Article Text

Abstract
Objective Previous studies suggested that microRNA-21 may be upregulated in the liver in non-alcoholic steatohepatitis (NASH), but its role in the development of this disease remains unknown. This study aimed to determine the role of microRNA-21 in NASH.
Design We inhibited or suppressed microRNA-21 in different mouse models of NASH: (a) low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed a high-fat diet and treated with antagomir-21 or antagomir control; (b) microRNA-21-deficient and wild-type mice fed a methionine-choline-deficient (MCD) diet; (c) peroxisome proliferation-activator receptor α (PPARα)-deficient mice fed an MCD diet and treated with antagomir-21 or antagomir control. We assessed features of NASH and determined liver microRNA-21 levels and cell localisation. MicroRNA-21 levels were also quantified in the liver of patients with NASH, bland steatosis or normal liver and localisation was determined.
Results Inhibiting or suppressing liver microRNA-21 expression reduced liver cell injury, inflammation and fibrogenesis without affecting liver lipid accumulation in Ldlr−/− fed a high-fat diet and in wild-type mice fed an MCD diet. Liver microRNA-21 was overexpressed, primarily in biliary and inflammatory cells, in mouse models as well as in patients with NASH, but not in patients with bland steatosis. PPARα, a known microRNA-21 target, implicated in NASH, was decreased in the liver of mice with NASH and restored following microRNA-21 inhibition or suppression. The effect of antagomir-21 was lost in PPARα-deficient mice.
Conclusions MicroRNA-21 inhibition or suppression decreases liver injury, inflammation and fibrosis, by restoring PPARα expression. Antagomir-21 might be a future therapeutic strategy for NASH.
- FATTY LIVER
- INFLAMMATION
- NONALCOHOLIC STEATOHEPATITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
MicroRNAs are attractive therapeutic targets since they can be modulated in patients.
Microarray studies suggested that microRNA-21 could be overexpressed in the liver in patients with non-alcoholic steatohepatitis (NASH).
Peroxisome proliferation-activator receptor α (PPARα) is a target of microRNA-21 and is implicated in NASH.
What are the new findings?
MicroRNA-21 contributes to key features of NASH, that is, cell injury, inflammation and fibrosis, through PPARα inhibition.
Using different mouse models of NASH, we demonstrate that microRNA-21 blockade prevents NASH development.
MicroRNA-21 is overexpressed in the liver of patients with NASH, but not in those with bland steatosis.
Liver microRNA-21 in NASH is primarily expressed in inflammatory and biliary cells.
How might it impact on clinical practice in the foreseeable future?
MicroRNA-21 antagonism might be useful in the future in the treatment of NASH.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a condition ranging from simple triglyceride accumulation in the liver (steatosis) to steatosis combined with lobular inflammation and hepatocyte injury (clarification and ballooning) with or without fibrosis. The latter is referred to as non-alcoholic steatohepatitis (NASH).1 NAFLD and NASH are increasingly relevant public health issues owing to their close association with the worldwide epidemics of diabetes and obesity.2 NAFLD/NASH is one of the most common chronic liver diseases. Indeed, in the USA, 39% of newly identified cases of chronic liver disease had NAFLD.3 NASH has potential for fibrosis, cirrhosis decompensation and hepatocellular carcinoma. Despite its prevalence and severity, therapeutic options for NASH are limited.1 Therefore, understanding the mechanisms leading to hepatic inflammation and fibrosis in NAFLD is of great importance for the diagnosis and treatment of NASH.
MicroRNAs are a family of short non-coding RNAs (19–22 nucleotides in length) that regulate gene expression at the post-transcriptional level via messenger RNA degradation or translational inhibition.4–6 MicroRNA microarray analysis identified microRNA-21 (miR-21) as one of the most upregulated micro-RNAs in the liver of patients with NASH.7 This upregulation was subsequently confirmed.8 In two mouse models of NASH (choline-deficient and amino acid-defined diet and high-fat diet (HFD)), miR-21 was also overexpressed in the liver.8 ,9 However, these studies did not address whether or not liver miR-21 upregulation was a cause or a consequence of NASH. In addition, the cellular localisation of miR-21 has not yet been elucidated.
Therefore, the present study aimed to determine the role of miR-21 in the pathogenesis of NASH. For this purpose, we used complementary approaches: (a) a pharmacological inhibition of miR-21 using antagomir-21 in a genetic model of NASH (low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed an HFD); and (b) a genetic deletion of miR-21 (miR-21−/− mice) in mice exposed to a dietary model of NASH (methionine-choline-deficient (MCD) diet). We also determined whether miR-21 effect implicates peroxisome proliferation-activator receptor α (PPARα), a known miR-21 target, using PPARα-deficient (Pparα−/−) mice.
Materials and methods
Mouse models of NASH
All experiments were performed in accordance with the guidelines formulated by the European Community for experimental animal use (L358-86/609EEC) and were approved by the French Ministry of Agriculture (agreement no. A75-15-32). All animals received human care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health.
All mice were on a C57BL/6 background. Different NASH models were used: Ldlr−/− mice fed an HFD, and miR-21−/− mice fed an MCD diet.
Eight-week-old male Ldlr−/− mice (Charles River, L'Isle d'Abresle, France) were put on an HFD containing 1.25% cholesterol,15% cacao butter and 0% cholate (Safe Diets) for 14 weeks (figure 1A). When 12 and 17 weeks old, mice received three retroorbital intravenous injections (once a day for three consecutive days) of antagomir-21 (sequence 5′-UCAACAUCAGUCUGAUAAGCUA-3′; 16 mg/kg body weight, VBC Biotech, Vienna, Austria), or antagomir control (sequence 5′-AAGGCAAGCUGACCCUGAAGUU-3′; 16 mg/kg body weight; VBC Biotech Vienna, Austria) or phosphate buffer saline (PBS). This design was adapted from previous reports showing that antagomirs are able to induce a potent and prolonged miR inhibition.10–12 Preliminary experiments showed that mir-21 expression was more robustly decreased by antagomir-21 given at a dose of 16 than 8 mg/kg body weight (data not shown). C57BL/6 wild-type (WT) untreated mice were used as controls. All mice groups were euthanased at the age of 22 weeks.

