Article Text

Abstract
Over the last 10–15 years, our understanding of the composition and functions of the human gut microbiota has increased exponentially. To a large extent, this has been due to new ‘omic’ technologies that have facilitated large-scale analysis of the genetic and metabolic profile of this microbial community, revealing it to be comparable in influence to a new organ in the body and offering the possibility of a new route for therapeutic intervention. Moreover, it might be more accurate to think of it like an immune system: a collection of cells that work in unison with the host and that can promote health but sometimes initiate disease. This review gives an update on the current knowledge in the area of gut disorders, in particular metabolic syndrome and obesity-related disease, liver disease, IBD and colorectal cancer. The potential of manipulating the gut microbiota in these disorders is assessed, with an examination of the latest and most relevant evidence relating to antibiotics, probiotics, prebiotics, polyphenols and faecal microbiota transplantation.
- INTESTINAL BACTERIA
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Imagine the scenario: a scientist at a conference claims to have found a new organ in the human body. It is comparable to the immune system in as much as it is made up of a collection of cells, it contains a 100 times more genes than the host, is host-specific, contains heritable components, can be modified by diet, surgery or antibiotics, and in its absence nearly all aspects of host physiology are affected. While this may seem far-fetched, it is the current situation in which we find ourselves. We now realise that the human microbiota is an overlooked system that makes a significant contribution to human biology and development. Moreover, there is good evidence that humans co-evolved a requirement for their microbiota.1
In the past decade, partly because of high resolution observational studies using next-generation sequencing technologies and metabolite profiling (see box 1), the gut microbiota has become associated with promotion of health and the initiation or maintenance of different GI and non-GI diseases. As we enter the postmetagenomic era, we need to move away from simple observations to determine what are merely correlations and what are causal links—and focus efforts and resources on the latter. This postmetagenomic era is starting to provide new therapeutic targets based on a better understanding of how the microbiota interacts with the host's physiology. Ultimately, we aim to integrate an individual's microbiota into some form of personalised healthcare and, by better understanding its role, treat an individual's diseases more efficiently and in a more targeted fashion. With a more complete understanding of the disease process, we will be able to more accurately stratify different disease states and determine whether or not the gut microbiota is a potential therapeutic target which we can modulate in order to treat specific diseases.
A short primer of microbiology (see also Lepage et al)4
Dysbiosis
A disturbance or imbalance in a biological system, for example, changes in the types and numbers of bacteria in the gut which may lead to developing different diseases, such as IBD.
Inflammasomes
Recently discovered multiprotein complexes that are involved in a wide range of inflammatory processes including programmed cell death (pyroptosis), in response to the recognition of microbial and danger signals.
Lipopolysaccharide (LPS)
A major component of the outer membrane of Gram-negative bacteria; an endotoxin. Now implicated as a driver of inflammation and associated with onset of certain diseases.
Lipoteichoic acid
A major component of the outer membrane of Gram-positive bacteria; an endotoxin. Now implicated as a driver of inflammation and associated with onset of certain diseases.
Metabonome/metabolome
A profile of the chemicals in a tissue or sample, for example the urine metabonome. This profile represents a snapshot in time of what chemicals are present in the sample.
Metagenomics
A method which allows us to create catalogues of what the bacteria can do based on the genes that they have.
Microbiome
A collection of different microbes and their functions or genes found in an environmental habitat. Different parts of the body have different microbiomes, for example, the skin microbiome is different to the gut microbiome, but they are all part of the human microbiome.
Microbiota
The types of organisms that are present in an environmental habitat, whether they are bacteria, viruses or eukaryotes.
‘Omic’ methods
A term which describes a set of methods, such as genomics, metabonomics, metagenomics, etc, which we use to explore the interactions between the bacteria in the gut and the host.
Pathobiont
A commensal organism that can cause disease when specific genetic or environmental conditions are altered in the host.
Phenome
A collection of measurable features that define an individual.
This review gives a much needed update on current understanding of the gut microbiota in GI diseases and metabolic disorders, and gives an insight into how this might impact on clinical practice. The evidence for the preventive and therapeutic benefit of different ways of modulating the gut microbiota, such as probiotics, prebiotics, antibiotics and faecal microbiota transplantation (FMT) (see box 2), is reviewed.
Potential therapies aimed at modulation of the gut microbiota
Probiotics: Live microorganisms that, when administered in adequate amounts, confer a health benefit on the host.107 ,108 Examples include strains of the genera Bifidobacterium and Lactobacillus. Probiotics can have multiple interactions with the host,109 including competitive inhibition of other microbes, effects on mucosal barrier function and interaction with antigen presenting dendritic cells.72
Prebiotics: A selectively fermented ingredient that results in specific changes in the composition and/or activity of the GI microbiota, thus conferring benefit(s) upon host health.110 Prebiotics are usually non-digestible carbohydrates, oligosaccharides or short polysaccharides, with inulin, oligofructose, galactofructose, galacto-oligosaccharides and xylo-oligosaccharides being some of the most intensively studied.
