hepatitis E, (2) RBV therapy was associated with an increase in viral heterogeneity which was reversible when treatment was stopped, (3) the G1634R mutant was detectable as a minor population prior to therapy in patients who subsequently failed to achieve a sustained virological response to RBV therapy and (4) in addition to G1634R further dominant variants in the polymerase region emerged, impacting HEV replication efficiency in vitro.
Conclusions In summary, this first investigation of intrahost HEV population evolution indicates that RBV causes HEV mutagenesis in treated patients and that an emergence of distinct mutants within the viral population occurs during RBV therapy. We also suggest that next-generation sequencing could be useful to guide personalised antiviral strategies.
- HEPATITIS E
- CHRONIC HEPATITIS
- ANTIVIRAL THERAPY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
RNA viruses like hepatitis E virus (HEV) establish populations with high intrahost variability, which enables them to rapidly adapt to changing immune responses.
HEV is the major cause of acute hepatitis, but can also establish chronic infections in immunocompromised patients. Ribavirin (RBV) is currently the only treatment option available.
RBV inhibits HEV replication in vitro by, among other mechanisms, increasing the error rate of the viral RNA-dependent RNA polymerase. A mutation (G1634R) in the polymerase region of HEV can lead to treatment failure during RBV therapy.
What are the new findings?
Viral diversity differed markedly between patients but did not show major intraindividual short-term variations in untreated patients with chronic hepatitis E.
RBV therapy was associated with an increase in viral heterogeneity in all open-reading frames, which was reversible when treatment was stopped.
The G1634R mutant was detectable as a minor population prior to therapy in patients who subsequently failed to achieve a sustained virological response to RBV therapy.
Additional dominant variants in the polymerase emerged during RBV therapy impacting HEV replication efficiency in vitro.
How might it impact on clinical practice in the foreseeable future?
Investigation of HEV intrahost population evolution indicates that RBV causes HEV mutagenesis in treated patients and that an emergence of distinct viral populations may occur during RBV therapy.
Next-generation sequencing methods could be diagnostically used to rapidly identify patients at risk for treatment failure and predict therapy outcomes of chronically infected patients in clinics and could be a useful tool for personalised antiviral strategies.
Introduction
Hepatitis E virus (HEV) is a non-enveloped single-stranded RNA virus and a common cause of acute hepatitis worldwide.1 ,2 More than 3 million symptomatic hepatitis E cases occur each year accounting for an estimated 70 000 deaths.1 Four different HEV genotypes infecting humans have been described. HEV genotypes 1 and 2 have been linked with water-borne outbreaks in low/middle-income countries and exclusively infect humans. In contrast, HEV genotypes 3 and 4 can be found in various animal species, with the major route of HEV transmission to humans via consumption of undercooked meat.1 ,3 ,4 It is now well established that prolonged HEV viraemia and even courses of chronic hepatitis E may occur in immunocompromised patients potentially leading to liver cirrhosis and liver failure.5 ,6
Pathogenesis, epidemiology and evolution of RNA viruses are influenced by the composition of the viral population.7 Genetic diversity is achieved by high mutation rates and as a consequence, quasi-species populations are generated which may allow adaptation to antiviral drugs, potentially inducing resistance or enhanced viral fitness.8 In addition, viral diversity represents a potential mechanism to escape a successful immune response while in turn immune pressure may drive viral evolution.9 For HEV, greater intrahost heterogeneity has been linked with evolution to chronicity.10 The immune pressure on HEV maybe weak in chronic hepatitis E where HEV-specific T-cell responses are barely detectable, but various cytokines and chemokines are elevated in acute and chronic hepatitis E correlating with disease activity and progression of liver disease.10 ,11 However, the mode and tempo of HEV evolution in persistently infected patients undergoing therapy is currently unknown.
Ribavirin (RBV) monotherapy is currently considered as the treatment of choice for patients with chronic hepatitis E.12 ,13 While the majority of patients clear HEV, after 3–5 months of RBV therapy cases of on-treatment failures or post-treatment relapses have been reported.14–16 Recently, the selection of a distinct HEV mutant in the C-terminal region of the HEV polymerase (G1634R) during RBV therapy was reported. The G1634R mutant was still sensitive to RBV but conferred an enhanced replication fitness to the virus in vitro.17 Consequently, the selection of the G1634R mutant likely contributed to treatment failure. It is currently unknown if a minor part of the viral population harboured this particular mutation prior to therapy commencement.
RBV inhibits HEV replication in vitro by a depletion of cellular GTP pools.18 Additional modes of actions of RBV might be important in patients as inosine-5′-monophosphate dehydrogenase (IMPDH) inhibition by mycophenolic acid does not always prevent chronicity in HEV-infected solid organ transplant (SOT) recipients.6 Intrahost populations of RNA viruses are believed to exist at the edge of a genomic error threshold, and exceeding the threshold by RBV-induced additional mutations could lead to a sequence of error catastrophes resulting in viral extinction.19 ,20 Here, we hypothesised that RBV-induced increased mutagenesis may be implicated in HEV treatment outcomes as has been shown for various other viruses.21
The aims of the current study were to investigate HEV genome evolution in all open-reading frames (ORFs) prior to and during RBV therapy in patients with chronic hepatitis by Illumina deep sequencing. Furthermore, we wanted to determine if the G1634R mutant was present already before therapy as a minor population in patients not achieving a sustained virological response to RBV therapy. Finally, we aimed to explore if additional amino acid changes were selected during therapy and if these mutations had an impact on HEV replication in vitro.