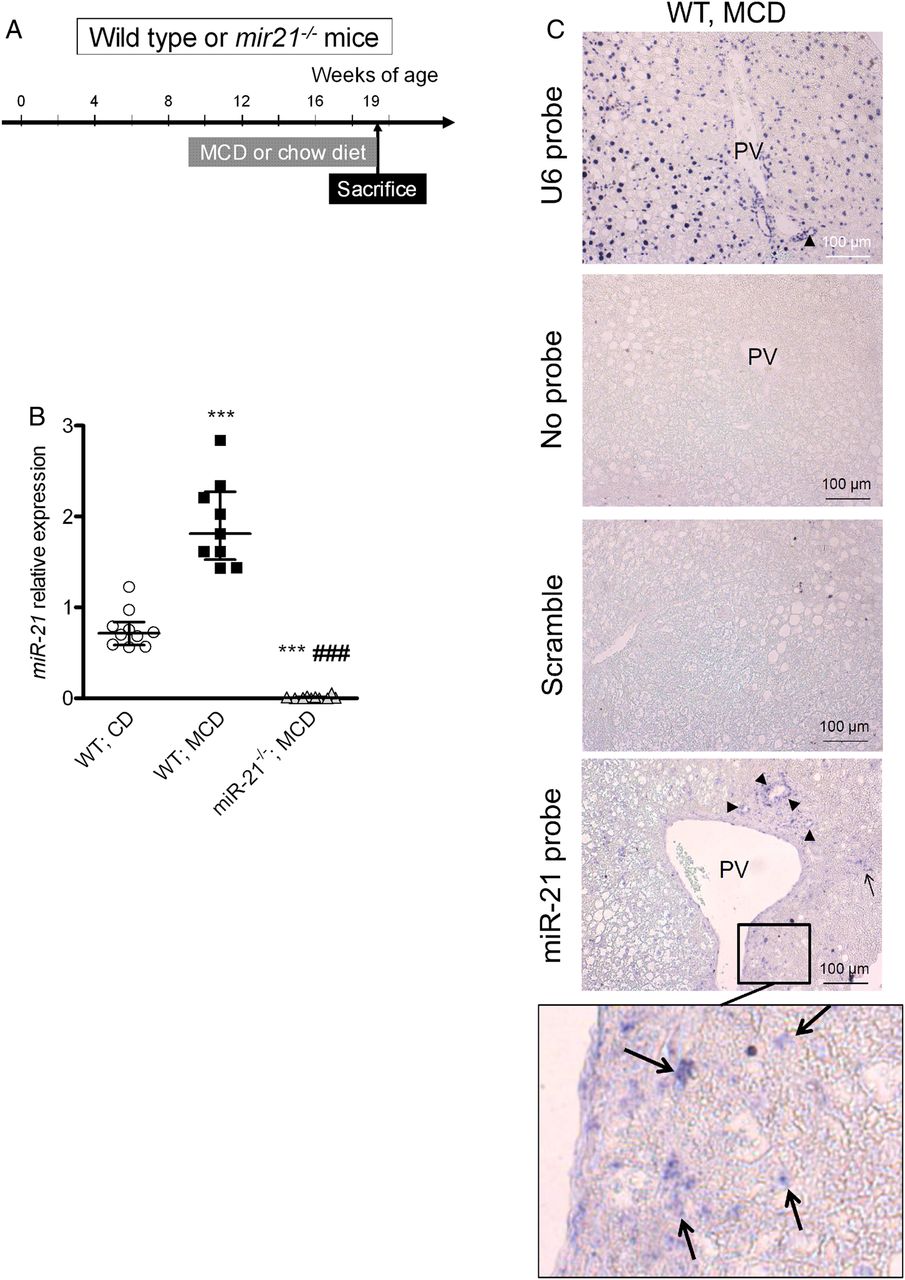
MicroRNA-21 (miR-21) was overexpressed in the liver of low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed a high-fat diet (HFD), primarily in inflammatory and biliary cells. (A) Mouse study design. (B) miR-21 expression in the liver of wild-type (WT) mice fed a chow diet (CD) and of Ldlr−/− mice fed an HFD treated with the vehicle (phosphate buffer saline (PBS)), antagomir control (AC) or antagomir-21 (A-21). **p<0.01 versus WT mice fed a CD; §§p<0.01 versus Ldlr−/− mice treated with PBS; ##, p<0.01 versus Ldlr−/− mice treated with AC; (C) in situ hybridisation. miR-21 was detected in liver inflammatory (arrow) and biliary cells (arrow head) from Ldlr−/− mice fed an HFD. Original magnification ×200. (D) miR-21 expression in sorted liver inflammatory (CD45+) and non-inflammatory (CD45−) cells from Ldlr−/− mice fed an HFD. (E) miR-21 expression in subpopulations of liver inflammatory (CD45+) cells from Ldlr−/− mice fed an HFD. *p<0.05; **p<0.01. Data are given as median (horizontal bar) and IQR (error bar). PV, portal vein.
Nine-week-old male WT or miR-21−/− mice were put for 10 weeks on MCD containing 18.3% methionine-deficient amino acid mix, 10% sucrose, 10% lard, 5% cellulose, 1% choline-deficient vitamin mix, 3.5% mineral mix and 53% cornstarch (SSNIFF Diet).13 C57BL/6 WT mice under chow diet served as controls. All mice were euthanased at the age of 19 weeks.
Sixteen-week-old male WT or Pparα−/− mice were put for 5 weeks on MCD diet. When 17 weeks old, mice received three retroorbital intravenous injections of antagomir-21 or antagomir control (16 mg/kg body weight). C57BL/6 (WT) mice under chow diet served as controls. All mice were euthanased at the age of 21 weeks.
On the day of the sacrifice, mice were sedated with 2.5% isofluorane and blood was collected from the inferior vena cava into a 25 gauge×1′ needle pre-coated with 3.8% sodium citrate. Blood was spun at 1500 g for 15 min to prepare platelet poor plasma that was then stored at −80°C. The mice were then euthanased. Livers were isolated and snap frozen in liquid nitrogen and stored at −80°C or fixed in formalin and paraffin-embedded.
RNA isolation and miR-21 quantification
Total RNA was extracted from snap frozen human or mouse liver samples with Trizol reagent according to the manufacturer's instructions (Invitrogen, Paris, France). MiR-21 levels were quantified using TaqMan assay (Tm 00397, Applied Biosystems) following the manufacturer's instructions. miR-21 expression was normalised to U6 snRNA (Tm 001973, Applied Biosystems) for mouse and human samples, together with RNU6B (Tm 001093; Applied Biosystems) for human samples and snoRNA234 (Tm001234; Applied Biosystems) for mouse samples as endogenous control. Relative expression was calculated using the 2-delta-delta CT method followed by geometric average, as recommended.14
In situ hybridisation
In situ hybridisation was adapted from a previously described method and is described in detail in the online supplementary methods.15 Liver samples from miR-21−/− mice served as negative control and failing myocardium induced by a myocardial infarction as a positive control (online supplementary figure S1).16
Hepatic inflammatory cells sorting and analysis by flow cytometry
Mouse livers were perfused in vivo with PBS and then collected, weighted and minced with sterile scissors in PBS supplemented with 1% fetal calf serum (100 mg liver tissue in 1 mL PBS). Liver cell suspension was filtered through a 40 µm membrane. Polymorphonuclear cells were then isolated by density-gradient centrifugation over endotoxin-free Ficoll 1083 (Sigma-Aldrich) for 30 min at 500 g without brake at room temperature. Pellets and polymorphonuclear cells rings were collected separately, washed and counted. Cells from pellets were stained with anti-Epcam allophycocyanin antibodies (eBiosciences anti-CD326, #17-5791-82) for 30 min at 4°C. Cells from the rings were stained with anti-mouse CD11b-fluorescein-isothiocyanate (eBiosciences, #130-081-201), anti-NK-1.1-phycoerythrin (BD Biosciences, #553165), anti-Ly6G-phycoerythrin (BD Biosciences, #551461), anti-CD45-PercP (BD Biosciences, #557235) and anti-CD3-PE-Alexa700 (eBiosciences, #17-0032-82) antibodies for 30 min at 4°C. Cells were then washed and resuspended in PBS supplemented with 1% fetal calf serum and extemporaneously with 1 µg/mL 4′,6-diamidino-2-phenylindole. Phenotyping of polymorphonuclear cells rings was performed on a LSR II flow cytometer (BD Biosciences, San Jose, California, USA). CD45+ subpopulations or Epcam+ cells were isolated for quantification of miR-21 expression using an ARIA II SORP cell sorter (BD Biosciences). Post-acquisition analysis was performed using FlowJo software.
