Article Text

Abstract
Objective The impact of glia cells during GI carcinogenesis and in cancer pain is unknown. Here, we demonstrate a novel mechanism how Schwann cells (SCs) become activated in the pancreatic cancer (PCa) microenvironment and influence spinal activity and pain sensation.
Design Human SCs were exposed to hypoxia, to pancreatic cancer cells (PCCs) and/or to T-lymphocytes. Both SC and intrapancreatic nerves of patients with PCa with known pain severity were assessed for glial intermediate filament and hypoxia marker expression, proliferation and for transcriptional alterations of pain-related targets. In conditional PCa mouse models with selective in vivo blockade of interleukin (IL)-6 signalling (Ptf1a-Cre;LSL-KrasG12D/KC interbred with IL6−/− or sgp130tg mice), SC reactivity, abdominal mechanosensitivity and spinal glial/neuronal activity were quantified.
Results Tumour hypoxia, PCC and/or T-lymphocytes activated SC via IL-6-signalling in vitro. Blockade of the IL-6-signalling suppressed SC activation around PCa precursor lesions (pancreatic intraepithelial neoplasia (PanIN)) in KC;IL6−/− (32.06%±5.25% of PanINs) and KC;sgp130tg (55.84%±5.51%) mouse models compared with KC mice (78.27%±3.91%). Activated SCs were associated with less pain in human PCa and with decreased abdominal mechanosensitivity in KC mice (von Frey score of KC: 3.9±0.5 vs KC;IL6−/− mice: 5.9±0.9; and KC;sgp130tg: 10.21±1.4) parallel to attenuation of spinal astroglial and/or microglial activity. Activated SC exhibited a transcriptomic profile with anti-inflammatory and anti-nociceptive features.
Conclusions Activated SC in PCa recapitulate the hallmarks of ‘reactive gliosis’ and contribute to analgesia due to suppression of spinal glia. Our findings propose a mechanism for how cancer might remain pain-free via the SC–central glia interplay during cancer progression.
- NERVE - GUT INTERACTIONS
- PANCREATIC CANCER
- NEUROGASTROENTEROLOGY
- VISCERAL NOCICEPTION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Neuropathy and neural invasion are independent prognostic factors for overall survival in pancreatic cancer (PCa).
Peripheral neuronal circuits are assumed to be activated in PCa and to thereby mediate pain.
What are the new findings?
Glia cells of peripheral nerves, that is, Schwann cells, are profoundly activated in PCa and secrete high amounts of interleukin (IL)-6.
Activation of Schwann cells in patients with PCa and in PCa mouse models is associated with diminished pain sensation due to suppression of spinal astroglial and microglial activity.
Depletion of IL-6 in PCa mouse models suppresses Schwann cells, restores spinal astroglial and/or microglial activity and results in increased mechanical allodynia in mice.
How might it impact on clinical practice in the foreseeable future?
Schwann cell activation in PCa seems to represent a potential reason for the lack of symptoms like pain during early stages of pancreatic carcinogenesis.
Introduction
The role of glia cells in the enteric nervous system is under-researched, and the potential contribution of glia cells to the pathogenesis and outcome of GI disorders is still highly neglected.1 In response to an insult (eg, ischaemia, tumour), glia cells in the central nervous system (CNS), for example, astrocytes, assume an activated state, encapsulate disease sites by the formation of a dense glial network and thereby regulate local tissue repair and homeostasis.2 However, it is not yet known whether peripheral glial cells, that is, Schwann cells (SCs), which are equally present in visceral nerve trunks innervating GI organs, can assume a similarly activated state in response to GI disease.
One GI disorder that exhibits a remarkable degree of neuroplastic and neuropathic alterations in the GI tract is pancreatic cancer (PCa).3 ,4 Notorious for its highly aggressive and lethal nature, human PCa is marked by a 100% frequency of invasion of nerves by cancer cells (neural invasion (NI)), pancreatic nerve damage, pancreatic neuroinflammation (neuritis), noticeable hypertrophy and sprouting of intrapancreatic nerves.5 These features correlate to the severity of the neuropathic abdominal pain syndrome in patients with PCa.4 Considering the critical role of SC in the generation of neuropathy,6 a potential mediator role for SC in the generation of pancreatic neuropathy becomes very likely.
