Article Text

Abstract
Objective We assessed the effectiveness and safety of daclatasvir (DCV) plus sofosbuvir (SOF), with or without ribavirin (RBV), in a large real-world cohort, including patients with advanced liver disease.
Design Adults with chronic HCV infection at high risk of decompensation or death within 12 months and with no available treatment options were treated in a European compassionate use programme. The recommended regimen was DCV 60 mg plus SOF 400 mg for 24 weeks; RBV addition or shorter duration was allowed at physicians' discretion. The primary endpoint was sustained virological response at post-treatment week 12 (SVR12).
Results Of the 485 evaluable patients, 359 received DCV+SOF and 126 DCV+SOF+RBV. Most patients were men (66%), white (93%) and treatment-experienced (70%). The most frequent HCV genotypes were 1b (36%), 1a (33%) and 3 (21%), and 80% of patients had cirrhosis (42% Child–Pugh B/C; 46% Model for End-Stage Liver Disease score >10). SVR12 (modified intention-to-treat) was achieved by 91% of patients (419/460); 1 patient had virological breakthrough and 13 patients relapsed. Virological failure was not associated with treatment group (adjusted risk difference DCV+SOF minus DCV+SOF+RBV: 1.06%; 95% CI −2.22% to 4.35%). High SVR12 was observed regardless of HCV genotype or cirrhosis, liver transplant or HIV/HCV coinfection status. Twenty eight patients discontinued treatment due to adverse events (n=18) or death (n=10) and 18 died during follow-up. Deaths and most safety events were associated with advanced liver disease and not considered treatment related.
Conclusions DCV+SOF with or without RBV achieved high SVR12 and was well tolerated in a diverse cohort of patients with severe liver disease.
Trial registration number NCT0209966.
- ANTIVIRAL THERAPY
- CHRONIC VIRAL HEPATITIS
- CIRRHOSIS
- HEPATITIS C
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Video abstract
Significance of this study
What is already known on this subject?
All-oral regimens have become the standard of care for treatment of chronic HCV infection.
In phase III studies, multiple HCV patient subgroups treated with daclatasvir (DCV) plus sofosbuvir (SOF), with or without ribavirin (RBV), achieved sustained virological response at post-treatment week 12 (SVR12) rates exceeding 90% after 12 weeks of treatment.
DCV+SOF has been well tolerated in clinical studies, with few treatment-related serious adverse events or treatment discontinuations.
What are the new findings?
This compassionate use programme provides clinically relevant information on the effectiveness and safety of DCV+SOF, with or without RBV, in a large, real-world cohort that included patients who would have been excluded from many phase III studies due to advanced disease or concomitant medical conditions.
SVR12 rates comparable with those reported in phase III studies were achieved, with similar virological efficacy regardless of liver disease stage or the presence of complicating medical conditions.
Treatment was well tolerated; most significant safety events were attributable to progression of advanced liver disease and not considered related to programme therapy.
How might it impact on clinical practice in the foreseeable future?
These findings support the use of DCV+SOF, with or without RBV, in a diverse spectrum of patients with chronic HCV infection, including those with severe liver disease or other medical complications.
Introduction
Oral combinations of direct-acting antivirals (DAAs) have become the standard of care for treating chronic HCV infection.1–4 In clinical trials, rates of sustained virological response at post-treatment week 12 (SVR12) exceeding 90% have been reported for several drug combinations, with safety profiles superior to those of peginterferon-based regimens. However, advanced liver disease and concomitant medical conditions can adversely affect therapeutic responses and complicate interpretation of results. Consequently, patients with such conditions are usually under-represented in clinical trials, and disease states encountered in clinical practice can differ in important ways from those permitted in randomised trials. Community-based programmes offer an important complement to registration studies by providing additional information concerning the therapeutic risk/benefit profile of a new regimen in a broader population.
Daclatasvir (DCV) is a potent, pan-genotypic inhibitor of the HCV NS5A protein; sofosbuvir (SOF) is a pan-genotypic nucleotide analogue inhibitor of the HCV NS5B RNA polymerase.5 ,6 In phase III studies, the 12-week, once-daily oral combination of DCV and SOF, with or without ribavirin (DCV+SOF±RBV), was well tolerated and achieved SVR12 rates exceeding 90% in patients who have been challenging to treat effectively, including those with advanced cirrhosis, HIV/HCV coinfection, HCV genotype 3 infection and HCV recurrence after liver transplant.7–9 These findings led to widespread approval of DCV and SOF for the treatment of chronic HCV infection.
Before the European approval of DCV, a compassionate use programme (CUP) was established to provide early access to DCV, in combination with SOF, with or without RBV, for patients with chronic HCV infection in urgent need of treatment and without therapeutic alternatives. This programme, conducted in a real-world setting, provides additional information concerning the efficacy and safety of DCV+SOF in a large, diverse population with minimal entry restrictions regarding liver disease stage or comorbidities.