Faecal microbiota transplantation: The introduction of gut bacteria from a healthy donor into a patient, through transfer of an infusion of a faecal sample via nasogastric tube, nasoduodenal tube, rectal enema or the biopsy channel of a colonoscope.111
Current understanding of the gut microbiota
In the last decade, several large-scale projects, for example, the human microbiome project, have investigated the microbiota of a variety of bodily niches, including the skin as well as the oral, vaginal and nasal cavities.2 While some of these are relatively easy to access, the GI tract remains a challenging environment to sample, and to describe. Currently the majority of research is focused on the gut microbiota, since this is where the greatest density and numbers of bacteria are found, with most data being derived from faecal samples and, to a lesser extent, mucosal biopsies. While it is relatively easy to obtain fresh faecal samples, the information obtained from them does not represent the complete picture within the gut. From a number of limited studies, we know that the small intestine contains a very different abundance and composition of bacteria, with much more dynamic variation compared with the colon.3 The colonic microbiota is largely driven by the efficient degradation of complex indigestible carbohydrates but that of the small intestine is shaped by its capacity for the fast import and conversion of relatively small carbohydrates, and rapid adaptation to overall nutrient availability. While faeces are not an ideal proxy for the GI tract, they do give a snapshot of the diversity within the large intestine. Furthermore, the majority of the data comes from North American and European studies with very few studies in Asia, Africa or South America. Hence we have a somewhat biased view of the gut microbiota.
This rapid increase in interest in the microbiome has also been driven by the application of multi-‘omic’ technologies; we refer the reader to Lepage et al4 for more detailed explanation of these (see also box 1).
What do we know about the gut microbiota?
Bearing in mind the limitations above, the GI tract is often seen as a two phylum system (the Firmicutes and Bacteroidetes) although it should be noted that members of at least 10 different phyla can also have important functional contributions (see box 3). We are also very bacteria-centric when we look at the gut microbiota; only a handful of papers have looked at the viral component (or virome) and micro-eukaryotes (protozoa and fungi). When the gut microbiota of relatively large cohorts of individuals (eg, more than 100) is analysed, it can be seen that the ratio of the Firmicutes:Bacteroidetes is not the same in all individuals. Currently we do not know the significance of being at either end of this continuum, especially as a large shift in the relative abundance of a group of organisms translates to a modest change in bacterial numbers. Yet there is evidence that depletion of a single species, for example, Faecalibacterium prausnitzii, belonging to the Firmicutes phylum, has been associated with IBD.5 But in the scientific literature, we see counterarguments for any involvement of this species in IBD.6 This disparity highlights the current status of understanding. We know that the gut microbiota is essential to the proper function and development of the host but we are unsure which are keystone species and whether the microbiota's function is more important than any individual member of the community. But this is too simplistic a view. In several cases, strain differences within a species can be the difference between being a pathogen/pathobiont and being a probiotic: for example, Escherichia coli is associated with IBD and colorectal cancer (CRC)7 ,8 yet an E. coli strain is used as a probiotic.
A primer in taxonomics
In order to classify bacteria we have adopted the Linnaean system, which comprises hierarchies into which an organism is placed.
For example humans are classified at the species level as Homo sapiens, which are members of the genus Homo, family Hominidae, order Primates, class Mammalia, phylum Chordata and finally kingdom Eukaryota. As one moves up through the different taxonomic levels, from species to kingdom, greater numbers of organisms become associated with each other.
In life there are three kingdoms, the Bacteria, Archaea and Eukaryota, with the majority of bacterial-like (or prokaryotes) being classified within the Bacteria and Archaea. For example the gut commensal and sometime pathogenic species Escherichia coli is found in the kingdom Bacteria; phylum Proteobacteria; class Gammaproteobacteria; order Enterobacteriales; family Enterobacteriaceae and finally genus Escherichia. Thus when we refer to phyla or a phylum, we are usually describing very large collections of related organisms.
In the large intestine of healthy adults the two most dominant phyla are the Firmicutes (comprised mainly of Gram-positive clostridia) and Bacteroidetes (comprised mainly of Gram-negative bacteria such as the species Bacteroides fragilis).
In fact, five phyla represent the majority of bacteria that comprise the gut microbiota. There are approximately 160 species in the large intestine of any individual9 and very few of these are shared between unrelated individuals. In contrast, the functions contributed by these species appear to be found in everybody's GI tract, an observation that leads us to conclude that function is more important than the identity of the species providing it. Yet differences in the gut microbiota may matter because these may result in differences in the effectiveness of a function. For example, while the ability to synthesise short chain fatty acids (SCFAs) is found in all humans,10 their amounts can vary.