Recently, we demonstrated that SCs are highly cancer-affine cells that emerge around precursor lesions of PCa both in mice and human disease.7 These observations demonstrated that pancreatic SCs initiate NI as opposed to the classical assumption that cancer cells primarily actively invade nerves first.7 Therefore, in the current study, we aimed to analyse how SCs become activated in PCa and what impact they exert on the disease course. For this purpose, human SCs (hSCs) were exposed to the hallmark components of PCa microenvironment, that is, tumour hypoxia, PCa cells and inflammatory cells. Furthermore, we show that hSCs recapitulate the features of reactive gliosis, which is primarily known for astrocytes of the CNS,8 in the PCa tumour microenvironment.
Furthermore, we demonstrate that this hSC activation is linked to the presence of interleukin (IL)-6 in vivo and to decreased pain sensation in patients with PCa and decreased mechanical hyperalgesia of mice with PCa due to concomitant suppression of spinal astrocytes and microglia.
Methods
For a detailed description of the applied methods, of hSC culture, and of the RT2 Profiler PCR array, please refer to the online supplementary methods. For a complete list of the applied antibodies, please refer to the online supplementary table S1.
Patients, tissues and pain severity classification
PCa tissue samples from the pancreatic head (n=26 patients), corpus (n=1 patient) and tail (n=3 patients) were collected from patients after informed consent and following tumour resection for pancreatic ductal adenocarcinoma. The degree of pain sensation prospectively registered and calculated prior to the operation (no pain/group 0, mild pain/group 1 and severe pain/group 2) as described in the online supplementary methods and also previously.9
Conditional knockout mice
The LSL-KrasG12D knockin,10 Ptf1a-Creex111 and sgp130tg12 strains were interbred to obtain compound mutant LSL-KrasG12D;Ptf1a-creex1;sgp130tg (KC;sgp130tg; n=10) mice. The transgenic expression of the glycoprotein 130 (sgp130) fusion protein under the control of the hepatic PEPCK promoter was previously reported to block IL-6 transsignaling via the specific binding of sgp130 to the IL-6/IL6R complex.12 Mutant LSL-KrasG12D;Ptf1a-Creex1 (KC, n=10) were used as control animals. LSL-KrasG12D knockin and IL-6−/− mice (C57BL/6 background, from Jackson Laboratory, Maine, USA) were interbred to generate LSL-KrasG12D;IL6−/−;Ptf1a-Creex1 mice (KC;IL-6−/−, n=10), as also described previously.13 To account for differences due to different pancreatic intraepithelial neoplasia (PanIN) progression rates of different genotypes, data from mice with histologically same degree of PanIN progression were included in the behavioural and spinal cord analyses. All animal procedures were in accordance with the regulatory standards and approved by the Regierung von Oberbayern, Bavaria, Germany (5.2-1-54-2532-79-2014 and 55.2-1-54-2532-95-2015).
Immunoblot analysis
Protein extraction and immunoblot analysis of cell culture monolayers were performed as described previously.14 30 µg of protein were separated, electroblotted and the membrane was exposed to glial fibrillary acidic protein (GFAP), nestin, vimentin, pSTAT3 or STAT3 antibodies at 4°C overnight or to mouse glyceraldehydes-3-phosphate-dehydrogenase (GAPDH) antibody for equal loading. The densitometric analysis of the blots was performed via the ImageJ Software (1.44p, Wayne Rasband, NIH, USA).9
Immunohistochemistry/IHC, immunofluorescence/IF, histopathological evaluation and quantitative assessment of immunoreactivity of nerves in human specimens
Consecutive 3 µm sections from paraffin-embedded normal pancreas (NPs) and PCa samples were analysed for hypoxia-inducible factor-1α (HIF-1α), carbonic anhydrase IX (CA-IX) and GFAP immunoreactivity of at least 10 nerves from different sections based on colorimetric quantification of immunostaining intensity via Threshold function of ImageJ software.15
Histological analysis of mouse pancreas and spinal cord
In the analysis of mouse pancreatic tissue, all visible PanIN lesions on one section per mouse were included in the analysis. Here, in addition to H&E staining, consecutive sections of the tumour were immunostained with antibodies against GFAP, S100, IL-6, ki67 or Sox10, followed by second antibody immunolabeling with Alexa Fluor Goat anti-rabbit IgG or anti-mouse antibodies. The nuclei were visualised by using 4′,6-diamidino-2-phenylindole (DAPI) counterstain (1:400 dilution, Invitrogen, Karlsruhe, Germany). Digital imaging was performed with the Keyence Biorevo BZ-9000 system (Keyence, Neu-Isenburg, Germany).