Methods
Patients and treatment
The DCV European CUP enrolled patients from 100 centres in Germany, Austria, the Netherlands, Sweden and Norway from April 2014 to April 2015. Eligible patients were ≥18 years of age with chronic HCV infection (any genotype), at high risk of hepatic decompensation or death within 12 months if left untreated and with no available treatment options. Patients with HIV/HCV or HBV/HCV coinfection, hepatocellular carcinoma (HCC) and decompensated cirrhosis were permitted with no restrictions based on Child–Pugh or Model for End-Stage Liver Disease (MELD) score. Patients with HCV recurrence after liver transplantation and patients with extrahepatic manifestations or other comorbidities in urgent need of viral clearance were permitted regardless of liver disease status. Key exclusions included creatinine clearance (CrCl) ≤30 mL/min, pregnancy and non-use of required contraception.
Liver disease stage was evaluated initially at each site. To maximise consistency across centres, cirrhosis was reassessed using a predefined algorithm with data from liver biopsy (Metavir >F3, Ishak >4 or the equivalent at any time prior to enrolment), FibroScan (>14.6 kPa at any time prior to enrolment) or Fibrosis-4 (FIB-4) score (>3.25 at baseline).
The recommended regimen was DCV 60 mg plus SOF 400 mg once daily for 24 weeks; at their discretion, physicians could add RBV to the regimen or reduce treatment duration. The DCV daily dose was reduced to 30 mg when coadministered with strong inhibitors of cytochrome P450 3A4 (CYP3A4) or P-glycoprotein, such as ritonavir-boosted HIV protease inhibitors (PI/r), and was increased to 90 mg when coadministered with moderate inducers of CYP3A4 or P-glycoprotein, such as efavirenz (EFV) or nevirapine (NVP). DCV could not be coadministered with strong inducers of CYP3A4 or P-glycoprotein.
Written informed consent was obtained from patients before enrolment. This programme was conducted in accordance with the Declaration of Helsinki.
Efficacy and safety assessments
All assessments were conducted at individual centres based on standard local practice and recommendations in the programme protocol. Blood samples for assessments of biochemical and haematological parameters and safety assessments were recommended at baseline; on-treatment weeks 4, 12 and 24 and post-treatment weeks 12 and (optional) 24. Efficacy assessments were based on serum HCV RNA determinations conducted by each centre using assay methods selected according to local preferences.
Endpoints
The primary efficacy assessment was SVR12, defined as HCV RNA below the assay's lower limit of quantitation (LLOQ), target detected (TD) or target not detected (TND), at post-treatment week 12. Virological failure categories included relapse (HCV RNA >LLOQ during any post-treatment visit in patients with HCV RNA <LLOQ, TD or TND at the end of treatment), virological breakthrough (HCV RNA ≥LLOQ on treatment following HCV RNA <LLOQ, TD or TND or a ≥1 log10 increase in HCV RNA from nadir) and other on-treatment virological failures (HCV RNA never <LLOQ or HCV RNA ≥LLOQ at the end of treatment but not meeting the breakthrough definition). Safety endpoints included graded adverse events (AEs), serious AEs, discontinuations due to AEs, deaths and clinical laboratory abnormalities.10
Statistical analyses
Enrolment was based on the clinical need for treatment rather than statistical considerations. The primary population for efficacy analyses (modified intention-to-treat (mITT)) included patients who received ≥1 dose of the programme regimen; those without virological failure who were lost to follow-up, withdrew informed consent or withdrew for undocumented reasons were excluded. Patients with missing data who died or discontinued treatment due to AEs were imputed as experiencing treatment failure.
Additional efficacy analyses were based on the ITT population (patients who received ≥1 dose of programme regimen) and on patients with available HCV RNA data at post-treatment week 12, excluding those with non-virological failure (as-observed population). The safety analysis population included all patients who received ≥1 dose of programme therapy.
Proportions of patients with SVR12 and two-sided 95% CIs were calculated by treatment group. Patients with missing HCV RNA data following virological failure were counted as treatment failures; missing data at post-treatment week 12 were imputed with HCV RNA data at a subsequent visit if available. Logistic regression analysis was performed to identify baseline factors associated with virological and treatment failure. Treatment and virological failure rates for patients receiving DCV+SOF versus DCV+SOF+RBV were compared using propensity scores with inverse probability weighting (IPW) to adjust for differences in demographic and disease characteristics between the two treatment groups.11
Results
Patients
Data were available for 485 enrolled patients who received therapy with DCV+SOF (n=359) or DCV+SOF+RBV (n=126) (figure 1). The median age was 57 years; most patients were men (66%), white (93%) and HCV treatment-experienced (70%). Patients were infected primarily with HCV genotypes 1b (36%), 1a (33%) or 3 (21%), and 27% had HCV RNA ≥2×106 IU/mL at baseline (table 1).