Subjects, materials and methods
Study subjects
All patients were recruited at Hannover Medical School between 2008 and 2015. Overall, 12 patients with chronic hepatitis E were included, for whom at least 2 stored plasma samples with detectable HEV RNA were available. In addition, for RBV-treated patients, we aimed to study at least one viraemic sample during treatment and in patients not achieving a sustained virological response, a post-treatment HEV RNA-positive sample was required to be available. RBV was administered orally twice daily with an initial daily dose of 600–1000 mg, depending on the patients' haemoglobin level and comorbidities, as previously described.15 Dose reductions were performed if haemoglobin levels declined and/or patients developed symptoms associated with anaemia. At each visit, aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyltranspeptidase (GGT) and HEV RNA viraemia were determined. Anti-HEV status was determined using Wantai HEV IgG ELISA (Beijing, China). The study protocol was in line with the ethical guidelines of the Institutional Review Committee. The study was approved by the ethics committee of Hannover Medical School in Hannover, Germany (record 930–2011), and it conforms to the ethical guidelines of the 1975 Declaration of Helsinki. All patients gave written informed consent to participate in this study. Healthy volunteers were also recruited from Hannover Medical School.
Cell culture
Human Huh7.5 hepatoma cells (kindly provided by Professor Charles Rice) were kept in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 1% non-essential amino acids (Invitrogen), 100 µg/mL of streptomycin (Invitrogen) and 100 IU/mL of penicillin. Cells were kept at 37°C in a 5% (v/v) CO2 incubator.
HEV constructs, cloning and in vitro transcription
A plasmid construct containing a subgenomic HEV Kernow-C1 p6 clone (GenBank Accession Number: JQ679013) sequence coupled with a Gaussia luciferase reporter gene (p6-Gluc) was used to generate HEV in vitro transcripts as previously described with an additional capping step (m7G Cap Analog, Promega, Madison, Wisconsin, USA).22 ,23 Single-nucleotide variations were introduced using site-directed mutagenesis PCR or gBlocks Gene Fragments (IDT, Coralville, Iowa, USA) (detailed information and primer sequences available on request).
HEV replication assay
HEV subgenomic RNAs were transfected into cells via electroporation as previously described.24 In brief, 5×106 cells/mL Huh7.5 cells in 400 µL Cytomix containing 2 mM ATP and 5 mM glutathione were mixed with 3 µg of p6-Luc subgenomic HEV RNA. Electroporation was carried out with a Gene Pulser system (Bio-Rad, Munich, Germany). Cells were immediately transferred to 12.1 mL of DMEM complete, and 50 µL containing 2×104 cells/well was seeded onto 96-well plates. After 4 h, 50 µL DMEM complete containing RBV at indicated concentrations, or DMEM complete only, was added. Viral replication was determined by measuring luciferase activity in the supernatant of 96-well plates 72 h after transfection.
Luciferase reporter assay
HEV subgenomic replicon (SGR) replication was assessed as described before.25 In brief, 20 µL of supernatant was added per well of a 96-well white, flat-bottom microplate followed by the detection of luminescence for HEV encoding Gaussia luciferase using a microplate reader (CentroXS3 LB960, Berthold Technologies, Bad Wildbad, Germany) using coelenterazine as a substrate.
Statistics
Data were analysed using GraphPad Prism V.6.0b (GraphPad Software, La Jolla, California, USA). Comparisons of single-nucleotide variants (SNV; a single-nucleotide variation among an individual) between RBV-treated patients and patients who did not receive RBV were performed using Mann–Whitney U test. p Values of <0.05 were considered to be significant (*p<0.05; **p<0.01; ***p<0.001 or ****p<0.0001). GraphPad Prism 5 software was used for dose–response curve calculation.
Results
Patient characteristics
We studied 12 selected patients with chronic HEV genotype 3 infections (8 patients with genotype 3c, 3 patients with genotype 3f and 1 patient with genotype 3e). The median age of the patient cohort was 46 years (range 14–70 years) and eight patients were male. Eleven of the 12 patients were organ transplant recipients (table 1). Three patients cleared HEV after reduction of immunosuppressive medication while antiviral therapy with RBV was initiated in nine subjects. RBV treatment was successful in five patients and failed in four organ transplant recipients (patient # 1–4). Initial immunosuppressive medications are listed in table 1 and included corticosteroids, proliferation inhibitors, calcineurin inhibitors and mechanistic Target of Rapamycin (mTOR) inhibitors. Immunosuppressive regimens were adjusted over time depending on the clinical parameters of the patients.
Characteristics of chronic infected hepatitis E virus (HEV) patients
Evolution of HEV intrahost populations in patients with chronic hepatitis E
RNA viruses exist as a diverse population of genetically related but distinct variants. For HEV, viral heterogeneity determined at a single time point has been associated with the outcome of infection.10 We here first performed an analysis of intrahost HEV viral population evolution by Illumina deep sequencing of the HEV ORF1 region encoding for the RNA-dependent RNA polymerase (RdRp) of at least two time points in patients not receiving antiviral treatment with RBV (figure 1). Significant interindividual variability of both synonymous (silent) as well as non-synonymous (amino acid replacement) SNVs was evident (figure 1). However, within a given patient (#1, #4, #8, #9, #10, #11), the HEV population composition was rather stable over several weeks or even months for the majority of patients before on-set of RBV treatment as well as in patients who never received antiviral therapy.