Subsequent to behavioural and hyperalgesia testing, mice were sacrificed and the thoracic spinal segments 8–11 (pancreas-innervating segments), together with the spinal cord, were removed en bloc, formalin-fixed, paraffin-embedded and subjected to EDTA-based demineralisation. Also, 3 µm consecutive sections of the spinal cord were immunostained for glial fibrillary acidic protein (GFAP), p75NTR, Iba-1, phospho-p38/p-p38, phospho-extracellular-signal regulated kinase/p-ERK and beta-III-tubulin. On the sections for astroglia and microglia analysis, colour thresholding function of the ImageJ software was used to determine the area covered by the activation markers (p75NTR for astrocytes and p-p38 for microglia), the total area of GFAP-stained astroglia and of Iba-1 stained microglia in both dorsal horns of each spinal cord section (including all Rexed laminae). The proportion of p75NTR to GFAP-covered and of p-p38 to Iba-1-stained areas were calculated to determine the percentage of astrocyte and microglia activation, respectively. To assess neuronal activation, cells that exhibited p-ERK staining in neuronal cytoplasma were counted and related to the total number of beta-III-tubulin-stained neurons in both dorsal horns. Results were expressed as mean±SEM %. Two spinal cord sections were analysed per mouse.
Assessment of mechanical abdominal hyperalgesia and open-field behaviour
Mechanical hypersensitivity was tested by means of von Frey filaments (Stoelting, Illinois, USA) of increasing tactile stimulus intensity by applying each filament type 10 consecutive times from the bottom of a grid on the abdomen of the mice. To assess the impact of pain on the exploratory behaviour, mice were placed in an open-field white plastic box (50×50×50 cm) made of polyvinyl chloride that was evenly illuminated, and then video-tracked for their locomotion in the open field.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 5 Software (La Jolla, California, USA). The unpaired t test was applied for two-group analysis. To compare more than two groups, the one-way analysis of variance was used, and the subsequent pairwise comparisons were performed via the unpaired t test. The growth assays were compared using the per cent growth/viability rates of hSC at 72 h. Spearman's r correlation coefficient was calculated to analyse the relationship between GFAP and HIF-1α or between GFAP and CA-IX neural immunoreactivity. Results are expressed as mean±SE of the mean (SEM). Two-sided p values were always computed, and an effect was considered statistically significant at a p value ≤0.05.
Results
Tumour hypoxia promotes reactive gliosis of SCs in PCa
Although tumour hypoxia was shown to trigger several pro-survival, pro-invasive and pro-metastatic pathways in human cancers,16 ,17 its influence upon tumour nerves has not yet been investigated. Therefore, we first assessed the amount of the three major glial intermediate filaments GFAP, nestin and vimentin in hSC under hypoxia. Here, exposure to hypoxia induced upregulation of the intermediate filaments GFAP and nestin in hSC, starting as early as after 15 min and lasting for the whole period of hypoxia exposure, with a similar yet statistically insignificant tendency for vimentin (GFAP: 98.23%±7.0%; nestin: 171.6%±43.2%; vimentin: 142.3%±52.3% of GAPDH at 6 h of hypoxia; figure 1A) compared with normoxic conditions (GFAP: 38.2%±7.2%; nestin: 35.6%±15.2%; vimentin: 38.7%±14.0% of GAPDH, figure 1A).

Hypoxia and human Schwann cell (hSC) activation in pancreatic cancer (PCa). (A) hSCs were exposed to varying periods of hypoxia, subsequently lysed, immunoblotted against the three major intermediate filaments glial fibrillary acidic protein (GFAP), nestin and vimentin, and compared via densitometry (bar graphs to the right). Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was used as loading control. Results were expressed as % of loading control (GAPDH). The asterisks (*) indicate significance in the unpaired t test compared with the 0 h group. (b) Immunolabeling of human normal pancreas (NP) and PCa tissues with the endogenous hypoxia markers hypoxia-inducible factor-1α (HIF-1α) and carbonic anhydrase IX (CA-IX) showed specific labelling of nerves by these markers. Nerves in pancreatic tissues were colorimetrically assessed for their immunoreactivity for these markers. ‘High GFAP’ denotes nerves in the upper 25%, ‘low GFAP’ those in the lower 25% range of all nerves analysed for GFAP immunoreactivity, Protein-gene-product-9.5 (PGP9.5) was used as pan-neural marker. The yellow scale bars indicate 50 µm. (C) Immunofluorescence labelling of PCa nerves against CA-IX showed co-localisation of CA-IX with nucleoli, the overwhelming majority of which are known to belong to SC. (D) Correlation of immunoreactivity of nerves for the hypoxia markers (HIF-1α and CA-IX) with GFAP in PCa nerves (left) and in nerves in NP (right). Spearman's rank correlation coefficient (r) test. All data were created from three independent experiments.