Metabolic analyses
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, and fasting glucose, total cholesterol, high-density lipoprotein (HDL)-cholesterol and triglyceride levels were determined using commercial enzymatic kits on a DxC800 analyzer (Beckman-Coulter, Villepinte, France).
Histology
Human liver samples
Paraffin-embedded liver sections (3 µm thick) were stained with H&E (haematoxylin, RAL diagnostics; and eosin, RAL diagnostics) and Picro-sirius (Sirius red, RAL diagnostics; picric acid, VWR Prolabo). The steatosis score assessed the quantities of large or medium-sized lipid droplets, but not foamy microvesicles, from 0 to 3:S0:<5%; S1:5–33%; S2:34–66%; and S3:>67%. Ballooning was graded from 0 to 2:0, normal hepatocytes with cuboidal shape and pink eosinophilic cytoplasm; 1, presence of clusters of hepatocytes with a rounded shape and pale cytoplasm usually reticulated; and 2, same as grade 1 with some enlarged hepatocytes, at least twofold that normal cells.17 Lobular inflammation was defined as a focus of two or more inflammatory cells within the lobule. Foci were counted at 20× magnification: 0, none; 1, ≤2 foci per 20×; and 2, >2 foci per 20× field. Stage of fibrosis was assessed using the score described by NASH-CRN as follows: stage 0 (F0), none; stage 1 (F1), perisinusoidal zone 3 or portal fibrosis; stage 2 (F2), perisinusoidal and periportal fibrosis without bridging; stage 3 (F3), bridging fibrosis; and stage 4 (F4), cirrhosis.18
Mice liver samples
Paraffin-embedded liver sections (3 µm thick) were stained with H&E (haematoxylin, RAL diagnostics; and Eosin, RAL diagnostics), Picro-sirius (Sirius red, RAL diagnostics; picric acid, VWR Prolabo), rabbit anti-CD3 antibody (DAKO, #A0452) and a monoclonal rat anti-mouse CD68 antibody (AbD Serotec, MCA1957). Steatosis was defined as the percentage of hepatocytes containing large or medium-sized lipid droplets. Steatosis was scored on H&E stained sections by a single experienced pathologist (VP) who was blinded to mice groups. The following scale was used: 0 (<5%), 1 (5–33%), 2 (34–66%) and 3 (>67%). The Sirius red positive area was measured in at least 10 fields without vessels or capsule (original magnification, ×200) and quantified using ImageJ software. Inflammatory cells (CD3+ or CD68+) were counted in ≥15 microscope views (original magnification, ×200).
Frozen liver sections (4 µm) were stained with the neutral lipid marker Oil Red O (Sigma-Aldrich) to confirm steatosis.
Hepatic collagen analysis
Hepatic total collagen content was determined using the total collagen assay (Quickzyme Biosciences, UK) according to the manufacturer's instructions.
Gene expression analysis
cDNA synthesis was performed with QuantiTect Reverse Transcription Kit (Qiagen). PCR was performed on an ABI StepOne Plus with the use of Power SYBR Green PCR Master Mix (Eurogentec). Liver expression of all genes has been normalised using three housekeeping genes (Gapdh, Hprt and Ppia). The primers used are described in online supplementary table S1. Relative expression was calculated using the 2-delta-delta CT method followed by geometric average, as recommended.19 ,20
Hepatic lipid analysis
Fifty micrograms of frozen liver tissue was homogenised by sonication in 1 mL of acetone. After overnight incubation at 4°C, samples were centrifuged at 4000 g for 10 min. Triglyceride (Diasys, Holzheim, Germany) and total cholesterol (Biomérieux, Craponne, France) contents were then measured in the supernatant using enzymatic kits according to manufacturers’ instructions. Triglycerides and total cholesterol content were normalised to protein content determined by Dc Protein Assay (BioRad, Marnes la Coquette, France).
Liver cytokine and chemokine protein level
Cytokines were quantified on total liver protein extracts. Frozen liver tissue samples were homogenised in TNE buffer (10 mmol/L Tris-HCl (pH 7.5), 1% Nonidet P40, 150 mmol/L NaCl, 1 mmol/L EDTA) containing a cocktail of protease and phosphatase inhibitors (Roche). The suspension was centrifuged at 20 000 g for 20 min at 4°C. The supernatant was then stored at −80°C before use. Levels of tumour necrosis factor (TNF)-α and monocyte chemoattractant protein-1 (MCP-1) in liver extracts were measured using a cytometric bead array (Mouse Flowcytomix, eBiosciences). Cytokines levels were normalised to protein content determined by Dc Protein Assay (BioRad, Marnes-la-Coquette, France).
Western blot
About 50 mg of frozen mouse liver tissue samples were homogenised in 650 µL of TNE buffer (10 mmol/L Tris-HCl (pH 7.5), 1% Nonidet P40, 150 mmol/L NaCl, 1 mmol/L EDTA) containing a cocktail of protease and phosphatase inhibitors (Roche). Lysates were incubated at 4°C for 45 min and then centrifuged at 12 000 g for 5 min. Protein content was quantified in the supernatant using BCA Protein Assay (Thermo Fisher). Lysates were mixed with the reducing sample buffer (Bio-Rad) for electrophoresis and subsequently transferred onto polyvinylidene fluoride membranes (Thermo Fisher). Equal loading (50 µg) was verified using Ponceau red solution. Membranes were incubated with anti-PPARα (1:1000, Ab24509, Abcam) antibody. After secondary antibody incubation (anti-rabbit, 1:3000; Amersham, GE Healthcare, Courtaboeuf, France), immunodetection proceeded using an enhanced chemiluminescence kit (Clarity Western ECl Substrate, BioRad, France) and bands were revealed using the Las-4000 Imaging System and Image MultiGauge software (Fujifilm, Tokyo, Japan). After initial immunodetection, membranes were stripped of antibodies and reprobed with mouse anti-GAPDH antibody (1:20 000; Millipore) and secondary antibody (anti-mouse, 1:3000; Amersham GE healthcare).
Measurement of hepatic total ketone bodies
Ketones were measured by the Williamson method.21 Briefly, frozen liver samples (200 mg) were deproteinised using perchloric acid 40%. Neutralised supernatants were used to measure acetoacetate and beta-hydroxybutyrate by a colorimetric method using beta-hydroxybutyrate deshydrogenase (Roche). The generation or the consumption of reduced nicotinamide adenine dinucleotide was measured by changes in absorbance at 340 nm. Results were expressed as micromole of ketone bodies per gram of liver tissue.
Patients
Twenty-five patients who underwent a percutaneous liver biopsy between 2008 and 2013 were selected from a pathological database: 6 had no or mild abnormalities at liver histological examination, 8 had bland steatosis and 11 had NASH as defined using the SAF (for steatosis, activity and fibrosis) score.17 None of the patients had detectable anti-hepatitis C virus antibodies or HBs antigen. Alcohol consumption was below 21 drinks on average per week in men and 14 drinks on average per week in women.1 None of the patients had a history of TIPS or of liver transplantation. Liver biopsy specimens were reviewed by a single experienced pathologist (VP) who was blinded to the clinical and laboratory data.
Statistics
Quantitative variables were expressed as median (interquartile range, IQR). Comparisons between groups of independent quantitative variables were performed using Mann–Whitney test. Spearman's correlation coefficient was used to test the correlation between liver miR-21 expression levels and pathological features. All tests were two-sided and used a significance level of 0.05. Data analysis was performed with SPSS V.17.0 (SPSS, Chicago, Illinois, USA) and figures were designed using GraphPad Prism 5 software.