Changes in total number of sites exhibiting variations in the hepatitis E virus (HEV) open-reading frame 1 (ORF1) region without the influence of ribavirin (RBV) in chronically infected HEV patients over time. The time course of the appearance of non-synonymous (black lines) and synonymous (grey lines) single-nucleotide variations (SNVs) in six chronically infected patients included in the study is depicted.
Effect of RBV on the heterogeneity in the HEV ORF1 encoding for the RNA-dependent RNA polymerase
To ascertain whether RBV therapy has as an influence on viral heterogeneity, we compared differences in the number of sites exhibiting non-synonymous and synonymous SNVs between RBV-treated and non-treated patients with chronic hepatitis E via next-generation sequencing methods. Indeed, patients receiving RBV showed higher frequencies of both synonymous (figure 2A) and non-synonymous (figure 2B) SNVs compared with samples drawn when patients were untreated. This finding indicates that RBV therapy leads to an increased nucleotide substitution rate in patients with HEV. We next analysed the long-term evolution of the total number of synonymous and non-synonymous SNV in three patients not achieving a sustained virological response (patients #1, #2 and #4). For these patients, samples were available before, during and after RBV medication (figure 3). HEV RNA copy numbers during the course of infection ranged for patient #1 from 5.6×105 to 1.09×107, for patient #2 from 1.66×104 to 1.2×107 and for patient #4 from 1.54×104 to 6.75×108 RNA copies/mL, respectively. Overall, there was a progressive increase in both synonymous and non-synonymous SNVs during RBV medication with higher changes in the number of sites exhibiting substitutions for synonymous SNV (figure 3). In patient #1, a marked SNV decline appeared after RBV was stopped and in patient #2 a gradual decline became evident 3–6 months after treatment for both, synonymous as well as non-synonymous SNV (figure 3A, B). Patient #4 showed a marked increase in SNVs in particular during a second course of RBV therapy; however, no post-treatment samples were available in this case (figure 3C). In all three patients, the occurrence of nucleotide substitutions was mainly caused by transitions, that is, a purine–purine or pyrimidine–pyrimidine exchange, and only to a lesser extent due to nucleotide transversions (purine–pyrimidine or pyrimidine–purine substitution, figure 3A–C). In summary, the analysis of viral evolution during and after therapy in individual patients shows that increasing SNVs over time correlate with continued administration of RBV. Furthermore, RBV-induced mutagenesis seemed to be reversible when treatment was stopped.
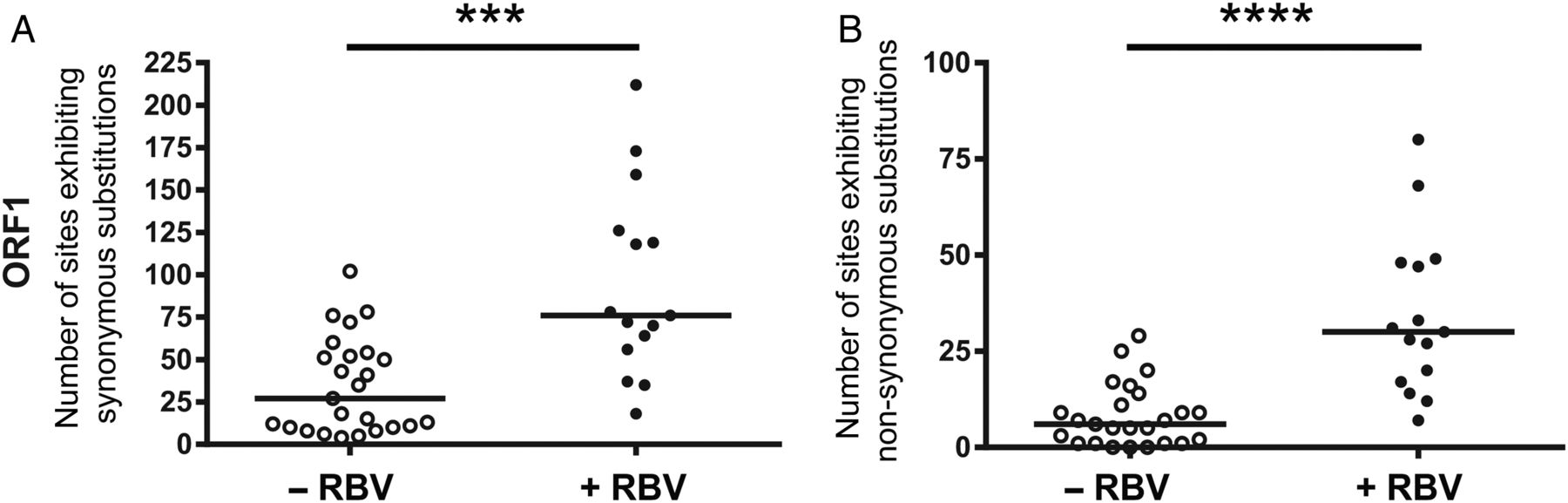
Effect of ribavirin (RBV) on the heterogeneity of hepatitis E virus (HEV) open-reading frame 1 (ORF1) region in the viral intrahost population in chronically infected patients. The total numbers of nucleotide sites exhibiting synonymous (A) as well as non-synonymous (B) single-nucleotide variations (SNVs) identified in the hepatitis E viral intrahost population in solid organ transplant patients are compared. Open circles represent analyses of available samples of patients before receiving RBV or before and during other antiviral treatment. Black dots show the number of SNVs in HEV ORF1 identified in patients during RBV treatment. Horizontal bars indicate the median, significance was tested with the Mann–Whitney U test (***p<0.001; ****p<0.0001).