Next, we assessed whether intrapancreatic nerves in PCa are also subject to hypoxia and similarly exhibit hypoxia-induced increase in intermediate filament expression. Here, immunolabeling of human NP and PCa tissues against the hypoxia markers HIF-1α, CA-IX, and against the intermediate filament GFAP revealed a specific immunolabeling of nerves not only with GFAP, but also with HIF-1α and CA-IX (figure 1B). Furthermore, immunofluorescence labelling of PCa tissues with CA-IX showed co-localisation of CA-IX with the nuclear (DAPI) staining, particularly in nucleoli (figure 1C), indicating CA-IX presence in hSC.
Next, we sought to identify whether a quantitative correlation between GFAP content and the expression of the hypoxia markers HIF-1α and CA-IX within nerves is detectable in the pancreas. Therefore, we compared the densitometric staining intensity of nerves for GFAP, HIF-1α and CA-IX, and in accordance with the in vitro upregulation of GFAP in hSC by hypoxia, there was a positive correlation between the intraneural GFAP content, HIF-1α (r=0.27, p<0.05) and CA-IX (r=0.77, p<0.0001) content in vivo (figure 1D).
Activated SCs, for example, after axotomy, secrete a large set of cytokines and chemokines that entail a pro-inflammatory milieu to ensure neural repair.6 However, the impact of hypoxia on the secretion of cytokines from hSC is unknown. Therefore, using Multiplex Luminex® technology, we quantified the levels of multiple cytokines (for a complete list, see online supplementary methods) in the serum-free growth medium of hSC exposed to hypoxia. Here, the majority of the studied cytokines, particularly the interleukins IL-6, IL-8, IL-9 and IL-12 (see online supplementary figure S1a), the pro-inflammatory mediators interferon-gamma, tumour necrosis factor-α and CCL2 (see online supplementary figure S1b), and the growth factors granulocyte colony stimulating factor/G-CSF and vascular endothelial growth factor/VEGF (see online supplementary figure S1c), were increasingly secreted from hSC under hypoxia, especially around 6–24 h of hypoxia exposure (see online supplementary figure S1). Other cytokines like IL-4 and IL-13 were also found to be increasingly secreted from hypoxic hSC, yet their maximum levels remained low at <10 pg/mL (see online supplementary figure S2).
PCa cells induce GFAP expression and hypertrophy of hSC
Second, we studied the potential glia-activating properties of PCa cells (PCCs). Similar to hypoxia, addition of PCC-conditioned media into the serum-free growth medium of hSC resulted in increased intracellular levels of GFAP in hSC, not affecting nestin and vimentin (figure 2A). However, GFAP upregulation was independent of the direct contact of PCC with nerves since increasing severity of NI (ie, no invasion vs perineural vs endoneural invasion) was not associated with any major change in the immunoreactivity for GFAP (no invasion/0: 21.4%±1.2%; perineural invasion/1: 23.7%±1.6%; endoneural invasion/2: 20.8%±0.6%, figure 2B).

Impact of pancreatic cancer cells (PCCs) on human Schwann cell (hSC) activation. (A) hSCs were cultivated in serum-free supernatants (SN) of the PCC-lines T3M4, SU86.86, Colo357 and Capan1 for 48 h and compared with hSC exposed to recombinant nerve growth factor/NGF in their serum-free growth medium (SFM) (positive control) or to SFM supplied with PCC growth medium (Roswell Park Memorial Institute/RPMI medium) (SFM-ctrl, negative control). The asterisks (*) indicate significance in the unpaired t test compared with the SFM-ctrl group. (B) Intrapancreatic nerves in human pancreatic cancer (PCa) specimens were classified as showing no neural invasion (score 0), perineural invasion (score I) and endoneural invasion (score II) and compared with regard to glial fibrillary acidic protein (GFAP) immunoreactivity.Protein-gene-product-9.5 (PGP9.5) as pan-neural marker. (C) hSCs were cultivated in PCC-SN for 48 h, subsequently immunostained against GFAP and measured for their mean cell area. SN of the PCC lines T3M4 and Colo357 induced star-like processes on the hSC surface, strongly reminiscent of ‘stellation’ of central glia. The asterisks (*) indicate significance in the unpaired t test compared with the control. Arrows point to the spindle-shaped processes of hypertrophic hSC. All data were created from at least three independent experiments.