Patient demographic and baseline characteristics

Patient disposition by treatment group. Patient disposition by treatment group and reasons for non-completion of 24 weeks of therapy and discontinuation of follow-up are shown. Data for patients who did not reach post-treatment week 12 due to virological failure (n=2 in each group) or who died after achieving sustained virological response at post-treatment week 12 (SVR12) (n=4) are not shown. Patients who discontinued treatment prematurely could continue to be followed. Discontinuations before follow-up week 12 include patients who stopped treatment prematurely and did not continue follow-up (on-treatment death, lost to follow-up and withdrew consent) and those who discontinued after completing treatment. In the daclatasvir (DCV)+sofosbuvir (SOF) group, three patients excluded from the modified intention-to-treat (mITT) population had HCV RNA<lower limit of quantitation at follow-up visits performed before post-treatment week 12. In the DCV+SOF+ribavirin (RBV) group, the mITT population includes two patients who were lost to follow-up; both prematurely discontinued treatment due to adverse events and were imputed as failures. D/C, discontinuation; EOT, end of treatment.
Patient medical histories were typically complex, with frequent concurrent medical conditions and prior hepatic decompensation events. Cirrhosis was diagnosed in 389 patients (80%); among them, 165 (42%) had a Child–Pugh score ≥7 and 31 (8%) had a MELD score >15. Low platelet counts (<100×109/L) and low albumin levels (<35 g/L) were present in 55% and 33% of patients, respectively. Twenty-six patients (5%) had HCC, 55 (11%) had HIV/HCV coinfection and 66 (14%) had moderate or severe renal impairment (CrCl <60 mL/min/1.73 m2), mostly secondary to associated comorbidities such as diabetes, hypertension or HCV-associated cryoglobulinemia. Four patients were kidney transplant recipients. Seventy-eight (18%) patients were liver transplant recipients; of them, 43% were cirrhotic (32% Child–Pugh B, 11% Child–Pugh C), 51% had MELD score >10 and 5 had fibrosing cholestatic hepatitis.
Despite the absence of randomised treatment assignment, baseline characteristics were comparable across treatment groups with few exceptions. Patients treated with DCV+SOF+RBV, compared with DCV+SOF, were more frequently infected with HCV genotype 3 (32% vs 17%) and had signs suggesting more advanced liver disease, including slightly higher proportions of patients with Child–Pugh class B (47% vs 33%) and MELD score ≥10 (64% vs 55%).
Of the 485 patients who initiated therapy, 418 (86%) completed ≥20 weeks of treatment, 43 (9%) completed 10 to <20 weeks and 24 (5%) completed ≤10 weeks. Ten patients stopped treatment after 12–16 weeks per physician choice, 18 discontinued treatment before week 24 due to AEs and 10 died during treatment. In 21 patients, DCV was added to an ongoing SOF+RBV regimen; 11 additional patients had HCV RNA ≤LLOQ at DCV initiation but prior SOF+RBV therapy was not documented. Most of these patients stopped therapy after receiving SOF for a combined 24 weeks; 5 received DCV+SOF±RBV for 14–20 weeks and 17 for <14 weeks. The daily dose of DCV was reduced to 30 mg in 23 HIV/HCV coinfected patients who received HIV PI/r and was increased to 90 mg in 7 coinfected patients who received concomitant EFV or NVP.
Twenty-five patients (18 DCV+SOF, 7 DCV+SOF+RBV) were excluded from the mITT population, of whom 12 were lost to follow-up, 4 withdrew consent and 9 withdrew for undocumented reasons (see online supplementary table S1). Twenty-one excluded patients received therapy for ≥12 weeks and had HCV RNA <LLOQ, TD or TND at their last available visits; the remaining four patients had only baseline HCV RNA data available.
Supplementary material
Efficacy outcomes
Overall, SVR12 was achieved by 91% of the 460 patients in the primary analysis (mITT), including 92% of patients treated with DCV+SOF and 89% of those treated with DCV+SOF+RBV. Response rates were higher (97% and 96%, respectively) after non-virological failures were excluded (as-observed analysis) (table 2). Five patients who initiated therapy with DCV+SOF added RBV to the regimen during treatment; all achieved SVR12. Conversely, 15 patients who initiated treatment with DCV+SOF+RBV discontinued RBV prematurely; 14 (93%) achieved SVR12. Additionally, RBV dose was reduced in 30 patients; 29 (97%) achieved SVR12 (see online supplemental table S2).
Efficacy outcomes and reasons for non-response
SVR12 rates were generally comparable across baseline characteristics, with no notable differences between subgroups after excluding non-virological failures (figure 2). In the mITT analysis, SVR12 was achieved by 96% of patients (149 of 155) infected with genotype 1a, 89% (150 of 169) with genotype 1b, 88% (82 of 93) with genotype 3 and all 22 patients (100%) with genotype 2, 4 or 5. Differences observed between subtypes 1a and 1b were driven primarily by non-virological factors (see online supplementary table S2), and SVR12 increased to 99% and 97%, respectively, after excluding patients who failed for non-virological reasons (as-observed analysis).