Dietary modulation of the gut microbiota
Metabolic activities of the gut microbiota
Carbohydrate fermentation is a core activity of the human gut microbiota, driving the energy and carbon economy of the colon. Dominant and prevalent species of gut bacteria, including SCFA-producers, appear to play a critical role in initial degradation of complex plant-derived polysaccharides,11 collaborating with species specialised in oligosaccharide fermentation (eg, bifidobacteria), to liberate SCFAs and gases which are also used as carbon and energy sources by other more specialised bacteria (eg, reductive acetogens, sulfate-reducing bacteria and methanogens).12 Efficient conversion of complex indigestible dietary carbohydrates into SCFA serves microbial cross-feeding communities and the host, with 10% of our daily energy requirements coming from colonic fermentation. Butyrate and propionate can regulate intestinal physiology and immune function, while acetate acts as a substrate for lipogenesis and gluconeogenesis.13 Recently, key roles for these metabolites have been identified in regulating immune function in the periphery, directing appropriate immune response, oral tolerance and resolution of inflammation, and also for regulating the inflammatory output of adipose tissue, a major inflammatory organ in obesity.14 In the colon, the majority of this carbohydrate fermentation occurs in the proximal colon, at least for people following a Western style diet. As carbohydrate becomes depleted as digesta moves distally, the gut microbiota switches to other substrates, notably protein or amino acids. Fermentation of amino acids, besides liberating beneficial SCFAs, produces a range of potentially harmful compounds. Some of these may play a role in gut diseases such as colon cancer or IBD. Studies in animal models and in vitro show that compounds like ammonia, phenols, p-cresol, certain amines and hydrogen sulfide, play important roles in the initiation or progression of a leaky gut, inflammation, DNA damage and cancer progression.15 On the contrary, dietary fibre or intake of plant-based foods appears to inhibit this, highlighting the importance of maintaining gut microbiome carbohydrate fermentation.16 Recognition of carbohydrate fermentation as a core activity of the gut microbiota provides the scientific basis for rational design of functional foods aimed at improving gut health and also for impacting on microbiota activities linked to systemic host physiology through newly recognised interkingdom axes of communication such as the gut:liver axis, the gut:brain axis and the gut:brain:skin axis.17
Three ‘P's’ for gut health: probiotics, prebiotics and polyphenols
A number of dietary strategies are available for modulating either the composition or metabolic/immunological activity of the human gut microbiota: probiotics, prebiotics and polyphenols are among the most well established.18
There are many examples of positive results with different probiotic strains against a range of disease states in animal models, however the human data are equivocal. This may partly be due to poor study design and poor choice of strain. However, there is also a persistent lack of understanding as to the very nature of probiotics, which cannot be considered a ‘class’ of bioactives, amenable to traditional efficacy assessments such as the meta-analysis (unless restricted to one strain), since they are all unique living organisms and their health-promoting traits are strain-specific. Rarely have probiotic strains been selected with specific mechanisms of effect in mind; this has led to conflicting observations and damaged the reputation of this area of science. A few exceptions do exist, most notably the work of Jones et al who selected a bile salt-hydrolysing Lactobacillus reuteri strain, to study its ability to reduce cholesterol levels in hypercholesterolaemic individuals. In two well powered, randomised, placebo-controlled and double-blinded studies, they demonstrated that ingestion of this strain significantly lowered total and low density lipoprotein (LDL)-cholesterol. Moreover, they suggested an underlying novel mechanism linked to reduced fat absorption from the intestine19 via the nuclear receptor farnesoid X receptor (FXR).20
Prebiotics represent a specific type of dietary fibre that when fermented, mediate measurable changes within the gut microbiota composition, usually an increase in the relative abundance of bacteria thought of as beneficial, such as bifidobacteria or certain butyrate producers. As with probiotics, despite convincing and reproducible results from animal studies showing efficacy in prevention or treatment of many diseases (eg, IBD, IBS, colon cancer, obesity, type 2 diabetes (T2D) and cardiovascular disease), the data in humans remain ambiguous. Fewer well powered or well designed clinical studies have been conducted with prebiotics compared with probiotics, and there may be an issue with prebiotic dose. Human studies rarely, if ever, employ prebiotics. A prebiotic is shown to be efficacious in animal studies: typically 10% w/w of the diet, which in humans equates to about 50 g per day.18 However, as we learn more about the ecology of the gut microbiota, it is becoming clear that the prebiotic concept has tapped into the underlying fabric of the gut microbiota as a primarily saccharolytic and fermentative microbes community evolved to work in partnership with its host's digestive system to derive energy and carbon from complex plant polysaccharides which would otherwise be lost in faeces.
Polyphenols are a diverse class of plant secondary metabolites, often associated with the colour, taste and defence mechanisms of fruit and vegetables. They have long been studied as the most likely class of compounds present in whole plant foods capable of affecting physiological processes that protect against chronic diet-associated diseases. The gut microbiota plays a critical role in transforming dietary polyphenols into absorbable biologically active species, acting on the estimated 95% of dietary polyphenols which reach the colon.21 Recent studies show that dietary intervention with polyphenol extracts, most notably dealcoholised red wine polyphenol extract and cocoa-derived flavanols, modulate the human gut microbiota towards a more ‘health-promoting profile’ by increasing the relative abundance of bifidobacteria and lactobacilli. These data again raise the possibility that certain functional foods tap into the underlying ecological processes regulating gut microbiome community structure and function, contributing to the health of the gut microbiota and its host.22
Obesity-related diseases and the gut microbiota
Starting around 2004, the hallmark studies of Gordon et al demonstrated a potential relationship between the gut microbiome and development of an obese phenotype. An increase in relative abundance of Firmicutes and a proportional decrease in Bacteroidetes were associated with the microbiota of obese mice,23 which was confirmed in a human dietary intervention study demonstrating that weight loss of obese individuals (body mass index, BMI>30) was accompanied by an increase in the relative abundance of Bacteroidetes.24 Nevertheless, based on most human studies, the obesity-associated decrease in the ratio of Bacteroidetes to Firmicutes (B:F) remains controversial.24 ,25 This is likely due to heterogeneity among human subjects with respect to genotype and lifestyle. Recent studies have identified diet, especially fat, as a strong modulator of the microbiota, particularly in inbred and age-standardised laboratory animals. The sources of variation in the microbiota are mainly limited to the experimental diets used, and there is growing evidence that the high fat intake rather than obesity per se had a direct effect on the microbiota and linked clinical parameters.26 However, in humans the microbiome is exposed to fundamentally different ‘environmental’ factors in obese and lean individuals that go beyond BMI alone, including diet26 and host hormonal factors.27 In addition, the aetiology of obesity and its metabolic complications, including low grade inflammation, hyperlipidaemia, hypertension, glucose intolerance and diabetes, reflect the complex interactions of these multiple genetic, behavioural and environmental factors.28 Lastly, the accuracy of BMI as an indicator for obesity is limited; 25% of obese people could in fact be regarded as metabolically ‘healthy’ (ie, with normal lipid and glucose metabolism).29 Therefore, linking GI tract microbial composition directly and exclusively to obesity in humans will remain challenging due to the various confounding factors within the heterogeneous population.