Activated astrocytes typically undergo a cellular hypertrophy and acquire a star-like conformation,8 yet a similar change in cellular shape has not yet been described for activated hSC. Indeed, treatment of hSC with PCC supernatants resulted in the acquisition of a star-like cellular conformation of hSC, highly reminiscent of stellation of astrocytes (figure 2C). Correspondingly, the cellular area of hSC cultivated in the supernatants of moderately to poorly differentiated PCC lines Colo357 (832.2±94.1 µm2) and T3M4 (780±70.0 µm2) was significantly greater than the control hSC (534±26.4 µm2, figure 2C). The cellular area of hSC cultivated in the supernatants of well-differentiated Capan1 (578.1±35.6 µm2) and SU86.86 (520.6±37.5 µm2) or in the CCa cell line HCT-116 (531.3±42.4 µm2) did not differ from the control (figure 2C; Capan1: well-differentiated, metastasis-derived/Colo357: moderately differentiated, lymph node-derived/SU86.86: poorly differentiated, metastasis-derived and T3M4: poorly differentiated, metastasis-derived18). These findings collectively indicate that PCC, although they differ with regard to their differentiation state and origin, exert different glia-activating effects.
Next, we investigated the influence of PCC-conditioned media on hSC proliferation. Colon cancer (CCa)-cell-conditioned media were used as control since CCa cells lack the neurotropic attributes of PCa cells.19 Addition of conditioned media of the moderately to poorly differentiated PCC Colo357, SU86.86 and T3M4 into the growth medium of hSC tended to slightly augment the viability of hSC compared with the addition of control medium after 72 h (Colo357: 114.4%±3.3%; SU86.86: 114.9%±3.6%; T3M4: 115.2%±2.5% of control hSC: 100.0%±8.2%; figure 3A), but this tendency was not present for the well-differentiated Capan1 cells (103.4%±3.8%; figure 3A). Furthermore, the viability rate of the CCa cell lines-SN-treated hSC did not differ from the level of control hSC (HCT-116: 99.4%±4.0%; HT29: 105.4%±3.6%; figure 3A). The tendency of the poorly differentiated SU86.86 cells to augment hSC proliferation was more prominent in a setting in which the number of ki67+or BrdU+hSC were counted (SU86.86-SN treatment: 55.3%±7.5% vs Ctrl.: 30.8%±4.3% ki67+ hSC, p=0.02; for BrdU+: 36.48%±3.9% BrdU+ in the Ctrl., 59.8%±4.2% after SU86.86-SN treatment, 75.6%±3.4% after T3M4-SN treatment; figure 3A,B). Moreover, quantification of ki67+ hSC in nerves within human PCa and NP specimens revealed greater amounts of proliferating hSC in PCa nerves (6.4%±1.1%) than in NP nerves (5.7%±1.3%, p=0.03; figure 3C), independent of NI (figure 3D). Therefore, human PCa nerves seem to contain greater amounts of proliferating SCs.

Influence of pancreatic cancer (PCa) on Schwann cell (SC) proliferation. (A) Left: effect of the supernantants (SN) of the pancreatic cancer cells (PCCs)-lines Capan1, Colo357, SU86.86, T3M4 and of the colon cancer cell (CCa) lines HCT-116 and HT29 on human Schwann cell (hSC) viability in the MTT test. Middle/right: human PCC SN were added into the serum-free growth medium of hSC, and the number of ki67+ hSC were proportioned to the total number of hSC on each coverslip. S100 was used as SC marker. Pos.Ctrl., recombinant human beta-NGF. Unpaired t test. (B) The number of proliferating hSC was assessed via BrdU immunolabeling in vitro after PCC (SU86.86 or T3M4) SN treatment compared with the serum-free growth medium of hSC (Ctrl.). DAPI: nucleus marker. Unpaired t test. (C) Human normal pancreas (NP) and PCa tissues were immunolabeled against the proliferation marker ki67 and the SC marker S100, and the number ki67+ SC (white arrows) was proportioned to the total number of SC in each nerve. (D) The proportion of ki67+, proliferative SC was additionally assessed in a subgroup analysis of nerves with and without neural invasion (NI). All yellow scale bars indicate 40 µm.