Results
Expression of miR-21 is increased in biliary and inflammatory cells in the liver of Ldlr−/− mice under HFD
MiR-21 expression was increased in the liver of Ldlr−/− mice fed an HFD compared with WT mice under chow diet (figure 1B). Using in situ hybridisation, we observed that miR-21 was primarily expressed in biliary and inflammatory cells in the liver of Ldlr−/− mice under HFD (figure 1C). In accordance with real-time PCR results, the staining frequency and intensity of miR-21 was higher in Ldlr−/− mice under HFD than in controls. In these controls, miR-21 staining was faint and restricted to biliary cells. To ascertain these observations, miR-21 expression was determined in liver inflammatory (CD45+) versus non-inflammatory (CD45−) cells from Ldlr−/− mice under HFD as well as in subpopulations of inflammatory cells. MiR-21 expression was overexpressed in CD45+ cells and particularly in T cells (figure 1D, E).
In order to determine the potential role of miR-21 in NASH, we used an antagomir strategy. As shown in figure 1B, antagomir-21 strongly decreased liver miR-21 expression in Ldlr−/− mice under HFD. When compared with antagomir control, antagomir-21 had no effect on serum fasting glucose, total cholesterol, HDL and triglyceride levels (see online supplementary table S2). These results thus establish the use of antagomir-21 in Ldlr−/− mice under HFD as a valid strategy to investigate the role miR-21 in NASH.
Antagomir-21 reduces liver injury, liver inflammation and liver fibrosis but not liver lipid accumulation in Ldlr−/− mice under HFD
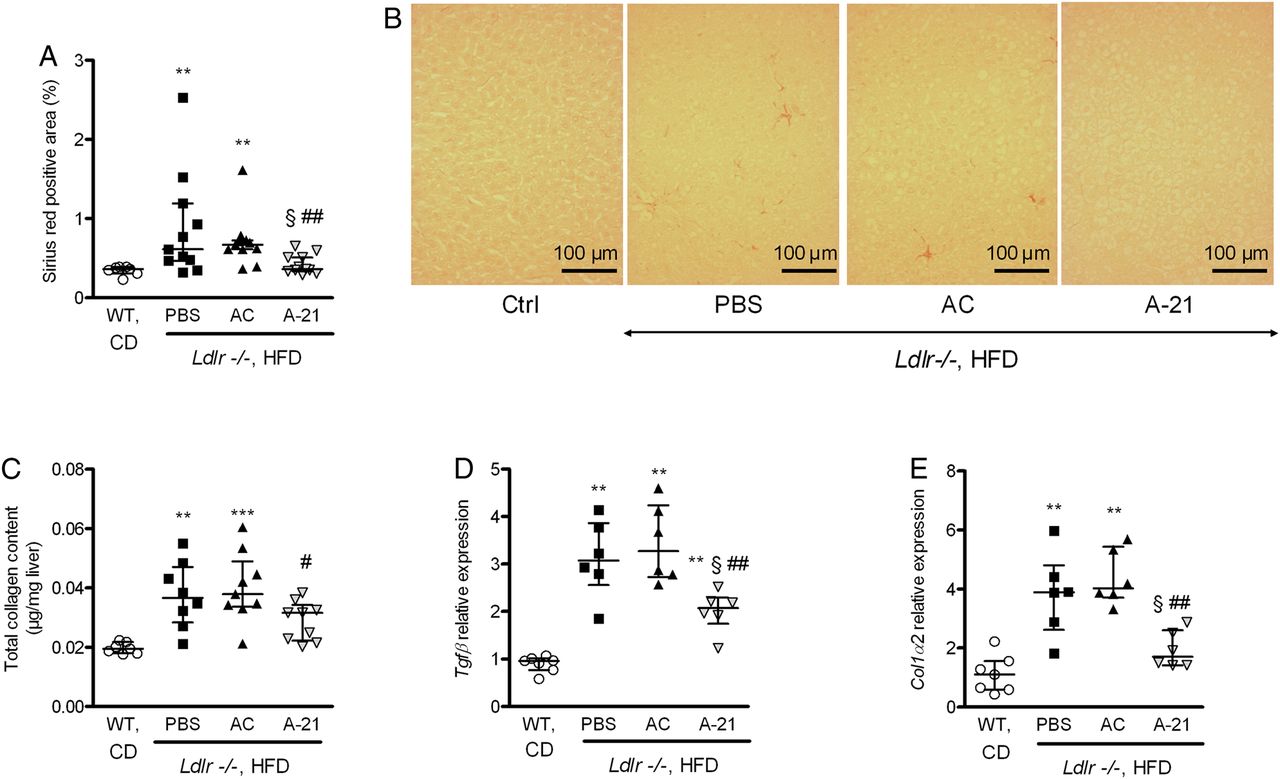
As expected in this mouse model of NASH, markers of liver injury (serum AST and ALT; figure 2A, B), of liver inflammation (liver MCP-1 and TNF-α gene and protein expression, liver CD3 and CD68 cell staining; figure 2C–J), of liver fibrosis (collagen staining with Sirius red, collagen quantification and gene expression of the fibrogenic-related genes Tgfβ and collagen-1α2; figure 3) and of liver lipid accumulation (liver triglycerides and total cholesterol levels and histological evaluation; see online supplementary figure S2) were higher in Ldlr−/− mice under HFD than in WT mice under chow diet.

Antagomir-21 reduced liver injury and liver inflammation in the liver of low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed a high-fat diet (HFD). Antagomir-21 (A-21) reduced serum aspartate aminotransferase (AST) (A) and alanine aminotransferase (ALT) (B) levels, liver monocyte chemoattractant protein-1 (MCP1) (C and D) and tumour necrosis factor (TNF)-α (E and F) gene and protein expressions, as well as CD3+ (G, quantification; H, representative images, original magnification ×200) and CD68+ (I, quantification; J, representative images, original magnification ×400) cell infiltrates. *p<0.05, **p<0.01 versus wild-type (WT) mice fed a chow diet (CD); §p<0.05, §§p<0.01 versus Ldlr−/− mice fed an HFD treated with phosphate buffer saline (PBS); #p<0.05, ##p<0.01 versus Ldlr−/− mice fed an HFD treated with antagomir control (AC). Data are given as median (horizontal bar) and IQR (error bar). Samples were randomly selected in each group of mice.

Antagomir-21 (A-21) reduced liver fibrosis and expression of fibrogenic-related genes in the liver of low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed a high-fat diet (HFD). A-21 reduced liver collagen staining (Sirius red; A, quantification; B representative images, original magnification ×200), collagen deposition (C), as well as liver normalised Tgfβ (D), and collagen-1α2 (E) mRNA expression. *p<0.05, **p<0.01, ***p<0.001 versus wild-type (WT) mice fed a chow diet (CD); §p<0.05 versus Ldlr−/− mice fed an HFD treated with phosphate buffer saline (PBS); #p<0.05, ##p<0.01 versus Ldlr−/− mice fed a HFD treated with antagomir control (AC). Data are given as median (horizontal bar) and IQR (error bar). Samples were randomly selected in each group of mice.
Antagomir-21 strongly reduced markers of liver cell injury (figure 2A, B), of hepatic inflammation (figure 2C–J) and of liver fibrosis (figure 3), but had no effect on liver lipid accumulation or on fatty acid ß-oxidation (liver gene expression of the ß-oxidation-related genes carnitine palmitoyltransferase 1 (Cpt1) and acyl-coenzyme A oxidase 1 (Acox1) and liver total ketone bodies) (see online supplementary figure S2).