Sustained virological response at post-treatment week 12 (SVR12) (as-observed) by baseline characteristics. SVR12 rates and 95% CIs by subgroups are shown for the as-observed population, which includes patients with data available on or after post-treatment week 12, including those with virological failure. Data not shown for patients infected with genotype 2 (n=2), genotype 5 (n=1), mixed genotypes (n=1) or unknown genotype (n=2) and patients with Model for End-Stage Liver Disease (MELD) score >20 (n=3); all achieved SVR12. CrCl, creatinine clearance; DCV, daclatasvir; RBV, ribavirin; SOF, sofosbuvir.
Response rates were high regardless of cirrhosis status or liver disease severity, as indicated by low platelet counts or albumin levels. SVR12 was achieved by 90% (331 of 368) of patients with cirrhosis (91% with DCV+SOF; 88% with DCV+SOF+RBV), 90% (225 of 250) of patients with platelet counts <100×109 cells/L and 87% (129 of 149) of those with albumin <35 g/L.
The SVR12 rate was lower in patients with more advanced disease (Child–Pugh C or MELD score ≥16). However, differences were driven mainly by more frequent liver disease-related discontinuations and deaths in patients with decompensated cirrhosis and/or high MELD score (figure 2). In the mITT analysis, SVR12 was achieved by 94% (200 of 213), 86% (115 of 134) and 76% (16 of 21) of patients with Child–Pugh classes A, B and C, respectively. Similarly, SVR12 was achieved by 92% (147 of 160), 93% (167 of 179) and 61% (17 of 28) of patients with MELD scores <10, 10–15 and ≥16, respectively. After patients with non-virological failure were excluded (as-observed analysis), SVR12 rates increased to 100% (16 of 16) and 89% (17 of 19) of patients with Child–Pugh C and a MELD score ≥16, respectively. However, these results must be interpreted with caution due to limited numbers of patients in these subgroups.
Comparable SVR12 rates were observed in HCV treatment-naive (93%, 125 of 135) and treatment-experienced (91%, 294 of 325) patients. Ten patients had a shorter duration of treatment, of whom nine achieved SVR12 and one died before post-treatment week 12. However, treatment duration was not randomised, and patients may have been selected for shorter treatment because of disease characteristics that suggested a high probability of response.
Efficacy in special populations
Genotype 3 infection
Advanced disease was more common in genotype 3-infected patients than in the overall population: 85% had cirrhosis, including 52% with decompensated liver disease; 44% had albumin levels <35 g/L and 63% had platelet counts <100×109/L. SVR12 was achieved by 88% (82 of 93) of genotype 3-infected patients in the mITT analysis, including 88% treated with DCV+SOF and 89% treated with DCV+SOF+RBV. The overall SVR12 rate was 92% after excluding four patients with non-virological failure. Response rates were slightly lower in treatment-experienced patients and those with decompensated cirrhosis (figure 3), although differences were driven mainly by non-virological failure (see online supplementary figure S1).

Sustained virological response at post-treatment week 12 (SVR12) (modified intention-to-treat (mITT)) in patients with HCV genotype 3 infection. SVR12 (mITT analysis) rates by treatment group in genotype 3-infected patients are shown according to baseline cirrhosis status and prior HCV therapy (A) and disease stage in patients with cirrhosis (B). Error bars indicate 95% CIs. Data for patients with cirrhosis status indeterminate (n=7, all achieved SVR12) or not reported (n=1, relapse) and for one patient with Model for End-Stage Liver Disease (MELD) score not reported (discontinuation due to adverse event, imputed as failure) are not shown. DCV, daclatasvir; RBV, ribavirin; SOF, sofosbuvir.
HIV/HCV and HBV/HCV coinfection
SVR12 was achieved by 92% of the HIV/HCV coinfected patients (48 of 52) included in the mITT population and by 98% of patients after excluding three patients with non-virological failure. All 12 patients (100%) with HBV/HCV coinfection achieved SVR12; one additional patient was lost to follow-up and excluded.
Post-liver transplant recurrence
SVR12 was achieved by 94% of the patients (80 of 85) with post-liver transplant HCV recurrence included in the mITT population; the SVR12 rate was 100% after excluding five patients with non-virological failure.
Renal impairment
Renal insufficiency had minimal impact on virological response; SVR12 was achieved by 96% of patients (103 of 107) with CrCl 60–89 mL/min, 89% (51 of 57) with CrCl 30–59 mL/min and 100% (5 of 5) with CrCl <30 mL/min.
Treatment failure
Forty-one patients did not achieve SVR12 (table 2, see online supplementary table S2). Fourteen patients (3%) experienced virological failure, including 13 relapses and one genotype 3-infected patient with virological breakthrough after serum HCV RNA decreased to 30 IU/mL. One additional patient relapsed at post-treatment week 24 after achieving SVR12. Virological failure occurred with similar frequency among patients treated with DCV+SOF with RBV (4 patients, 3%) or without RBV (10 patients, 3%).