Course of number of sites exhibiting nucleotide substitutions over time in three chronically hepatitis E virus (HEV) infected solid organ transplant recipients experiencing ribavirin (RBV) treatment failure. Shown are the total numbers of synonymous (solid black lines) and non-synonymous (dashed black lines) single-nucleotide variations (SNVs), as well as the numbers of nucleotide transitions (solid grey lines) and transversions (dashed grey lines) in the HEV open-reading frame 1 (ORF1) identified in three chronically infected patients (A, patient #1; B, patient #2; C, patient #4). Black bars indicate duration of RBV treatment starting with the first RBV dose at month 0.
Time course of distinct non-synonymous SNVs in patients not achieving a sustained virological response to RBV therapy
We next analysed, if viral heterogeneity and detection of the G1634R mutant via deep sequencing would be predictive for subsequent responses to RBV treatment. There was no significant difference between the numbers of synonymous and non-synonymous SNVs detected in responding patients and patients not achieving a sustained virological response to RBV therapy (see online supplementary figure S1). In most patients who cleared the HEV infection, the G1634R mutation could not be identified or was present at very low levels in the viral population. In two patients, the arginine at position 1634 was already the dominant strain from the beginning onwards. However, the G1634R mutant that has been selected during therapy in patients with treatment failure was detectable as minor population before therapy in patient #1 and patient #2 (figure 4). Deep sequencing revealed a gradual increase in the G1634R variant with continued RBV therapy until month 11, where it comprises more than two thirds of the HEV population (figure 4A). In patient #2, the G1634R variant also gradually increased with progression of RBV therapy until the ninth month (figure 4B). Of note, the relative dominance of G1634R declined around 3 months after RBV medication was stopped and decreased to one-third of the population (figure 4B). G1634R was the dominant strain in patient #3 already before therapy which did not change during treatment (data not shown), while in patient #4 the G1634R variant was only detectable in the population at month 11 and only at as very low frequency of about 3.4% (data not shown). Thus, the overall HEV heterogeneity does not seem to be associated with response to RBV therapy. However, variants possibly being associated with treatment failure may already be detectable before antiviral therapy as minor populations.
Supplemental material

Time course of non-synonymous single-nucleotide variations at certain positions of the hepatitis E virus (HEV) open-reading frame 1 (ORF1) in patient samples. Changes in nucleotide frequencies (x-axes) resulting in alterations of the predominant amino acids over monitoring time (y-axes) of chronically infected patients are depicted at amino acid positions indicated above the plots. The altered nucleotides in the coding triplets are underlined. White bars indicate proportions of adenine, light grey bars indicate proportions of guanine, dark grey bars represent cytosine and black bars show amount of thymine. (A) Three positions with a change in the dominant amino acid were identified for patient #1, four non-synonymous SNVs were found in patient #2 (B) and four in patient #4 (C). Black vertical lines indicate course of administration of ribavirin (RBV).
In addition to G1634R, two further mutations (K1383N and Y1587F) appeared in patient #1 in the C-terminal region of the HEV ORF1 at month 5 of therapy (figure 4A). The variant Y1587F did not exist and variant K1383N was present at extremely low frequencies prior to initiation of RBV medication (figure 4A). For the second non-responder (patient #2), we also found three additional amino acid changes in the HEV polymerase over time (K1383N, D1384G and V1479I; figure 4B). Similar to G1634R, the selection of these variants was reversible when RBV was stopped. Figure 4C shows the time course of SNV of the only non-responder not harbouring the G1634R (patient #4). In patient #4, we observed three amino acid changes previously detected in patient #1 (K1383N and Y1587F) and patient #2 (K1383N and D1384G). Additionally, one variant (K1398R) was identified which was not detectable in the other patients (figure 4). A linkage analysis using clonal sequencing methods employing amplicons obtained from time points with highest viral heterogeneity revealed different combinations of the identified variations on individual viral genomes (see online supplementary figure S2) that were present at different frequencies. In viral populations of all three patients, the wild-type sequences were no longer detectable anymore. For patient #1, viral genomes harbouring all three mutations could be detected (see online supplementary figure S2A), whereas for patient #2 and patient #4 several combinations of mutations were detected, but no viral genomes containing all four amino acid changes (see online supplementary figure S2B, C). In summary, the G1634R mutation was already detectable as a minor variant prior to therapy and became dominant during RBV administration. Furthermore, additional amino acid changes in ORF1 could be identified that emerged in various combinations in the viral population.