T-lymphocytes potentiate PCC-induced hSC activation
Nerves in PCa are not only subject to invasion by PCC, but also to targeted infiltration by inflammatory cells like CD8+-T-lymphocytes (‘pancreatic neuritis’).20 Whether a concomitant infiltration of nerves by cancer cells and inflammatory cells augments glial activation by cancer is not known. Therefore, in the current study, we performed a triple co-culture of PCC, hSC and Jurkat-T-cells and compared the intracellular GFAP content in hSC under these conditions. Co-culture of hSC with T3M4 alone resulted in increased GFAP content of hSC at 48 h of co-culture compared with control (mono-cultured) hSC (with Capan1: 43.7%±13.4%, with T3M4: 55.1%±10.5% vs control hSC: 25.3%±4.4% of GAPDH, figure 4A). However, the additional presence of Jurkat-T-cells in the triple co-culture setting led to a further GFAP-upregulation in hSC compared with control hSC and hSC-PCC dual co-cultures (hSC+Capan1+Jurkat: 76.9%±13.5%, hSC+T3M4+Jurkat: 74.2%±10.8% of GAPDH, figure 4A).
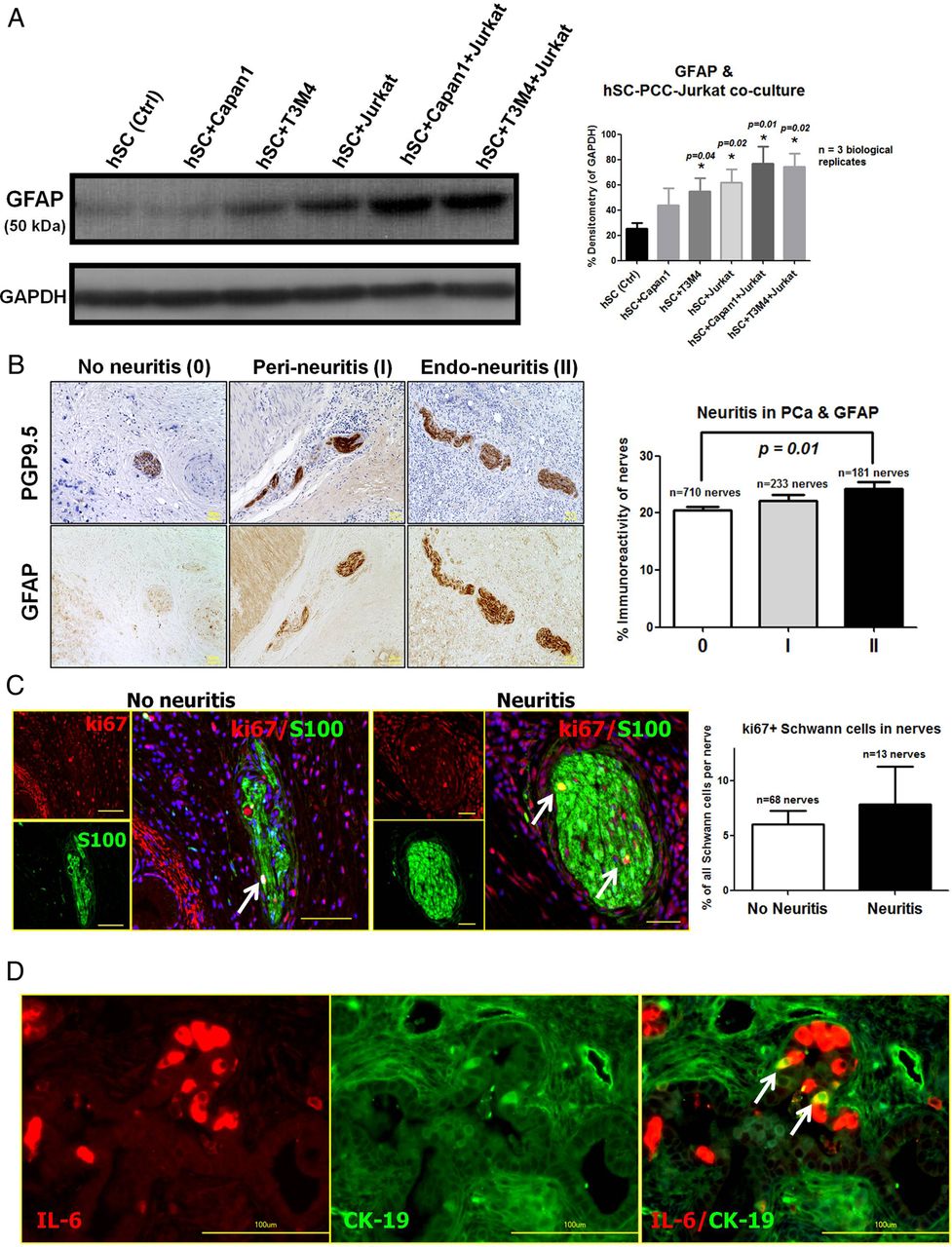
Impact of inflammatory cells and pancreatic neuroinflammation (‘neuritis’) on human Schwann cells (hSCs) activation. (A) To assess the impact of inflammatory cells upon pancreatic cancer cells (PCCs)-induced hSC activation, hSCs were either grown in dual co-culture with one of the PCC lines Capan1 or T3M4 (hSC+Capan1 or hSC+T3M4) or in triple co-culture with the addition of the Jurkat human T-lymphocyte cell line and assessed by relative densitometry of glial fibrillary acidic protein (GFAP) after immunoblotting of hSC lysates (bar chart to the right). One-way analysis of variance, followed by pairwise comparisons with the unpaired t test. The *p value corresponds to t test p values compared with the hSC Ctrl. group. All data were created from three independent experiments. (B) Nerves in human pancreatic cancer (PCa) tissues were classified as showing no pancreatic neuritis (score 0), perineuritis (score I) and endoneuritis (score II). PGP9.5, protein-gene-product-9.5 as pan-neural marker. Here, nerves with endoneural-neuritis (score II) exhibited a greater GFAP immunoreactivity than nerves without pancreatic neuritis (score 0). The asterisks (*) correspond to p<0.05 in the unpaired t test compared with the 0 group. Scale bars: 50 µm. (C) PCa tissues were double-immunolabeled against the