Twenty-seven patients (6%) failed to achieve SVR12 due to death or treatment discontinuation without evidence of virological failure, including 10 on-treatment deaths, 14 deaths during follow-up and 3 treatment discontinuations due to AEs with subsequent loss to follow-up.
We examined baseline factors that might predict treatment and virological failure. Univariate logistic regression analysis (see online supplementary figure S2) indicated significantly higher (p<0.05) risks of treatment failure among patients with more advanced liver disease, as indicated by baseline MELD score ≥16, Child–Pugh class B or C, albumin levels <35 g/L or total bilirubin ≥1.2 mg/dL. After excluding non-virological failures, these associations became less significant (p>0.05). No significant difference in the risk of treatment or virological failure was observed between patients who received DCV+SOF versus DCV+SOF+RBV. A slightly higher incidence of virological failure was observed among patients receiving DCV+SOF+RBV (3.64% vs 3.10%; risk difference −0.54%, 95% CI −4.52% to 3.44%). Because treatment assignment was not randomised, this might have been caused by physicians' common practice of adding RBV to regimens for harder-to-treat patients.
The effect of RBV on risk of virological failure was further evaluated using IPW with propensity scores (see online supplementary table S3). After propensity score weighting, disease and treatment parameters that could affect failure risk were well balanced across treatment groups. Adjusted virological failure rates were 2.35% and 3.41% with and without RBV, respectively. The adjusted risk difference (DCV+SOF minus DCV+SOF+RBV) of 1.06% (95% CI −2.22% to 4.35%) was not conclusive, suggesting a clinically non-significant treatment effect on the probability of virological failure.
Changes in liver function
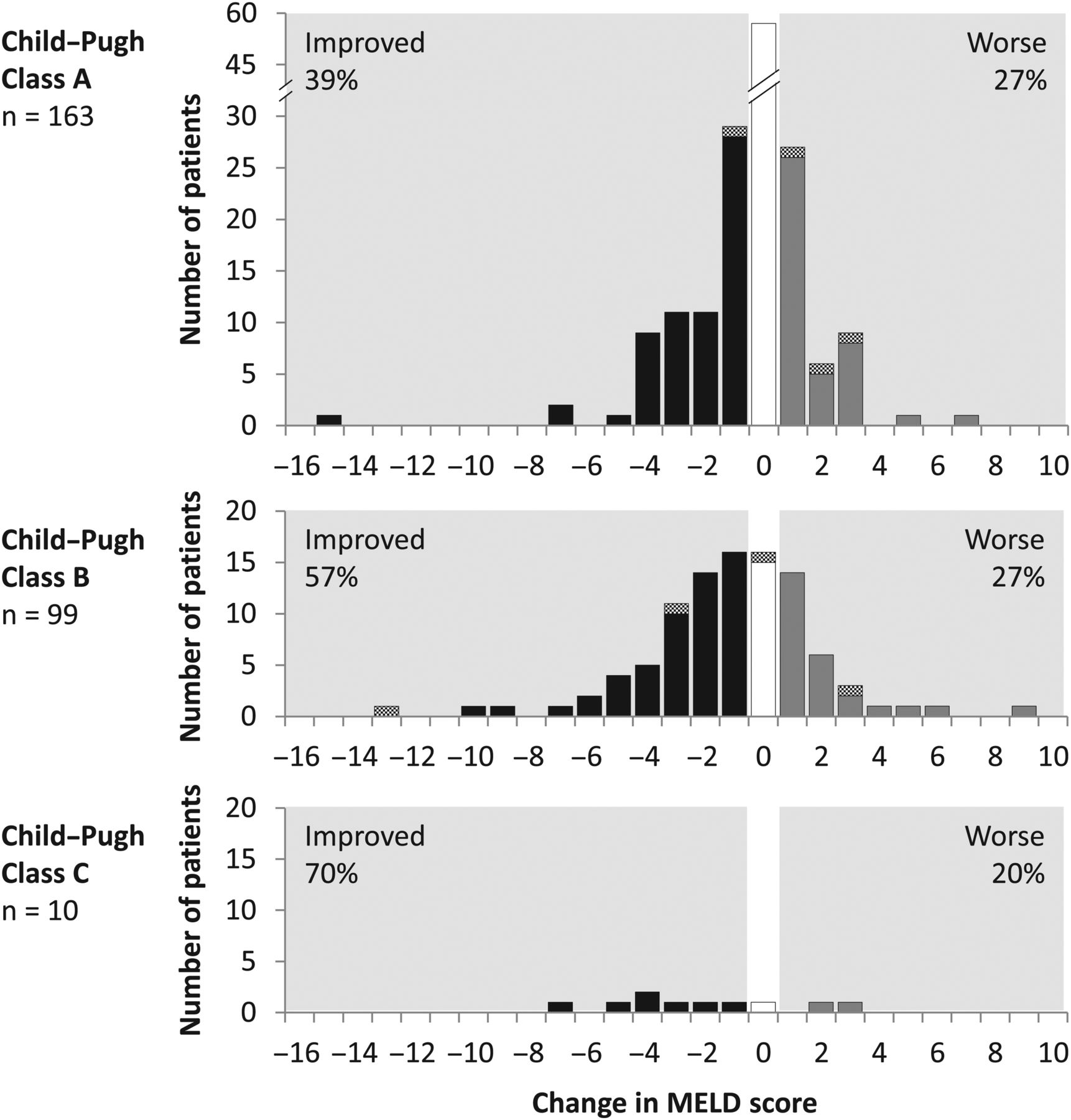
Laboratory parameters associated with liver function were assessed at baseline and post-treatment week 12. Among patients with samples at both time points, total bilirubin decreased by a median 0.2 mg/dL (IQR 0.60), alanine aminotransferase (ALT) decreased by 37 IU/L (IQR 54.0), albumin increased by 2.0 g/L (IQR 6.0) and platelets increased by 7.0×109 cells/L (IQR 32.0). Among 272 patients with available data, MELD score improved or remained unchanged in 47% and 28% of patients, respectively; improvements were observed in 58% of patients (63 of 109) with decompensated cirrhosis (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in Model for End-Stage Liver Disease (MELD) score from baseline to post-treatment week 12. Changes in MELD score from baseline to post-treatment week 12, by baseline Child–Pugh class, are shown. Each panel indicates the numbers of patients according to the magnitude of change in MELD score. Solid bars indicate patients who achieved sustained virological response at post-treatment week 12; hatched bars indicate patients with virological failure.
Safety and tolerability
Treatment was discontinued prematurely in 28 patients, including 10 who died on treatment. Sixteen DCV+SOF recipients (4%) discontinued treatment; the most common events leading to discontinuation were multiorgan failure (n=4), sepsis (n=2) and hepatic encephalopathy (n=2). The discontinuation rate was higher (n=12, 10%) in DCV+SOF+RBV recipients; the most common events leading to discontinuation were general physical health deterioration (n=3), acute kidney injury (n=3) and hepatic failure (n=2) (table 3; see online supplementary table S4).