MiR-21−/− mice under MCD diet have lower liver injury, liver inflammation and liver fibrogenic genes expression, but no change in liver lipid accumulation
We next analysed a second complementary model of NASH induced by MCD diet in miR-21−/− versus WT mice (figure 4A). As shown in figure 4B, liver miR-21 was increased by MCD diet in WT mice but remained undetectable in miR-21−/− mice. Fasting serum glucose, total cholesterol, HDL and triglyceride levels were not different between miR-21−/− and WT mice fed an MCD diet (see online supplementary table S3).

MicroRNA-21 (miR-21) was overexpressed in the liver of wild-type (WT) mice fed a methionine-choline-deficient (MCD) diet. (A) Mouse study design. (B) miR-21 expression in the liver of WT mice fed a chow diet (CD) or an MCD diet as well as in miR-21−/− mice fed an MCD diet. ***p<0.001 versus WT mice fed a chow diet (CD); ###p<0.001 versus WT mice fed an MCD diet. Data are given as median (horizontal bar) and IQR (error bar). (C) In situ hybridisation. Arrows point to liver inflammatory cells and arrow heads point biliary cells. Original magnification ×200. PV, portal vein.
As expected in this mouse model of NASH, markers of liver injury (serum AST and ALT; figure 5A, B), of liver inflammation (liver MCP-1 and TNF-α expression, liver CD3 and CD11b cell number; figure 5C–H), of liver fibrosis (collagen deposition and Tgfβ and collagen-1α2 gene expression; figure 6) and of liver lipid accumulation (liver triglycerides and total cholesterol levels and histological evaluation; see online supplementary figure S3) were increased in WT mice fed an MCD diet compared with WT mice fed a chow diet.
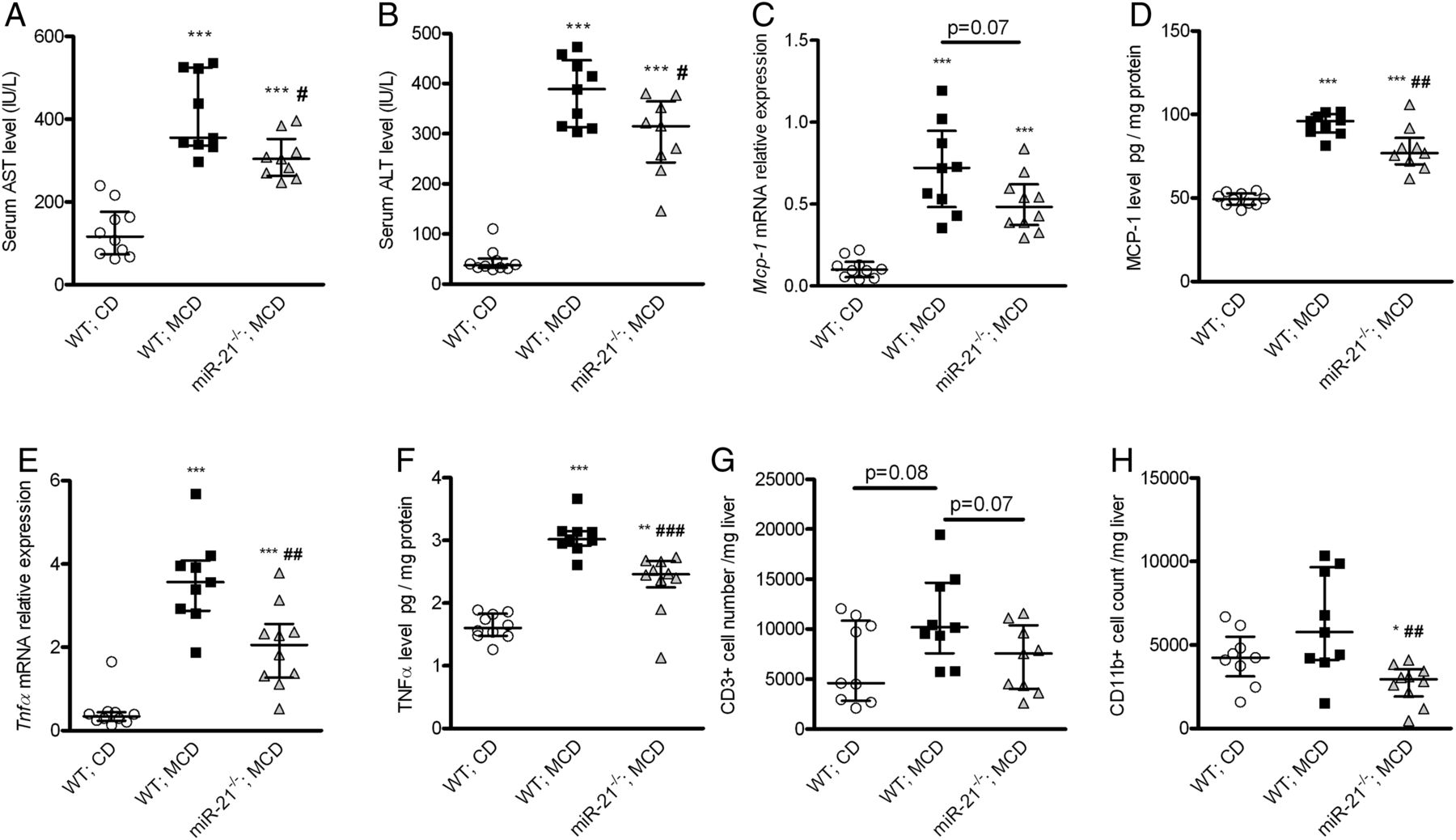
MiR-21−/− mice fed a methionine-choline-deficient (MCD) diet had reduced liver injury and liver inflammation. When compared with wild-type (WT) mice, miR-21−/− mice had reduced serum aspartate aminotransferase (AST) (A) and alanine aminotransferase (ALT) (B) levels, liver monocyte chemoattractant protein-1 (MCP-1) (C, D) and tumour necrosis factor (TNF)-α (E, F) gene and protein expressions, as well as lower CD3+ (G, not significant) and CD11b+ (H) cell infiltrates. *p<0.05, **p<0.01, ***p<0.001 versus WT mice fed a chow diet (CD). #p<0.05, ##p<0.01, ###p<0.001 versus WT mice fed an MCD diet. Data are given as median (horizontal bar) and IQR (error bar).