RBV sensitivity and viral replication of HEV subgenomic replicon harbouring identified SNVs
In our previous study, it was demonstrated that the G1634R mutant was still sensitive to RBV but conferred an enhanced replication fitness to the virus in vitro.17 Here, we first confirmed that the previously described G1634R mutant (EC50=12. 9 µM, 95% CI 11.91 to 13.98) introduced into a genotype 3 replicon has comparable RBV sensitivity to the HEVp6 wild-type (wt) replicon (EC50=13.61 µM, 95% CI 12.35 to 15.00) (figure 5A). However, the G1634R mutation resulted in an increase in luminescence signal compared with the wild-type construct suggesting increased viral RNA replication (figure 5B). RBV was further used as control for active viral RNA replication in this assay setup. Consequently, after revealing two additional dominant amino acid changes close to the G1634R site in patient #1 using deep sequencing, we also introduced all three mutations (K1383N/Y1587F/G1634R) into the HEVp6 replicon to investigate the RBV sensitivity and viral replication compared with the HEVp6 wild-type replicon and HEVp6 G1634R replicon. Interestingly, the K1383N/Y1587F/G1634R combination showed an increased sensitivity to RBV when compared with the wild-type or G1634R mutant (EC50=4.95 µM, 95% CI 4.47 to 5.49) (figure 5A). Similar viral replication could be detected for the K1383N/Y1587F/G1634R construct compared with the wild-type construct after transfecting Huh7.5 cells (figure 5B). A comparable phenotype could be observed for the mutations found in patient #2 (K1383N/D1384G/V1479I/G1634R, EC50=3.03 µM, 95% CI 2.80 to 3.28), whereas a HEV construct harbouring the mutations identified in patient #4 (K1383N/D1384G/K1398R/Y1587F, EC50=11.98 µM, 95% CI 10.39 to 13.82) showed replication levels as well as RBV sensitivity like the wild-type replicon (figure 5A, B). In summary, next to the G1634R additional ORF1 mutations could be identified that abrogated the improved replication fitness and showed an increased RBV sensitivity.

Ribavirin (RBV) sensitivity and replication fitness of hepatitis E virus (HEV) subgenomic replicon SGR wild-type (wt) and mutants harbouring the single-nucleotide variations identified in patients experiencing RBV treatment failure. (A) Antiviral activity of RBV against HEVp6 wt replicon (solid line, EC50=13.61 µM, 95% CI 12.35 to 15.00), the G1634R mutant (dashed line, EC50=12.9 µM, 95% CI 11.91 to 13.98), the K1383N/Y1587F/G1634R mutant (dotted line, EC50=4.95 µM, 95% CI 4.47 to 5.49), the K1383N/D1384G/K1398R/Y1587F mutant (dotdashed line, EC50=11.98 µM, 95% CI 10.39 to 13.82) and the K1383N/D1384G/V1479I/G1634R mutant (dotdotdashed line, EC50=3.03 µM, 95% CI 2.80 to 3.28) replicons in Huh7.5. (B) Luminescence read-out of Huh7.5 cells transfected with the five constructs 72 h post-transfection. As a control for replication-specific read-out, the wt replicon was treated with 62.5 µM RBV.