On-treatment safety and tolerability
Ninety-four patients (19%) experienced serious AEs on treatment (table 3). Most events were directly or indirectly related to advanced liver disease; those that occurred in >5 patients included hepatic encephalopathy (n=12), HCC (n=8) and hepatic failure (n=6) (see online supplementary table S5). Twenty-eight patients died during treatment or follow-up, including four who died after achieving SVR12. Most deaths occurred in patients with advanced liver disease and were liver related; causes of death in ≥2 patients included non-HCC liver-related events (n=9), multiorgan failure (n=5) and sepsis (n=4, including one with concomitant multiorgan failure). No deaths were considered treatment related (see online supplementary table S2).
The most common AEs were non-specific, such as fatigue, headache, arthralgia and gastrointestinal events (table 3). Liver-related grade 3–4 laboratory abnormalities were infrequent; ALT elevations were reported in 3 patients (1%) and elevated total bilirubin in 22 (5%). Twenty-four patients experienced reduced haemoglobin levels, most frequently those receiving RBV. RBV dose was reduced or stopped in 45 patients, mainly due to AEs; 11 of these patients were liver transplant recipients and 19 had decompensated cirrhosis. Among patients with HBV/HCV coinfection, there were no reports of HBV reactivation during or after HCV therapy.
Among the six patients with CrCl <30 mL/min/1.73 m2 at baseline, two patients had SOF dose reductions to 200 mg due to further decline in renal function, one patient who started treatment with a reduced dose of SOF experienced further decline in CrCl after increasing the daily dose of SOF to 400 mg and the remaining three patients maintained stable CrCl levels despite receiving full-dose SOF (see online supplementary figure S3). Among the 60 patients with moderate renal insufficiency at baseline, only one patient discontinued treatment due to renal events. In three additional patients, CrCl decreased below 30 mL/min/1.73 m2 and remained stable without dose adjustment.
Discussion
This European CUP provided clinically relevant information on the effectiveness and safety of DCV+SOF, with or without RBV, in a large cohort of patients with advanced liver disease, including many with decompensated cirrhosis. This population, which is often under-represented in clinical trials, is less likely to respond satisfactorily to treatment and may suffer more frequent treatment-related AEs.
Treatment resulted in an overall SVR12 rate of 91%. Similarly, high SVR12 rates were observed in patient subgroups with characteristics regarded as more difficult to cure, such as decompensated cirrhosis and genotype 3 infection with cirrhosis. Consistent with previous studies, the SVR12 rate was lower in patients with Child–Pugh C,9 and correspondingly, indicators of advanced liver disease such as low platelet count or low albumin level were associated with increased risk of failure. However, much of this difference was related to pre-existing advanced liver disease rather than to inadequate virological efficacy. After excluding patients with non-virological failure, most of whom had died from advanced liver disease, a 97% SVR12 rate was observed in patients with cirrhosis. Rates were similar across Child–Pugh classes, and indicators of advanced liver disease were not significantly associated with a higher risk of virological failure.