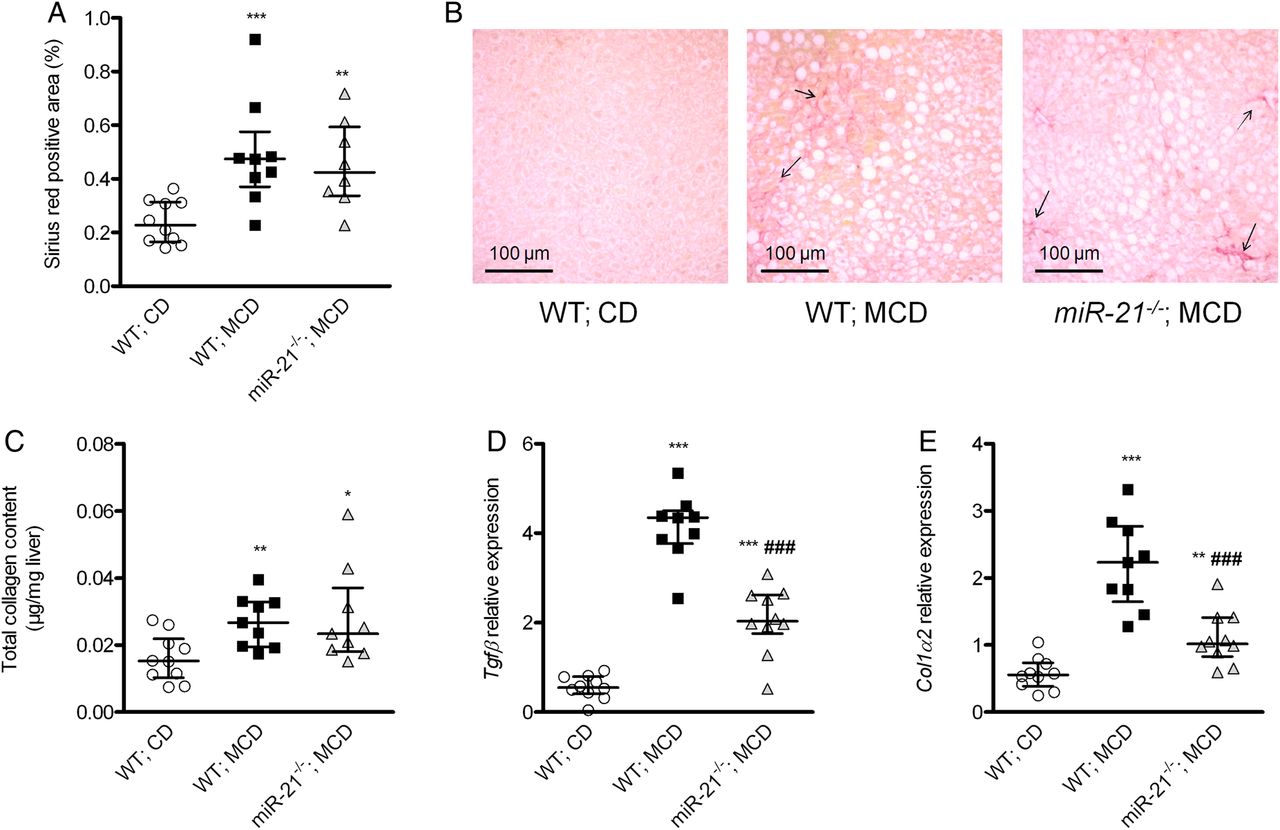
MiR-21−/− mice fed a methionine-choline-deficient (MCD) diet had reduced expression of fibrogenic-related genes. When compared with wild-type (WT) mice, miR-21−/− mice had similar liver collagen staining (Sirius red; A, quantification; B representative images, original magnification ×200) and collagen deposition (C), but reduced liver normalised Tgfβ (D) and collagen-1α2 (E) mRNA expression. *p<0.05, **p<0.01, ***p<0.001 versus WT mice fed a chow diet (CD). ##p<0.01 versus WT mice fed an MCD diet. Data are given as median (horizontal bar) and IQR (error bar).
When compared with WT mice fed an MCD diet, miR-21−/− mice fed an MCD diet had decreased liver injury and hepatic inflammation (figure 5). Although liver expression of the fibrogenic-related genes Tgfβ and collagen-1α2 was lower in miR-21−/− than in WT mice fed an MCD diet, there was no significant difference in liver collagen deposition (figure 6). Liver lipid accumulation and liver expression of the fatty acid ß-oxidation-related genes Cpt1 and Acox1 were not different between miR-21−/− and WT mice fed an MCD diet (see online supplementary figure 3A–F). Liver total ketone bodies were slightly lower in miR-21−/− than in WT mice fed an MCD diet (see online supplementary figure S3G).
PPARα is responsible for miR-21 effect in NASH
To explore the molecular mechanisms through which miR-21 exerts its function in NASH, we used the miRWalk algorithm and identified 231 genes validated as having a miR-21 target site. We then searched MEDLINE to select among these genes those relevant to NASH, that is, those with a known downregulated liver expression in NASH. We used each one of the 231 gene names together with the terms “steatohepatitis OR NASH OR fatty liver”. We identified phosphatase and tensin homologue deleted on chromosome 10 (PTEN), caspase 9 and PPARα. Out of these three potential targets, we screened those that would fit with our observation of increased miR-21 expression in biliary and inflammatory cells (figures 1 and 4). Only PPARα fulfilled this criteria.22–25 In both mouse models, liver PPARα gene and protein expressions were decreased in NASH and were restored by pharmacological inhibition or genetic suppression of miR-21 (figure 7).

MicroRNA-21 (miR-21) inhibition or suppression restored liver peroxisome proliferation-activator receptor α (PPARα) expression. (A–C) Antagomir-21 (A-21) restored liver PPARα gene (A) and protein (B and C) expression. *p<0.05, **p<0.01 versus wild-type (WT) mice fed a chow diet (CD); §p<0.05; §§p<0.01 versus low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed a high-fat diet (HFD) treated with phosphate buffer saline (PBS); ##p<0.01 versus Ldlr−/− mice fed an HFD treated with antagomir control (AC). (D–F) MiR-21−/− mice fed a methionine-choline-deficient (MCD) diet had restored liver PPARα gene (D) and protein (E and F) expression. *p<0.05, **p<0.01, versus WT mice fed a chow diet (CD). ###p<0.001 versus WT mice fed an MCD diet. Data are given as median (horizontal bar) and IQR (error bar). Liver samples randomly selected in each group of mice.
To firmly establish that the effect of miR-21 implicates PPARα, WT and Pparα−/− mice fed an MCD diet for 5 weeks were treated with antagomiR-21 or antagomir control (figure 8A). Liver miR-21 expression was increased by MCD diet both in WT and in Pparα−/− mice and was dramatically decreased upon antagomiR-21 treatment (figure 8B). In accordance with results obtained in Ldlr−/− mice, miR-21 was overexpressed in CD45+ cells, particularly in T cells, and was also detected in biliary cells (figure 8C, D).

Antagomir-21 (A-21) did not improve liver cell injury or liver inflammation in Pparα−/− mice fed a methionine-choline-deficient (MCD) diet. (A) Mouse study design. (B) Liver microRNA-21 (miR-21) expression. (C) miR-21 expression in sorted liver inflammatory (CD45+) and non-inflammatory (CD45−) cells from wild-type (WT) mice fed an MCD diet for 5 weeks. (D) miR-21 expression in subpopulations of liver inflammatory (CD45+) cells and in biliary cells (Epcam+) from WT mice fed an MCD diet for 5 weeks. A-21 did neither reduce serum aspartate aminotransferase (AST) (E) or alanine aminotransferase (ALT) (F) levels, nor liver Mcp1 gene expression (G) or CD3+ (H) cell infiltrates in Pparα−/− mice. *p<0.05, **p<0.01, ***p<0.001 versus WT mice fed a chow diet (CD). #p<0.05, ##p<0.01, ###p<0.001 versus WT mice fed an MCD diet and treated with antagomir control (AC). Data are given as median (horizontal bar) and IQR (error bar).