Effect of RBV on the heterogeneity of HEV in ORF2 and ORF3 genome regions
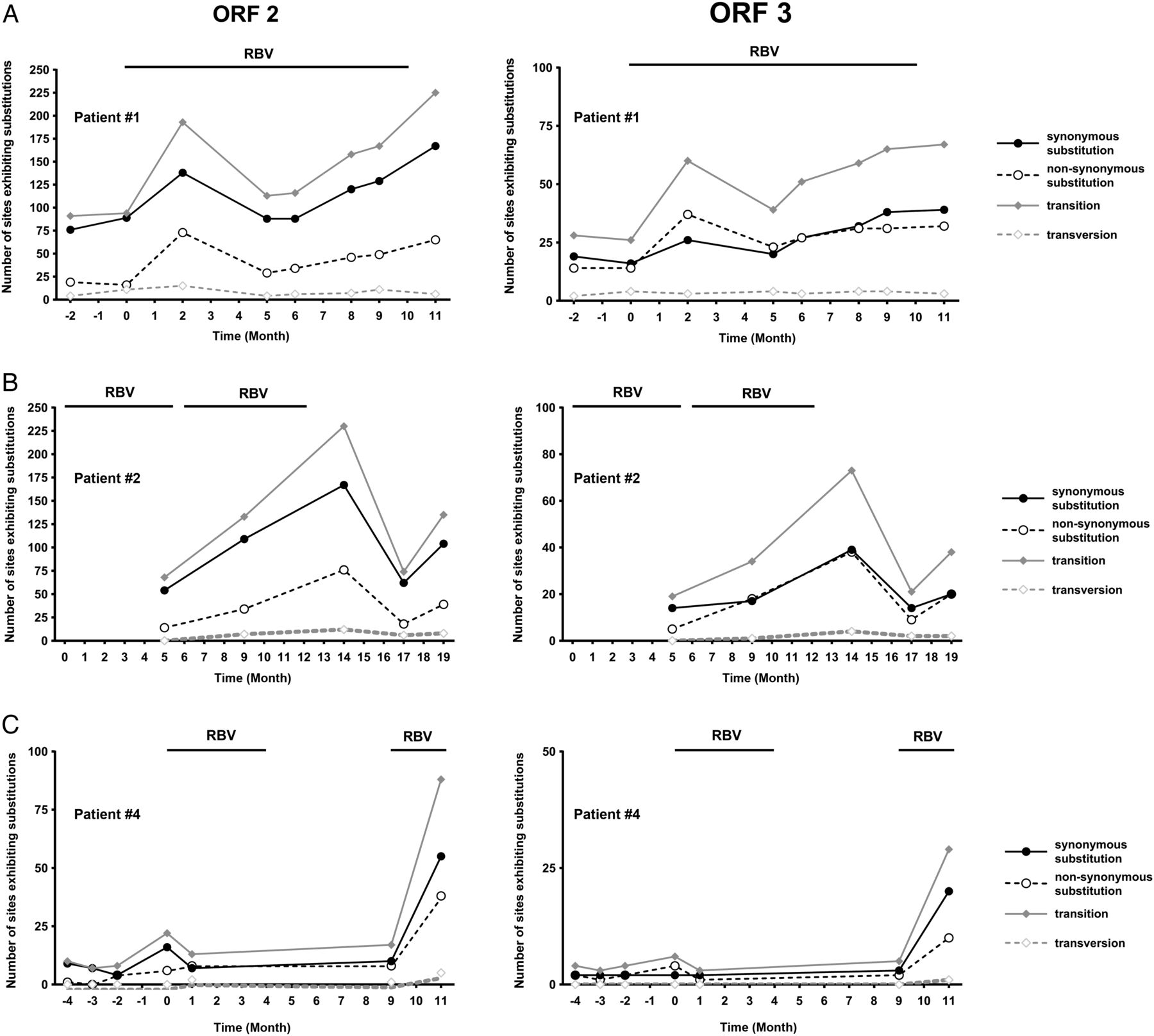
To extend the RBV-induced mutagenesis analysis to HEV genome regions other than the RdRp in ORF1, we next performed deep-sequencing approach of ORF2 and ORF3 regions with RBV-treated and non-treated patients with chronic hepatitis E (figure 6). In line with the findings made for ORF1, patients receiving RBV showed higher numbers of sites exhibiting both non-synonymous (figure 6A, C) and synonymous (figure 6B, D) in ORF2 and ORF3, respectively. The long-term evolution of the total number of SNVs in ORF2 and ORF3 for the three patients not receiving a sustained virological response demonstrated an increase in SNVs over time which also correlated with the administration of RBV and findings made for ORF1 (figure 7; compare figure 3). As the amplicon size for each ORF reference was different, we performed a normalised comparison of the number of sites exhibiting SNVs as well as average frequencies of SNVs in percent between ORF1, 2 and 3 (see online supplementary figure S3A–D). These results show that in all genome regions comparable mutagenic effects of RBV can be observed. Only the average frequencies of synonymous substitutions in ORF1 under RBV treatment were not significantly increased compared with the non-treated patient samples (see online supplementary figure S3C). Monitoring the changes in nucleotide frequencies in ORF2 and ORF3 resulting in alterations of the predominant amino acid over time in the three chronically infected patients identified four mutations in the ORF2 for patient #2 (P25S, G71R, P95S and V245I) and patient #4 (G38S, A64T, P79S and T324S) and additionally one mutation in the ORF3 of patient #4 (S82N) (see online supplementary figure S4). In summary, in line with the results described for ORF1, the increase of SNVs in individual patients receiving RBV could also be observed in the HEV ORF2 and ORF3.
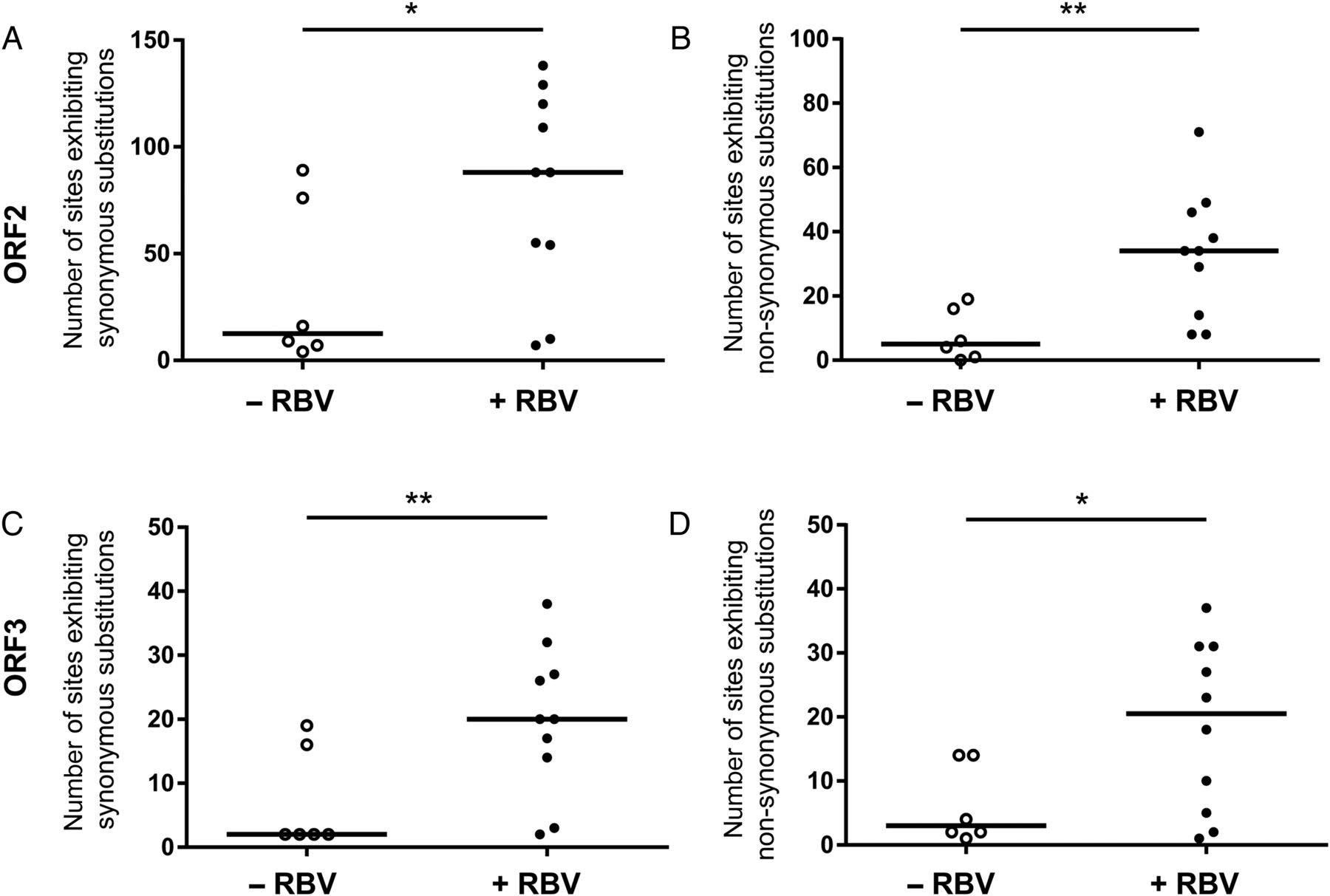
Effect of ribavirin (RBV) on the heterogeneity of hepatitis E virus (HEV) open-reading frame 2 (ORF2) and ORF3 in the viral intrahost population in chronically infected patients experiencing RBV treatment failure. The total numbers of sites exhibiting synonymous (A and C) as well as non-synonymous (B and D) single-nucleotide variations (SNVs) identified in ORF2 (A and B) and ORF3 (C and D) of the hepatitis E viral intrahost population found in patient #1, patient #2 and patient #4 at time points of available serum samples before (open circles) and during RBV treatment (black dots) are compared. Horizontal bars indicate the median, significance was tested with the Mann–Whitney U test (*p<0.05; **p<0.01).