This complexity has led to a shift from treating obesity as a single phenotype, to attempts at correlating microbial signatures to distinct or multiple features associated with (the development of) metabolic syndromes such as T2D. Recently, two (meta)genome-wide association studies were performed, with 345 Chinese individuals30 and 145 European women.31 In both studies, de novo generated metagenomic species-level gene clusters were employed as discriminant markers which, via mathematical modelling, could better differentiate between patients and controls with higher specificity than a similar analysis based on either human genome variation or other known risk factors such as BMI and waist circumference. At the functional level, membrane transporters and genes related to oxidative stress were enriched in the microbiota of patients,31 while butyrate biosynthesis was decreased.30 Although both studies observed high similarities in microbial gene-encoded functions, the most discriminant metagenomic species-level gene clusters differed between the cohorts (Akkermansia did not contribute to the classification in the European cohort whereas Lactobacillus showed no contribution in the Chinese study population), indicating that diagnostic biomarkers could be specific to the population studied.
In another metagenomic study, a bimodal distribution of microbial gene richness in obese individuals was observed, stratifying individuals as High Gene Count or Low Gene Count (HGC and LGC).32 HGC individuals were characterised by higher prevalence of presumed anti-inflammatory species such as F. prausnitzii, and an increased production potential of organic acids (including butyrate). In contrast, LGC individuals showed higher relative abundance of potentially proinflammatory Bacteroides spp and genes involved in oxidative stress response. Remarkably, only biochemical obesity-associated variables, such as insulin resistance, significantly correlated with gene count while weight and BMI did not, underscoring the inadequacy of BMI as an indicator for ‘Obesity and its Associated Metabolic Disorders’ (OAMD).33 An accompanying paper demonstrated that a diet-induced weight-loss intervention significantly increased gene richness in the LGC individuals which was associated with improved metabolic status.34 Although gene richness was not fully restored, these findings support the reported link between long-term dietary habits and the structure of the gut microbiota.31 It also suggests permanent adjustment of the microbiota may be achieved through diet.
Most studies involving the microbiome have been solely correlative but recently a causal relationship was established between host glucose homoeostasis and gut microbial composition. FMT from lean donors to individuals with metabolic syndrome significantly increased their insulin sensitivity.33 The transplant produced an increase in faecal butyrate concentrations, microbial diversity and the relative abundance of bacteria related to the butyrate-producing Roseburia intestinalis.
Together, these studies produce a body of evidence that the microbiome plays a role in host energy homoeostasis and the establishment and development of OAMD, although the exact mechanisms remain obscure. Previous contradictory findings might be attributed to miscellaneous approaches,35 and also heterogeneity in genotype, lifestyle and diet of humans combined with the complex aetiology of OAMD. Nonetheless, a clearer picture is emerging. The gut of individuals with OAMD is believed to harbour an inflammation-associated microbiome, with a lower potential for butyrate production and reduced bacterial diversity and/or gene richness. Although the main cause of OAMD is excess caloric intake compared with expenditure, differences in gut microbial ecology might be an important mediator and a new therapeutic target or a biomarker to predict metabolic dysfunction/obesity in later life.
Liver disease and the gut microbiota
The liver receives 70% of its blood supply from the intestine via the portal vein, thus it is continually exposed to gut-derived factors including bacterial components, endotoxins (lipopolysaccharide, flagellin and lipoteichoic acid) and peptidoglycans. Multiple hepatic cells, including Kupffer cells, sinusoidal cells, biliary epithelial cells and hepatocytes, express innate immune receptors known as pathogen-recognition-receptors that respond to the constant influx of these microbial-derived products from the gut.36 It is now recognised that the gut microbiota and chronic liver diseases are closely linked. Characterising the nature of gut dysbiosis, the integrity of the gut barrier and mechanisms of hepatic immune response to gut-derived factors is potentially relevant to development of new therapies to treat chronic liver diseases.37 Furthermore the field of bile acid signalling has thrown open the concept of the gut:liver axis as being active and highly regulated.38
Non-alcoholic fatty liver disease
The pathophysiology of NAFLD is multifactorial with strong genetic and environmental contributions. Recent evidence demonstrates that gut microbiota dysbiosis can result in the development of obesity-related non-alcoholic fatty liver disease (NAFLD), and patients with NAFLD have small intestinal bacterial overgrowth and increased intestinal permeability.39 In the 1980s, development of non-alcoholic steatohepatitis (NASH) and small intestinal bacterial overgrowth was observed in humans after intestinal bypass and, interestingly, regression of hepatic steatosis after metronidazole treatment, suggesting a possible role for the gut bacteria in NAFLD.40 Disruption of the murine inflammasomes (see box 1) is associated with an increase in Bacteroidetes and reduction in Firmicutes and results in severe hepatic steatosis and inflammation.41 Faecal microbiota analysis of patients with NAFLD and NASH has produced variable results due to significant variation of patient demographics, severity of liver disease and methodology. A lower proportion of Ruminococcaceae was noted in patients with NASH compared with healthy subjects42 and a study which characterised gut microbiota of children with NASH, obesity and healthy controls showed that patients with NASH had a higher proportion of Escherichia compared with other groups.43 Patients with NAFLD also have increased gut permeability suggesting that translocation of bacteria or microbe derived products into the portal circulation contributes to the pathogenesis.39