When compared with WT mice fed an MCD diet, Pparα−/− fed an MCD diet had lower serum HDL cholesterol and glucose levels, more steatosis and lower liver expression of the ß-oxidation genes Cpt1 and Acox1, but no significant change in liver cell injury, hepatic inflammation or liver fibrosis (see online supplementary table S4, figure S4 and figures 8 and 9). Similarly to what we observed in Ldlr−/− mice, antagomir-21 also improved liver cell injury, hepatic inflammation and liver fibrosis in WT mice fed an MCD diet. Importantly, this effect was lost in Pparα−/− mice fed an MCD diet (figures 8 and 9).
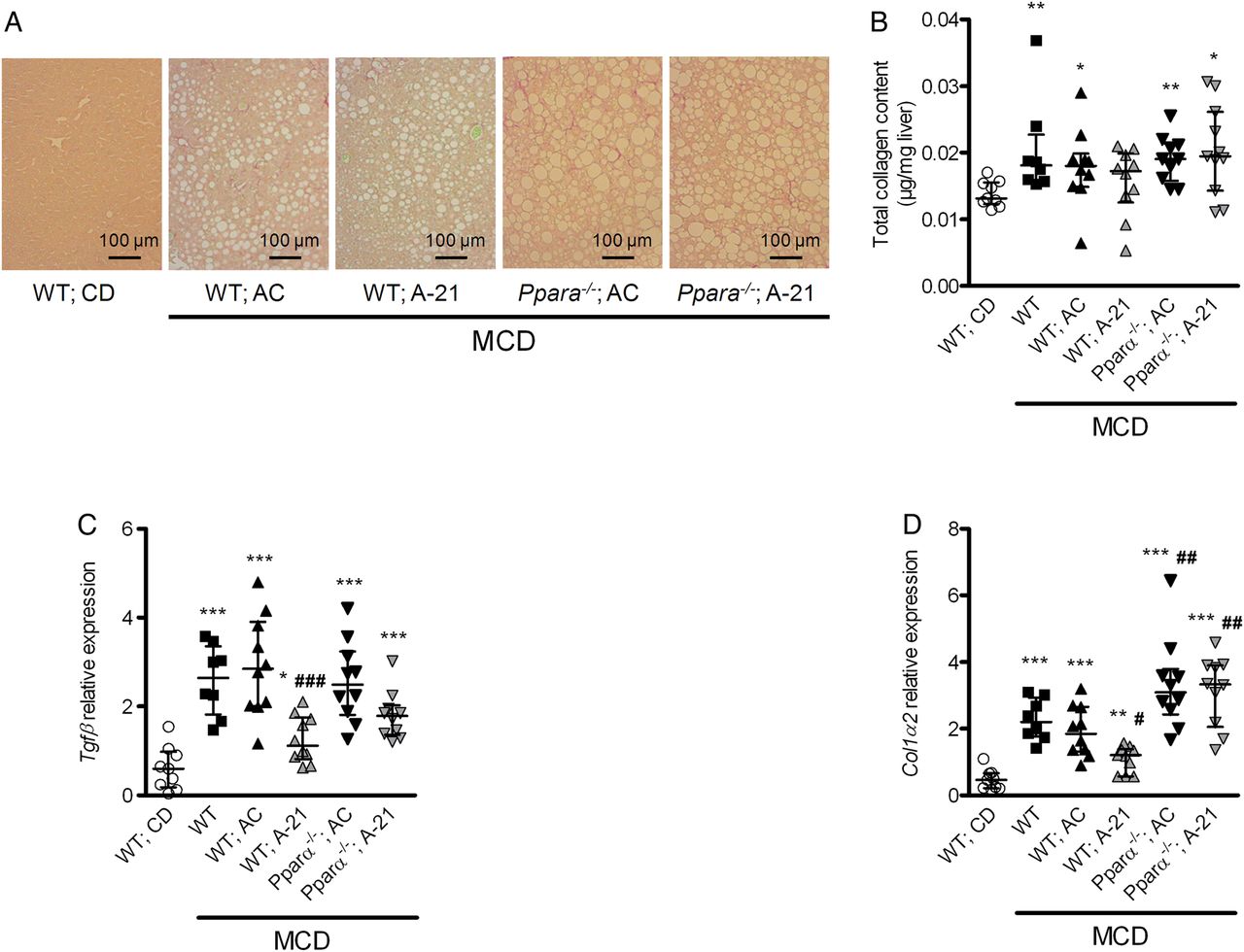
Antagomir-21 (A-21) did not improve liver fibrosis in Pparα−/− mice fed a methionine-choline-deficient (MCD) diet. A-21 did not reduce liver collagen staining (A; Sirius red, representative images, original magnification ×200), collagen deposition (B) or liver normalised Tgfβ (C) and collagen-1α2 (D) mRNA expression in Pparα−/− mice fed an MCD diet. *p<0.05, **p<0.01, ***p<0.001 versus wild type (WT) mice fed a chow diet (CD). #p<0.05, ###p<0.001 versus WT mice fed an MCD diet and treated with antagomir-21. Data are given as median (horizontal bar) and IQR (error bar).
miR-21 is overexpressed in the liver of patients with NASH primarily in inflammatory cells
Online supplementary table S5 summarises the features of the 25 patients included in this study. As shown in figure 10A, miR-21 was overexpressed in the liver of patients with NASH compared with patients with no or mild abnormalities at histological examination or, interestingly, with bland steatosis. Liver miR-21 expression correlated better with liver activity (ballooning, r=0.713, p<0.001; lobular inflammation, r=0.735, p<0.001) than with steatosis (r=0.539, p=0.005) or fibrosis (r=0.461, p=0.021). Using fluorescent in situ hybridisation, we observed that miR-21 was primarily expressed in human biliary (CK19+) and inflammatory (CD3+) cells in the liver from patients with NASH, similarly to what was observed in Ldlr−/− mice fed an HFD (figure 10B, C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MicroRNA-21 (miR-21) was overexpressed in the liver of patients with non-alcoholic steatohepatitis (NASH), primarily in inflammatory and biliary cells. (A) miR-21 expression normalised to U6 snRNA in the liver of patients with no or mild abnormalities at histological examination (control) or with bland steatosis or with NASH. *p<0.05, **p<0.01. (B) In situ hybridisation. miR-21 was detected in inflammatory (arrow) and biliary cells (arrow head). U6 probe is the positive control and no probe the negative control. Original magnification ×200. (C and D) Fluorescence in situ hybridisation (FISH). Combined FISH was performed for miR-21 and CD3 (lymphocyte marker) or CK19 (biliary cells marker) in liver sections from controls or patients with NASH. miR-21 expression (red) was detected in CD3-positive cells (C, green) from patients with NASH and in CK19-positive cells from patients with NASH and from controls (D, green). Original magnification ×630. Representative of three experiments. PV, portal vein.
Discussion
This study demonstrates that miR-21 inhibition or suppression decreases liver injury, inflammation and fibrosis, in different murine models of NASH, by restoring PPARα expression. These findings support the hypothesis that miR-21 is involved in the pathogenesis of NASH. We also show that miR-21 is primarily expressed in liver inflammatory and biliary cells in murine models and in patients with NASH, thereby enhancing the clinical relevance of our findings. Taken all together, these findings point to the potential of antagomir-21 as a possible treatment for NASH.
A first major finding in this study was that miR-21 inhibition or suppression reduced liver cell injury, liver inflammation and liver fibrogenesis in complementary models of NASH. The effect of the pharmacological inhibition of miR-21 appeared stronger than the genetic inhibition. Indeed, Ldlr−/− mice fed an HFD and treated with antagomir-21 had normal markers of liver injury, liver inflammation and liver fibrosis, while these parameters were reduced but not normalised in miR-21−/− mice fed an MCD diet. There was even no change in collagen deposition in miR-21−/− mice despite a reduction in liver expression of fibrogenic-related genes. We can speculate that genetic deletion of miR-21 occurring in early life leads to the development of compensatory mechanisms that could account for that observation.