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Course of number of sites exhibiting nucleotide substitutions over time in three chronically hepatitis E virus (HEV) infected solid organ transplant recipients experiencing ribavirin (RBV) treatment failure. Shown are the total numbers of synonymous (solid black lines) and non-synonymous (dashed black lines) single-nucleotide variations (SNVs), as well as the numbers of nucleotide transitions (solid grey lines) and transversions (dashed grey lines) in the HEV ORF2 (left panels) and ORF3 (right panels) identified in three chronically infected patients not achieving a sustained virological response (A, patient #1; B, patient #2; C, patient #4). Black bars indicate duration of RBV treatment starting with the first RBV dose at month 0.
Discussion
Chronic hepatitis E is an increasing problem in immunocompromised patients, with RBV being the only treatment option in many cases. Recently, the emergence of a distinct HEV polymerase mutation (G1634R) during RBV therapy of chronic HEV was reported demonstrating enhanced replication dynamics, which likely contributed to treatment failure and a poor clinical long-term outcome.17 In that study, selection of the HEV-G1634R mutant was identified by standard population sequencing. Here, we performed a detailed analysis of intrahost HEV viral population evolution via deep sequencing of the HEV genome in a larger cohort of patients at multiple time points. We show that (1) the HEV population composition was stable over several weeks or even months in most patients with chronic HEV although it was variable between patients, (2) RBV therapy was associated with a marked increase in viral population heterogeneity and (3) in addition to G1634R further mutants comprising the majority of the viral population emerged during RBV therapy, which impacted HEV replication efficiency.
HEV generally causes acute infections with rather short durations of viraemia in infected hosts. Prolonged courses of HEV infections or even chronicity only occur with the introduction of immunosuppressive medical therapies. Yet, the evolution of the HEV population during prolonged viraemia has never been previously studied. The potential importance of HEV heterogeneity for both development of chronic infections as well as fibrosis progression has been suggested previously in an elegant study from Toulouse.10 In line with that study, we here also observed extensive variability in the complexity of the HEV population between different patients. However, HEV diversity remained rather stable within a given patient followed for several months. This observation indicates a low level of immune pressure in these patients with different types of immunosuppressive medications. This is in agreement with our earlier finding that HEV-specific T-cell responses are rather weak in chronically infected solid organ transplant recipients while much stronger T-cell responses can be observed in immunocompetent patients with acute HEV genotype 3 infections.11 More and larger studies are required to better define the importance of viral diversity for disease progression once chronic hepatitis E infection has been established.