Alcoholic liver disease
Since not all alcoholics develop liver injury, it appears that chronic alcohol abuse is necessary but not sufficient to cause liver dysfunction. Numerous animal model and human observational studies indicate that gut bacterial products like endotoxin may mediate inflammation and function as cofactors for the development of alcohol-related liver injury.44 Serum endotoxin levels are elevated in humans and rats with alcoholic liver disease, and monocytes from alcoholics are primed to produce cytokines after endotoxin exposure. Alcohol causes intestinal bacterial overgrowth in humans and bacterial numbers were significantly higher in jejunal aspirates from patients with chronic alcohol abuse compared with controls, with similar findings in patients with alcohol-induced cirrhosis.45 The degree of overgrowth correlates with the severity of cirrhosis. Tsukamoto-French model mice fed intragastrically with alcohol for 3 weeks showed increased relative abundance of Bacteroidetes and Akkermansia spp and a reduction in Lactobacillus, Leuconostoc, Lactococcus and Pediococcus while control mice showed a relative predominance of Firmicutes.46 Patients with alcoholic liver disease also show increased gut permeability, allowing translocation of bacteria and bacterial products to the liver.47
Autoimmune liver diseases
These consist of primary sclerosing cholangitis (PSC), primary biliary cirrhosis (PBC) and autoimmune hepatitis and represent at least 5% of all chronic liver diseases. They are presumed autoimmune conditions but the expectation is that the gut microbiota is relevant to pathogenesis, particularly because (A) PSC is associated with IBD and aberrant lymphocyte tracking, and (B) significant gut:liver axes exist through bile acid signalling. Patients with PSC develop a distinct form of IBD thus understanding the relationship between PSC and IBD is essential in uncovering the pathogenesis of PSC, which remains largely undetermined. However, it is likely that in genetically susceptible individuals, intestinal bacteria could trigger an abnormal or inadequate immune response that eventually leads to liver damage and fibrosis. Recently it was shown that patients with PSC have distinct gut microbiota. Analysis of colon biopsy microbiota revealed that patients with PSC-IBD and IBD showed reduced abundance of Prevotella and Roseburia (a butyrate-producer) compared with controls.48 ,49 Patients with PSC-IBD had a near-absence of Bacteroides compared with patients with IBD and control patients, and significant increases in Escherichia, Lachnospiraceae and Megasphaera. Randomised controllled trials (RCTs) investigating antibiotic therapy in PSC have shown these to be superior in improving biochemical surrogate markers and histological parameters of disease activity compared with ursodeoxycholic acid alone.50 In a recent prospective paediatric case series, oral vancomycin was shown to normalise or significantly improve liver function tests.51 There is evidence that mucosal integrity is compromised in patients with PSC, supporting the traditional leaky gut hypothesis of microbe-derived products translocating to the liver and biliary system to trigger an inflammatory reaction.52 It was also demonstrated that tight junctions of hepatocytes were impaired in patients with PSC and infusion of non-pathogenic E. coli into portal circulation caused portal fibrosis in animal models.53 These findings collectively suggest that bacterial antigens translocate across a leaky and possibly inflamed gut wall into the portal and biliary system to induce an abnormal immune response and contribute to PSC pathogenesis.
PBC is a chronic cholestatic liver disease with an uncertain aetiology. It is generally believed to be an autoimmune disease triggered by environmental factors in individuals with genetic susceptibility. As yet, there have been no studies directly characterising the gut microbiota in patients but molecular mimicry has been suggested as a proposed mechanism for the development of autoimmunity in PBC, with serum antibodies of patients cross-reacting with conserved bacterial pyruvate dehydrogenase complex component E2 (PDC-E2) homologues of E. coli, Novosphingobium aromaticivorans, Mycobacterium and Lactobacillus species. Hence it has been speculated that these bacteria (of possible GI origin) may initiate molecular mimicry and development of PBC in genetically susceptible hosts.54
Modulation of the microbiota as a therapy in liver disease
Probiotics have shown promise in ameliorating liver injury by reducing bacterial translocation and hepatic inflammation.55 A recent meta-analysis concluded that probiotics can reduce liver aminotransferases, total cholesterol, tumour necrosis factor α and improve insulin resistance in patients with NAFLD.56 A recent study in patients with cirrhosis with ascites showed that the probiotic VSL#3 significantly reduced portal hypertension.57 A further study evaluated the role of FMT in modulating liver disease by transferring the NAFLD phenotype from mice with liver steatosis to germ-free mice.58 There remains a need for detailed descriptive and interventional studies focused on bacterial diversity and mechanisms linking gut dysbiosis with inflammatory, metabolic and autoimmune/biliary liver injury.