A second major finding in this study was that miR-21 expression was elevated in the liver in patients with NASH compared with those with bland steatosis or with no or mild abnormalities at liver histological examination. The difference between patients with NASH and normal liver had been previously suggested by microRNA microarray assay, but not demonstrated by real-time quantitative PCR, and patients with bland steatosis had not been analysed.7 This result contrasts with the normal circulating miR-21 levels reported in patients with NASH.26 Importantly, we showed here in mice and in patients that this augmented liver miR-21 expression resides predominantly in inflammatory and biliary cells, and not in hepatocytes. This result is reinforced by the very significant correlation between liver miR-21 expression and activity score in the liver of patients. Liver infiltration with inflammatory cells is a key feature of NASH since this is one of the minimal criteria for the histological diagnosis of adult NASH.17 ,27 MiR-21 has been reported to be upregulated in many inflamed states, including lungs in lipopolysaccharide-treated mice,28 allergic airway inflammation,29 osteoarthritis,30 psoriasis and atopic eczema,31 disease-active UC tissue,32 Helicobacter pylori-associated gastric cancer,33 cardiac muscle injury34 and cardiac hypertrophy.35 Interestingly, miR-21 expression is upregulated by lipopolysaccharide in macrophages and human peripheral blood mononuclear cells.36 This feature might explain the upregulation of miR-21 in NASH since plasma lipopolysaccharide levels are higher in patients and mice with NAFLD than controls.37 ,38
Biliary cells also seem to play a key role in NASH. In patients with NASH, there is an increased ductular reaction compared with simple steatosis, which correlates with the stage of fibrosis, with the grade of NASH activity, as well as with the grade of portal inflammation.39 MiR-21 is expressed in cholangiocytes in a basal manner and overexpressed following exposure to lipopolysaccharide.40 We can speculate that endotoxemia associated with NASH could increase miR-21 expression in biliary cells.
To explore the molecular mechanism involving miR-21 in NASH, we first identified 231 genes validated as having an miR-21 target site. When selecting those relevant to NASH, that is, those for which liver expression was downregulated in NASH, we identified phosphatase and tensin homologue deleted on chromosome 10 (PTEN), caspase 9 and PPARα. However, in fatty liver, PTEN expression is decreased specifically in fatty hepatocytes.41 This did not fit with our findings, showing that miR-21 is overexpressed specifically in biliary and inflammatory cells. Similarly, caspase 9 has a reduced liver expression only in ballooned hepatocytes in NASH.42 Therefore, it was unlikely that PTEN and caspase 9 were responsible for miR-21 effect in NASH. PPARα is a well-known target of miR-21.43 ,44 Previous studies reported that PPARα expression is decreased in the liver in patients and rats with NASH, and inversely correlates with NASH severity.22 ,23 As PPARα is expressed not only by hepatocytes, but also by biliary cells and inflammatory cells, this prompted us to test the hypothesis that miR-21 contributes NASH by targeting PPARα.24 ,25 ,45 We first observed in the murine models of NASH that changes in liver PPARα mRNA and protein expression mirrored those of miR-21. Pharmacological inhibition and genetic suppression of miR-21 restored liver PPARα expression in the murine NASH models. Second, although not specific, changes in liver PPARα levels were accompanied by opposite changes in the downstream molecules of the PPARα pathway, including TNF-α and MCP-1.44 ,46 Third, PPARα is known to be expressed in inflammatory and biliary cells, that is, the cells where we observed an miR-21 upregulation.24 ,25 Interestingly, a recent report has shown that PPARα inhibits hepatic inflammatory responses leading to liver injury and progression to fibrosis by a mechanism independent of its effect on lipid accumulation in hepatocytes.45 This latter study suggests that steatosis is related to decreased expression of PPARα in hepatocytes and that inflammation and fibrosis are related to decreased expression of PPARα in inflammatory cells. Fourth and most importantly, we observed that the beneficial effect of antagomir-21 on liver cell injury, hepatic inflammation and liver fibrosis was lost in Pparα−/− mice fed an MCD diet. Altogether, this suggests that miR-21, expressed mainly in the liver in inflammatory cells, regulates liver inflammation and fibrosis via PPARα inhibition in these cells, but has no effect on steatosis because miR-21 is weakly expressed in hepatocytes.
MiR-21 is considered a druggable target microRNA.5 The present results suggest that inhibition of the miR-21 pathway might be an interesting new avenue for treating patients with NASH in the future, similarly to miR-122 inhibition in patients with chronic hepatitis C virus infection.47 Such a strategy may overcome the limitations of PPARα pharmacological agonists in NASH. Indeed, even though the novel dual PPARα/δ agonist GFT505 seems promising, trials with fibrates, that is, PPARα activators, have yielded mixed results in humans so that they are currently not recommended in patients.1 ,48 ,49 Yet, while hepatitis C virus can be eradicated, one would speculate that NASH would recur after treatment interruption as long as the risk factors for NASH persist. Therefore, long-term miR-21 antagonism treatment would be necessary with inherent risks of long-term side effects. Careful study design and safety monitoring would be critical.
In conclusion, we demonstrated here using complementary mouse models of NASH that inhibition and suppression of miR-21 strongly decreased liver injury, liver inflammation and fibrogenesis, through liver PPARα normalisation. We also showed that miR-21 was overexpressed in the liver of patients with NASH primarily in inflammatory and in biliary cells. Antagomir-21 might be a possible therapeutic strategy for NASH.
Acknowledgments
We wish to acknowledge Marion Dalloz for excellent technical assistance and Adrien Laigle for his helpful contribution. We also thank the members of the INSERM U970 ERI facility. We gratefully acknowledge the tumour biobank ‘Tissutheque Beaujon’ (Pathology Department, Beaujon Hospital) for technical support. BS is a member of the Institut Universitaire de France.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Correction notice This article has been corrected since it published Online First. The Open Access licence has been added.
Contributors XL, P-ER, and AT obtained funding, conceived and designed the study, and interpreted the data. XL and P-ER drafted the manuscript. P-ER collected the patients clinical and laboratory data. CMB and AT contributed to the writing of the manuscript. XL, A-CV, CH, NC, CLG, CD, SO, JS, MR and J-LP carried out experiments and analysed the data. MER provided miR-21−/− mice. CPB carried out hepatic ketonic bodies measurement and discussed data and manuscript. EB and BS provided Pparα−/− mice and discussed the data and the manuscript. PM, FD, PB and VP provided liver samples and contributed to the analysis of the data. All authors critically revised the manuscript and approved its final version.
Funding This work was supported by ‘appel à projets scientifiques de Sorbonne Paris Cité 2012’, ANR 11-META-004-02 MIRA, Fondation de France (2012-00029497) and Institut National de la Santé et de la Recherche Médicale (INSERM). XL was supported by ‘Fondation pour la Recherche Médicale’ (FRM), ‘Fondation Lefoulon-Delalande’ and ANR-Meta-2011-miR-A. P-ER was supported by ‘bourse Robert Tournut’ of the Société Nationale Française de Gastroentérologie (SNFGE) and the Association Française pour l'Etude du foie (AFEF), and A-CV by a grant from Région Ile-de-France (CODDIM).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We agree to share all primary data and protocols employed in this study.