Efficacy outcomes are consistent with results of phase III studies of this regimen (ALLY programme) and community-based expanded access programmes, despite the high proportion of patients with advanced disease.7–9 ,12 ,13 This finding contrasts with results of earlier studies of telaprevir, boceprevir and SOF+RBV,14 ,15 which demonstrated suboptimal safety and/or efficacy in a real-world setting, particularly in patients with advanced disease. Efficacy outcomes were also generally comparable with those achieved after 12 or 24 weeks of treatment with SOF plus ledipasvir (LDV) or velpatasvir (VEL), with or without RBV, in studies of patients with advanced disease.16–18 However, results for SOF+LDV are limited to genotypes 1 and 4, and data for the combination SOF+VEL without RBV suggest lower response rates in genotype 3 infection regardless of treatment duration.18
Response rates were similar in the two treatment groups, suggesting that RBV may not confer an efficacy benefit with this regimen when treatment is extended to 24 weeks. After adjustment for differences in baseline characteristics in the IPW analysis, treatment with or without RBV had no significant effect on the probability of virological failure. However, definitive conclusions in this regard should be confirmed in randomised clinical trials.
Optimising therapy for patients with cirrhosis with genotype 3 infection remains an important objective. In this cohort, 89% of genotype 3-infected patients with cirrhosis achieved SVR12 after 24 weeks of treatment. In a previous phase III study of genotype 3 infection, SVR12 was achieved by 96% of patients without cirrhosis but by only 63% of patients with cirrhosis after 12 weeks of treatment with DCV+SOF.8 Subsequently, addition of RBV to the regimen for 12 or 16 weeks increased SVR12 rates to 83% and 89%, respectively, in patients with advanced fibrosis or compensated cirrhosis.19 However, SVR12 rates remained suboptimal (71%) with shorter duration of treatment in patients with decompensated cirrhosis.20 Thus, 12 weeks of treatment may be adequate for patients with genotype 3 infection without cirrhosis, whereas patients with cirrhosis may benefit from addition of RBV and/or extension of treatment beyond 12 weeks. The incremental benefit of adding RBV is most evident with 12-week regimens and uncertain when treatment is extended to 24 weeks. The results of a French expanded access programme support this interpretation.12 SVR12 was achieved by 70% of patients with genotype 3 infection with cirrhosis treated for 12 weeks with DCV+SOF. With 24 weeks of treatment, SVR12 rates were 81% and 86% for DCV+SOF with and without RBV, respectively. Similarly, in our cohort where most patients were treated for 24 weeks, comparably high SVR12 rates were achieved with RBV (88%) and without RBV (89%).
The short follow-up precludes definitive conclusions regarding treatment-related changes in liver disease. Our data suggest a gradual improvement in MELD score and other liver disease markers, with the greatest changes in MELD score generally observed in patients with the highest scores at baseline. Further follow-up is needed to assess long-term improvements in liver disease parameters following viral clearance.
Virological failure was infrequent, occurring in 14 patients (3%) overall. Thirteen were post-treatment relapses; there was a single case of virological breakthrough in a genotype 3-infected patient who never had undetectable HCV RNA during treatment. Relapse was slightly more common in patients with genotype 3 versus other genotypes. Nevertheless, 92% of genotype 3-infected patients achieved SVR12 after excluding non-virological failures, even though 85% had cirrhosis and 52% of patients with cirrhosis had evidence of hepatic decompensation. Logistic regression analysis found no other baseline characteristics associated with an increased risk of failure.
Most patients who failed to achieve SVR12 had adequate virological responses but did not complete the programme due to AEs or death; most such events were associated with advanced liver disease that was present at programme entry. However, most patients with advanced liver disease completed the programme successfully. Consistent with other studies in similar populations, this finding confirms that HCV suppression is not always capable of arresting clinical deterioration in patients with very advanced disease.21 ,22 Recent observations suggest that the risk of HCC in patients with cirrhosis remains after SVR12 is achieved.23 ,24
Overall, DCV+SOF with or without RBV was well tolerated, exhibiting a safety profile consistent with data reported in phase III studies. No unique safety events were reported even though a high proportion of patients had advanced disease—a population that often exhibits reduced tolerability to HCV therapies, especially those containing interferon. There were few discontinuations due to AEs, and not unexpectedly in a population with advanced liver disease, most serious AEs and treatment discontinuations were attributable to continued disease progression. Safety outcomes were generally similar between the two treatment groups except for a higher frequency of generally mild haematological events in patients receiving RBV.