IBD and the gut microbiota
Early studies implicating bacteria in IBD pathogenesis focused on identifying a potential culprit that could initiate the inflammatory cascade typical of IBD. Many organisms have been proposed: Mycobacterium avium subsp paratuberculosis and a number of Proteobacteria including enterohepatic Helicobacter, non-jejuni/coli Campylobacter and adherent and invasive E. coli. The focus has recently shifted with the realisation that the gut microbiota as a whole is altered in IBD. The concept of an altered gut microbiota or dysbiosis is possibly the most significant development in IBD research in the past decade. A definitive change of the normal gut microbiota with a breakdown of host-microbial mutualism is probably the defining event in IBD development.59
Changes in the gut microbiota have been repeatedly reported in patients with IBD, with certain changes clearly linked to either Crohn's disease (CD) or UC: the most consistent change is a reduction in Firmicutes.60 This has been balanced by reports of increased levels of Bacteroidetes phylum members,61 although a reduction in Bacteroidetes has also been reported.62 There is a suggestion that there may be spatial reorganisation of the Bacteroides species in patients with IBD, with Bacteroides fragilis being responsible for a greater proportion of the bacterial mass in patients with IBD compared with controls.63
Reduction in the Firmicutes species F. prausnitzii has been well documented in patients with CD, particularly those with ileal CD, although an increase in F. prausnitzii has been shown in a paediatric cohort, suggesting a more dynamic role for the species that merits further study.64 Other studies have also demonstrated a decrease in Firmicutes diversity, with fewer constituent species detected in patients with IBD compared with controls.65 Changes in the two dominant phyla, Firmicutes and Bacteroidetes, are coupled with an increase in abundance of members of the Proteobacteria phylum, which have been increasingly found to have a key role in IBD.66 Studies have shown a shift towards an increase in species belonging to this phylum, suggesting an aggressor role in the initiation of chronic inflammation in patients with IBD.67 More specifically, increased numbers of E. coli, including pathogenic variants, have been documented in ileal CD.68 The IBD metagenome contains 25% fewer genes than the healthy gut with metaproteomic studies showing a correlative decrease in proteins and functional pathways.69 Specifically, ileal CD has been shown to be associated with alterations in bacterial carbohydrate metabolism and bacterial-host interactions, as well as human host-secreted enzymes.69 A detailed investigation of functional dysbiosis during IBD built on this by including inferred microbial gene content from 231 subjects and an additional 11 metagenomes.70 This study identified enrichment in microbial pathways for oxidative stress tolerance, immune evasion and host metabolite uptake, with corresponding depletions in SCFA biosynthesis and typical gut carbohydrate metabolism and amino acid biosynthetic processes. Intriguingly, similar microbial metabolic shifts have been observed in other inflammatory conditions such as T2D,30 suggesting a common core gut microbial response to chronic inflammation and immune activation. In addition, recent work suggests a role for viruses in IBD, with a significant expansion of Caudovirales bacteriophage in patients.71
Modulation of the microbiota as a therapy in IBD
Several clinical trials have examined the approach of modulating the microbiota in patients with IBD, many of which predate the ‘omics’ era. Such trials provide a ‘proof of concept’ for the importance of the role of the gut microbiota in IBD, but marrying up individual approaches with the complex multifactorial nature of IBD remains a challenge, particularly in addressing the different phenotypes and genotypes of disease and the different ‘phases’ of the disease process: for example, prophylaxis, maintenance of remission, treatment of relapses.
Ulcerative colitis
In terms of probiotic research, one of the largest clinical trials in IBD was the use of E. coli Nissle 1917 in the setting of remission maintenance in UC. Patients (n=327) were assigned to a double-blind, double-dummy trial to receive either the probiotic or mesalazine.72 Both treatments were deemed equivalent with regards to relapse. E. coli Nissle is now considered an effective alternative to 5-aminosalicylate for remission maintenance in UC.73 There are two published clinical trials of the multistrain probiotic VSL#3 in the setting of mild to moderate flares of UC.74 Both demonstrate that high doses improve disease activity scores but whether such improvements in scores are clinically meaningful for patients, particularly compared with other treatment options, remains to be clarified. An alternative approach is transplantation of the whole gut microbiota from a healthy donor: FMT. In IBD, a recent systematic review and meta-analysis has shown that of nine cohort studies, eight case studies and one randomised controlled trial, overall 45% (54/119) achieved clinical remission. When only cohort studies were analysed 36% achieved clinical remission.75 Since that meta-analysis, two randomised controlled trials in UC show discrepant results. One trial, in which two faecal transplants were given via the upper GI route, showed no difference in clinical or endoscopic remission between the faecal transplant group and the control group (given autologous stool).76 A second trial, in which patients with UC were randomised to weekly faecal enemas from healthy donors or placebo enemas for 6 weeks, demonstrated remission in a greater percentage of patients given FMT compared with the control group (given water enema).77 There are unanswered questions regarding mode of delivery, frequency of delivery and optimal donor/host characteristics.