proliferation marker ki67 and the SC marker S100, and the proportion of ki67+ SC (arrows) to all SC in each nerve was compared between nerves with neuritis and those without neuritis. Yellow scale bars indicate 40 µm. (D) PCa tissues were double-immunolabeled against interleukin-6 (IL-6) and the cancer cell marker cytokeratin-19 (CK19). IL-6 was detected in co-localisation with a subset of PCa cells (arrows). Scale bars: 100 µm.
Moreover, in accordance with the in vitro GFAP upregulation in hSC, increasing severity of pancreatic neuritis correlated to stronger GFAP immunoreactivity of intrapancreatic nerves in vivo (no neuritis: 20.4%±0.6%; perineural-neuritis: 22.2%±1.1%; endoneural-neuritis: 24.2±1.3%, p=0.0023, figure 4B). Still, presence of neuritis alone did not considerably affect the proportion of proliferating (ki67+) hSC in nerves within PCa (figure 4C).
IL-6 is responsible for PCC-induced intermediate filament induction in hSC
The promotion of an activated hSC phenotype by hypoxia, PCC and neuroinflammation suggests a role for secreted factors in the PCa microenvironment that activate glia cells. Here, we hypothesised that pro-inflammatory cytokines like IL-1β and IL-6 may induce hSC activation in PCa. To test this possibility, we first compared the GFAP, nestin and vimentin content of hSC that were exposed to hypoxia in the presence or absence of specific blocking antibodies against IL-1β and IL-6. Blockade of these cytokines in hypoxic hSC supernatants did not significantly affect the intermediate filament expression within hSC (see online supplementary figure S3a).
In contrast, GFAP or nestin content of hSC was prominently affected when IL-6, but not IL-1β was blocked in PCC supernatants. In particular, the addition of IL-6 blocking antibodies to the supernatants of SU86.86 diminished the nestin content of hSC (see online supplementary figure S3c). Furthermore, blockade of IL-6 from T3M4 SN reduced both the GFAP and nestin content of hSC (see online supplementary figure S3c). Treatment of hSC with PCC SN resulted in increased amounts of the IL-6 downstream mediator phospho-STAT3 in vitro (see online supplementary figure S3b). In accordance with these observations, IL-6 was specifically detected in cancer cells of PCa tissues in double-immunolabeling experiments with IL-6 and the cancer cell marker cytokeratin-19 (figure 4D).