Data from this cohort have several limitations. Treatment allocation was not randomised; RBV use was at physicians' discretion, potentially resulting in imbalanced groups that could complicate assessments of the role of RBV. To mitigate this limitation, an IPW analysis was performed to further explore the role of RBV in efficacy outcomes. Laboratory tests were conducted using the standard technology that was available at each centre. Consequently, assay differences may have caused inconsistencies in laboratory-based efficacy and safety assessments. As with other real-world cohorts, the limited requirements for data capture may have led to under-reporting of safety events despite close monitoring. In this regard, although most patients lost to follow-up had HCV RNA <LLOQ at their last available visit, the possibility of subsequent deaths among this group cannot be completely excluded. Additionally, the potential contribution of drug-related hepatotoxicity to disease progression and mortality can likewise not be fully excluded.25 Comprehensive analyses of drug resistance polymorphisms were not performed; therefore, the potential contribution of pre-existing polymorphisms to virological outcome cannot be determined. Despite these limitations, this cohort represents one of the largest cohorts of patients with advanced liver disease treated with an oral DAA combination in a real-world setting. The findings are consistent with the results of clinical trials evaluating DCV+SOF with or without RBV despite the inclusion of a broad spectrum of patients.
In summary, DCV+SOF, with or without RBV, achieved high SVR12 rates in a large, diverse cohort of patients with potentially life-threatening liver disease. Treatment was well tolerated and was associated with improvements in liver function.
Acknowledgments
The authors thank the patients, treating physicians and research staff for their time and contributions to this programme.
References
Footnotes
Collaborators All collaborators are listed in online supplementary table S6.
Contributors All programme participants are listed in online supplementary table S6. MJJ-E developed the concept and design of the programme. TMW, JP, KH, PF, MG, HW, TB, US, OW, MvdV, JR, MP-R and SZ acquired the data. YZ and MJJ-E analysed the data. All authors participated in data interpretation, manuscript preparation and critical review and approved the final version of the manuscript.
Funding This CUP was funded by Bristol-Myers Squibb. Editorial assistance was provided by Richard Boehme of Articulate Science and funded by Bristol-Myers Squibb.
Competing interests TMW—consultant: Novartis, Janssen, Gilead, AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb. JP—grant: Roche, GlaxoSmithKline; consultant: Bristol-Myers Squibb, Gilead, Novartis, Merck; speaking and teaching: Abbott, Tibotec, Merck. PF—consultant: Idenix, Gilead, Merck, Janssen, Salix, AbbVie, Bristol-Myers Squibb; patent held/filed: Madaus Rottapharm; speaking and teaching: Gilead, Roche. MG—consultant: Janssen, Bristol-Myers Squibb, Gilead, AbbVie; speaking and teaching: Janssen, Bristol-Myers Squibb, Gilead, AbbVie. HW—grant: Merck, Novartis, Gilead, Roche, Abbott, AbbVie; speaking and teaching: Bristol-Myers Squibb, Merck, Novartis, Italfarmaco, AbbVie, Gilead; advisory committee or review panel: Transgene, Merck, Roche, Gilead, Abbott, AbbVie, Bristol-Myers Squibb, Falk, Novartis, GlaxoSmithKline. TB—grant: Gilead, Bristol-Myers Squibb, Roche, Tibotec, Vertex, Janssen, Merck, Boehringer Ingelheim, Novartis, AbbVie; consultant: Gilead, Bristol-Myers Squibb, Roche, Tibotec, Vertex, Janssen, Novartis, Abbott, Merck, AbbVie; speaking and teaching: Gilead, Bristol-Myers Squibb, Roche, Tibotec, Vertex, Janssen, Merck, Novartis, Bayer, AbbVie. OW—speakers' bureau: Merck, Roche, Bristol-Myers Squibb, Novartis, Janssen, Medivir, Gilead, AbbVie; consultant: Merck, Bristol-Myers Squibb, Medivir, Gilead, AbbVie. MvdV—consultant: Gilead, Merck, Bristol-Myers Squibb, AbbVie, Janssen, ViiV Healthcare, Roche. JR—grant: Merck; consultant: AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb, Merck, Roche, Tibotec, Bionor, Tobira, ViiV Healthcare, Gilead, Janssen, Novartis; speaking and teaching: Abbott, Boehringer Ingelheim, Bristol-Myers Squibb, Merck, Roche, Tibotec, Gilead, Janssen, ViiV Healthcare. MP-R—grant: Bayer, Roche, Gilead, Merck, AbbVie; consultant: Bayer, Boehringer Ingelheim, Jennerex, Eli Lilly, AbbVie; advisory committee or review panel: Bayer, Gilead, Janssen, Bristol-Myers Squibb, AbbVie; speaking and teaching: Bayer, Roche, Gilead, Merck, Eli Lilly, AbbVie. YZ—employee: Bristol-Myers Squibb. MJJ-E—employee: Bristol-Myers Squibb. SZ—consultant: AbbVie, Bristol-Myers Squibb, Gilead, Merck, Janssen.
Patient consent Obtained.
Ethics approval This programme was approved by national health authorities for all participating countries. Ethics committee approval was managed by participating sites on an individual basis in accordance with local legislation regulating CUPs.
Provenance and peer review Not commissioned; externally peer reviewed.