Crohn's disease
Antibiotics demonstrate efficacy in particular groups of patients with CD but some antibiotics may be detrimental, showing a complex interplay between host and microbiota. Patients who have had a resection for CD have a decreased rate of endoscopic and clinical recurrence when metronidazole or ornidazole are used as prophylactic therapies.78 Several studies have assessed the specific role of antimycobacterial therapies in CD treatment but overall results are disappointing. There is no clinically relevant evidence base for the use of probiotics in CD and in terms of prebiotics, although an open label trial of fructo-oligosaccharide in CD showed promise,79 and a subsequent randomised placebo-controlled trial of fructo-oligosaccharide did not support any clinical benefit.80
Pouchitis
Restorative proctocolectomy with ileal-pouch anal anastomosis is the operation of choice for patients with UC requiring surgery. Pouchitis has an incidence of up to 50% of patients although it is a significant clinical problem for only about 10%. Antibiotics are used as primary therapy; if single antibiotics fail, dual antibiotics used for longer periods of time or antibiotics tailored to the microbiota in an individual patient can be used. VSL#3 reduced the risk of disease onset and maintained an antibiotic-induced disease remission in pouchitis.81 A meta-analysis has shown that VSL#3 significantly reduced the clinical relapse rates for maintaining remission in patients with pouchitis.82
CRC and the gut microbiota
Many microbiome studies have focused on colitis-associated cancers83 or rodent preclinical models.84 Despite this, there is increasing evidence that the colonic microbiota plays an important role in the cause of sporadic CRC.85 Reduced temporal stability and increased diversity has been shown for the faecal microbiota of subjects with established CRC and polyposis,86 and now metagenomic and metatranscriptomic studies have identified an individualised oncogenic microbiome and specific bacterial species that selectively colonise the on-tumour and off-tumour sites.87
Several competing theories of the microbial regulation of CRC have emerged (figure 1) to explain these observations. The keystone-pathogen hypothesis88 and the α-bug hypothesis both state that certain, low abundance microbiota members (such as enterotoxigenic B. fragilis) possess unique virulence traits, which are pro-oncogenic and remodel the microbiome and in turn promote mucosal immune responses and colonic epithelial cell changes.89 Tjalsma et al90 have also proposed the ‘driver-passenger’ model for CRC: a first hit by indigenous intestinal bacteria (‘bacterial drivers’), which drive the DNA damage that contributes to CRC initiation. Second, tumorigenesis induces intestinal niche alterations that favour the proliferation of opportunistic bacteria (‘bacterial passengers’). For example, CRCs have an increased enrichment of opportunistic pathogens and polymicrobial Gram-negative anaerobic bacteria91 but it is not yet clear whether these opportunistic pathogens merely benefit from the CRC microenvironment or influence disease progression. However, colonic polyps demonstrate higher bacterial diversity and richness when compared with control patients, with higher abundance of mucosal Proteobacteria and lower abundance of Bacteroidetes.92 This may in part be explained by the mucosal defensive strategies designed to manage the commensal microbiota. For example, α-defensin expression is significantly increased in adenomas resulting in an increased antibacterial activity compared with normal mucosa.93

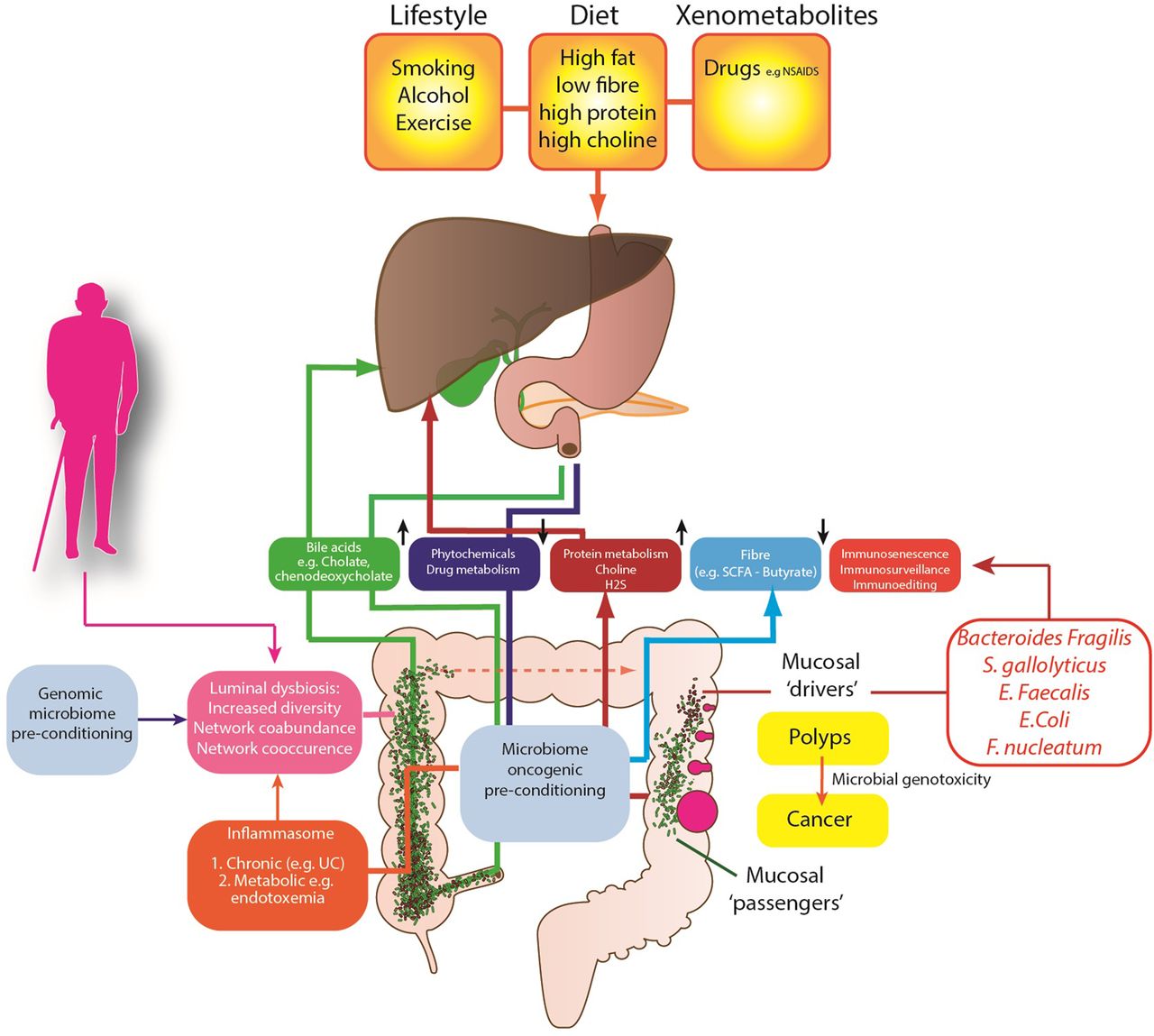{kind=link}
Proposed mechanisms of the gut microbiome in colon cancer aetiology.