RBV displays antiviral activities against a broad range of RNA and DNA viruses.26 Inhibition of HEV replication in vitro by RBV has been shown to be mediated by a depletion of cellular GTP pools.18 However, additional modes of actions of RBV might be important as patients can develop chronic hepatitis E even in the presence of mycophenolic acid, which also inhibits IMPDH and thereby alters cellular GTP pools.6 We here suggest that RBV-induced viral mutagenesis is important for HEV therapy outcomes. Various in vitro studies showed that RBV exhibits mutagenic properties against different viruses such as GB virus B, poliovirus, Hantaan virus and foot-and-mouth disease virus.27–30 Yet, few studies presented in vivo evidence that RBV-induced mutagenesis is of relevance. The best studied example is hepatitis C where partially contradicting results were observed by direct sequencing of PCR products of about 30 clones per time point.31–34 However, deep sequencing of patients being treated with RBV monotherapy revealed that, even though no overall increase of nucleotide substitutions of hepatitis C occurred during therapy, a mutagenic effect was demonstrated with more frequent G-to-A and C-to-U nucleotide transitions.35 We here demonstrate that RBV therapy is associated with an increased HEV heterogeneity indicating that nucleotide substitutions accumulate over time during treatment. Of note, this diversity partially declined again in two patients upon RBV therapy cessation, further supporting that RBV is inducing HEV mutagenesis and that this effect is reversible. Similar to the findings for HCV, we also observed an excess of transitions compared with transversions in the HEV polymerase and, in addition, we observed a surplus of synonymous versus non-synonymous substitutions in the majority of the cases (figures 1, 3 and 7).35 Nucleotide changes occurred throughout the studied HEV regions in ORF1, ORF2 and ORF3 with no preferential location of SNVs—which is also in line with findings in hepatitis C where respective SNVs were not limited to specific non-structural proteins as suggested previously but occurred ubiquitously in the HCV genome.32 ,35 Overall, our findings could indicate that the RBV-induced mutagenesis of HEV may lead to an exceeding of a genomic error threshold followed by lethal mutagenesis as it has been shown for other viruses.19 Still, further studies in more patients are needed which should try to determine correlations between viral declines and RBV-induced viral diversity.
The HEV G1634R mutation was selected during RBV therapy in two patients not achieving a sustained HEV clearance and this mutant had a better replication capacity in vitro.17 We here provide a more detailed analysis of how this variant became dominant in the viral population over time. Importantly, deep sequencing revealed that the G1634R mutant was already present as a minor population before therapy in patient #1 and the proportion gradually increased until treatment month 11, when G1634R was the dominant strain representing more than two thirds of the HEV population (figure 4A). The mutant was also detectable before treatment in patient #2, who failed to achieve a sustained virologic response (SVR), and also in the third patient G1634R was the dominant strain throughout follow-up. Overall, these data extend on our previous observation and suggest that the selection of G1634R may have contributed to treatment failure. Next-generation sequencing maybe used diagnostically to early identify patients at risk for treatment failure who may require alternative treatment approaches.
Importantly, next-generation sequencing revealed that the situation might be even more complex, as various additional amino acid changes were detected in some patients (figure 4). These variants may have additional implications for the replication efficiency as demonstrated in figure 5. Viral constructs harbouring additional mutations in the ORF1 region next to the G1634R mutation abrogated the improved replication fitness of G1634R viruses and showed an increased RBV sensitivity. One has to keep in mind that the p6 HEV clone is a cell culture adapted isolate, which might not reflect the individual dominant patient strain found in vivo in a given patient. Furthermore, due to the limitations of Illumina sequencing, whereby no linkage between polymorphisms can be assigned, we also performed a linkage analysis via clonal sequencing methods, where single viral amplicons are cloned into a vector and subsequently sequenced by standard Sanger sequencing. Here, we were able to identify viral genomes harbouring all three SNVs in patient #1. Although no genomes with all four SNVs for patient #2 and patient #4 were detected, different combinations with up to three SNVs were also present in these populations.
The study has several strengths as this was the very first study evaluating viral evolution over time in patients with chronic hepatitis E by deep sequencing. We could study a cohort of patients with and without sustained response to RBV treatment and as single patients could be studied for up to nine different time points. However, limitations need to be considered. Blood sampling was performed in a routine clinical setting and samples were retrospectively analysed based on availability. The number of patients is still limited and a larger cohort needs to be investigated. Moreover, different immunosuppressive regimens were used in different patients and specific drugs may interfere with viral RNA replication and thereby also affect the viral population composition.36 ,37 Finally, the dose of RBV varied between patients and was even adapted during treatment based on tolerability, haemoglobin levels and kidney function. Thus, drug exposure may have differed between individuals.
In summary, this first investigation of HEV intrahost evolution revealed that viral diversity differs between patients but does not show major intraindividual short-term variations in untreated patients indicating that there is no major immune selection pressure on HEV in immunocompromised patients with chronic hepatitis E. Furthermore, we provide strong evidence that RBV causes HEV mutagenesis in treated patients and that an emergence of distinct viral populations may occur during RBV therapy. Next-generation sequencing could be useful to rapidly identify patients at risk for not achieving a sustained virological response. Overall, this study gives novel insights to better understand the pathophysiology of chronic hepatitis E and may guide development of personalised antiviral strategies.
Acknowledgments
The authors thank Charles Rice for Huh7.5 cells, Suzanne Emerson for the HEV p6 clone and Julia Hinzmann for excellent technical support. The authors thank all physicians and nurses involved at Hannover Medical School in treating patients with hepatitis E. The authors are most grateful to all patients who agreed to provide materials for this study.
References
Footnotes
DT and AG contributed equally.
ES, CTB and HW share senior authorship.
Contributors Conception and design of the study: ES and HW, generation, collection, assembly, analysis and interpretation of the data: DT, AG, AR, PB, PVS, SP, AN, BB, RJPB, MPM, MC, CTB, drafting of the manuscript: DT, AG, RJPB, CTB, ES, HW.
Funding This work was supported by a grant from the Robert Koch Institute Berlin, Germany. ES was supported by the Helmholtz Centre for Infection Research and by the German Ministry for Education and Research (BMBF) through a GINAICO grant 16GW0105.
Competing interests None declared.
Ethics approval Ethics committee of Hannover Medical School in Hannover, Germany (record 930-2011).
Provenance and peer review Not commissioned; externally peer reviewed.