At present, human studies have involved small patient numbers, with evidence of sampling heterogeneity, limited tumour phenotyping and oncological data. Despite this, a small number of specific pathobionts have now been linked with adenomas and CRC including Streptococcus gallolyticus,94 Enterococcus faecalis95 and B. fragilis.84 E. coli is also overexpressed on CRC mucosa; it expresses genes that confer properties relevant to oncological transformation including M cell translocation, angiogenesis and genotoxicity.96 Enrichment of Fusobacterium nucleatum has also been identified in adenoma versus adjacent normal tissue and is more abundant in stools from CRC and adenoma cases than in healthy controls. F. nucleatum's fadA, a unique adhesin, allows it to adhere to and invade human epithelial cells, eliciting an inflammatory response97 and stimulating cell proliferation.98 Novel mechanisms from previously unassociated bacteria are also being described to explain how bacterial proteins target proliferating stem-progenitor cells. For example, AvrA, a pathogenic product of Salmonella, has been shown to activate β-catenin signals and enhance colonic tumorigenesis.99
Work has also focused on the metabolic function of the gut microbiome and dietary microbiome interactions in the aetiology of CRC. It is likely that the metabolism of fibre is critical to this. Metagenomic analyses have consistently identified a reduction of butyrate-producers in patients with CRC,100 a finding replicated in animals.101 The microbiome also plays an important role in the metabolism of sulfate, through assimilatory sulfate-reduction to produce cysteine and methionine, and dissimilatory sulfate-reduction to produce hydrogen sulfide (H2S). H2S is likely to contribute to CRC development, as colonic detoxification of H2S is also reduced in patients with CRC; it also induces colonic mucosal hyperproliferation.102 There is also evidence that differences in host genotype, which affect the carbohydrate landscape of the distal gut, interact with diet to alter the composition and function of resident microbes in a diet-dependent manner.103 Therefore it is possible that patients genetically predisposed to CRC have a modified metabolically active microbiome, which is determined by their genes and by their family environment and dietary habits. There is other evidence from global studies of cancer risk, that the microbiome is important in cancer risk.
African Americans possess a colon dominated by Bacteroides, while in Africans Prevotella are more abundant.104 African Americans, who are at high risk of CRC, may have evolved a CRC-microbiota moulded by dietary habits and environmental exposures. Critically, mucosal Ki67 expression (a biomarker for cancer risk) may decrease or increase within 2 weeks of either a high fibre (>50 g/day) dietary intervention in African Americans or a high fat, high protein low fibre Westernised diet in African subjects. This short-term intervention leads to reciprocal changes in luminal microbiome co-occurrence network structures that overwhelm interindividual differences in microbial gene expression. Specifically, an animal-based diet increases the abundance of bile-tolerant microorganisms (Alistipes, Bilophila and Bacteroides) and decreases the levels of Firmicutes that metabolise dietary plant polysaccharides (Roseburia, Eubacterium rectale and Ruminococcus bromii).105 ,106
Conclusions
In the past decade, interest in the human microbiome has increased considerably. A significant driver has been the realisation that the commensal microorganisms that comprise the human microbiota are not simply passengers in the host, but may actually drive certain host functions as well. In sterile rodents, we see the dramatic impact that removing the microbiota has on nearly all aspects of the host's ability to function normally. This review highlights some key disease areas in which the microbiota and its microbiome are thought to have not just an association, but also a key modulatory role (table 1). By better understanding the mechanisms and contribution the microbiota make to these diseases, we hope to develop novel therapeutics and strategies to modulate the microbiota to treat or prevent disease. Additionally, in some instances it may be possible to use the microbiome to detect gut-related diseases before conventional diagnostics can. In the future we hope to use this information to stratify patients more accurately and for more efficient treatment. A body of evidence also points to the gut microbiota being an environmental factor in drug metabolism, for example, inactivation of the cardiac drug digoxin by Eggerthella lenta in the gut. Thus, if we are to realise the vision of a personalised healthcare revolution, we must explore how the microbiome fits with this notion.
Key insights into the influence of the gut microbiota on GI and liver diseases
Acknowledgments
This review was commissioned by the Gut Microbiota for Health expert panel of the British Society of Gastroenterology.
References
Footnotes
Contributors AH, JRM, DHA, GDAH, LVT, FF, GMH and GH, MNQ, HS, KMT, EGZ and JK contributed to the conception/design of the work, drafting the work and revising it critically for important intellectual content and final approval of the version published.
Competing interests AH has lectured for Yakult.
Provenance and peer review Not commissioned; externally peer reviewed.