Disruption of classical IL-6 signalling, and less of IL-6 transsignaling, reduces the accumulation of SC around murine PanIN lesions
In order to investigate the in vivo impact of IL-6 blockade in murine PCa, we bred mice with murine PCa (Ptf1a-Cre;LSL-KrasG12D/KC mice) with IL-6 knockout mice to generate classical IL-6 signalling knockouts (KC;IL6−/−) or with sgp130tg transgenic mice (that overexpress the soluble sgp130 fusion protein), resulting in blockade of IL-6 transsignaling (KC;sgp130tg).12 ,13 IL-6 transsignaling is characterised by complexing of IL-6 with its soluble receptor IL-6R and the subsequent binding of this complex to the gp130 receptor on the cell surface.12 ,13 SCs are known to express gp130 and need it for their integrity.21 In all these mice, SC were detected by double-immunolabeling with the SC markers Sox10 (glial identity marker)/S100 and additionally on consecutive sections with GFAP/S100. In KC mice, specific multiple immunolabeling with these markers revealed the presence of a SC niche specifically around the PCa precursors, the so-called PanIN lesions (figure 5A7). In contrast with the abundant presence of glial cells around KC PanINs, there were hardly any detectable GFAP-immunoreactive or Sox10-immunoreactive glial cells around KC;IL-6−/− PanINs (figure 5B). However, glial cells were again detectable around KC;sgp130tg PanINs, yet at a lower frequency than in KC mice (figure 5C). The quantification of the frequency of PanIN-SC associations in these mice accordingly revealed prominently reduced numbers of glial cells in KC;IL6−/− mice compared with KC or KC;sgp130tg strains (figure 5D, frequency of PanINs surrounded by GFAP/S100 immunolabeled SC in KC mice: 78.27%±3.91%; KC;IL-6−/− mice: 32.06%±5.25%; KC;sgp130tg mice: 55.84%±5.51%; Sox10/S100 immunolabeling: KC mice: 80.627%±3.37%; KC;IL-6−/− mice: 26.91%±2.85%; KC;sgp130tg mice: 64.93%±8.08%).

Disruption of interleukin (IL)-6 signalling in murine pancreatic cancer (PCa) reduces the frequency of Schwann cells (SCs) around murine pancreatic intraepithelial neoplasia (PanIN) lesions. (A) Double-immunolabeling of consecutive pancreatic tissue sections from KC (Ptf1a-Cre;KrasG12D), KC;IL-6−/− and KC;sgp130tg mice against the SC markers glial fibrillary acidic protein (GFAP)/S100 or Sox10/S100 to detect the frequency of SC (arrows) around the PCa precursor PanIN lesions (demarcated by yellow dashed lines in the uppermost H&E image of each row). (b) Quantification of the proportion of PanINs that are surrounded by SC in each model revealed prominent reduction of SC around PanINs of KC;IL-6−/− mice. Unpaired t test. Yellow scale bars: 200 µm.
Activated SCs in PCa are associated with decreased pain sensation due to suppression of spinal astroglia and microglia
Ablation of IL-6 in KC mice was previously reported to result in delayed cancer progression, that is, delayed transformation from PanIN to overt cancer.13 However, the impact of SC activation on the clinical course of PCa has not yet been investigated. Here, we first compared the immunoreactivity of GFAP and another glial activation marker, the low-affinity NGF receptor p75NTR, in nerves among PCa patients with varying severity of abdominal pain. Here, intriguingly, increasing pain degree was associated with decreased neural GFAP expression (pain degree 0: 26.5%±3.0%; degree 1: 17.38%±3.0%; degree 2: 15.76%±2.5%), with a similar but statistically insignificant tendency for p75NTR (pain degree 0: 49.4%±7.3%; degree 1: 33.3%±10.3%; degree 2: 33.3%±10.7%; figure 6A). To further understand the association between glial activation and pain in PCa, we compared the mechanical abdominal hypersensitivity and the locomotion of PanIN-stage matched KC, KC;IL6−/− and KC;sgp130tg in open-field tests. Using von Frey filament testing as a valid method of abdominal mechanosensitivity assessment in murine pancreatitis,22–26 we observed considerably greater von Frey scores among KC;IL6−/− (5.9±0.9) and KC;sgp130tg (10.21±1.4) mice than in KC mice (3.9±0.5, p=0.04 for KC vs KC;IL6−/−; and p=0.01 for KC vs KC;sgp130tg, figure 6B), suggesting increased mechanosensitivity after IL-6 blockade. Accordingly, the total track covered in the defined open field was on average longer for KC mice (393±49 cm) compared with KC;IL6−/− mice (324±46 cm), yet both were below the high open-field track length of KC;sgp130tg mice (2811±238.5 cm, figure 6C).

Impact of interleukin (IL)-6 disruption and Schwann cell (SC) suppression on pain, mechanical hypersensitivity and pain-related behaviour. (A) Patients with pancreatic cancer (PCa) were immunostained for the glial activation markers glial fibrillary acidic protein (GFAP) and p75NTR, and the immunostaining intensity of all nerves in these tissues were correlated to the degree (0, 1 or 2) of pain sensation. Unpaired t test. (B) The extent of abdominal mechanical hypersensitivity was assessed in KC (Ptf1a-Cre;KrasG12D), KC;IL-6−/− and KC;sgp130tg mice via von Frey filaments. Unpaired t test. (c) Mice were tracked in an open field (50×50 cm) for their exploratory behaviour in consecutive recordings and compared for the total length of their locomotion path.
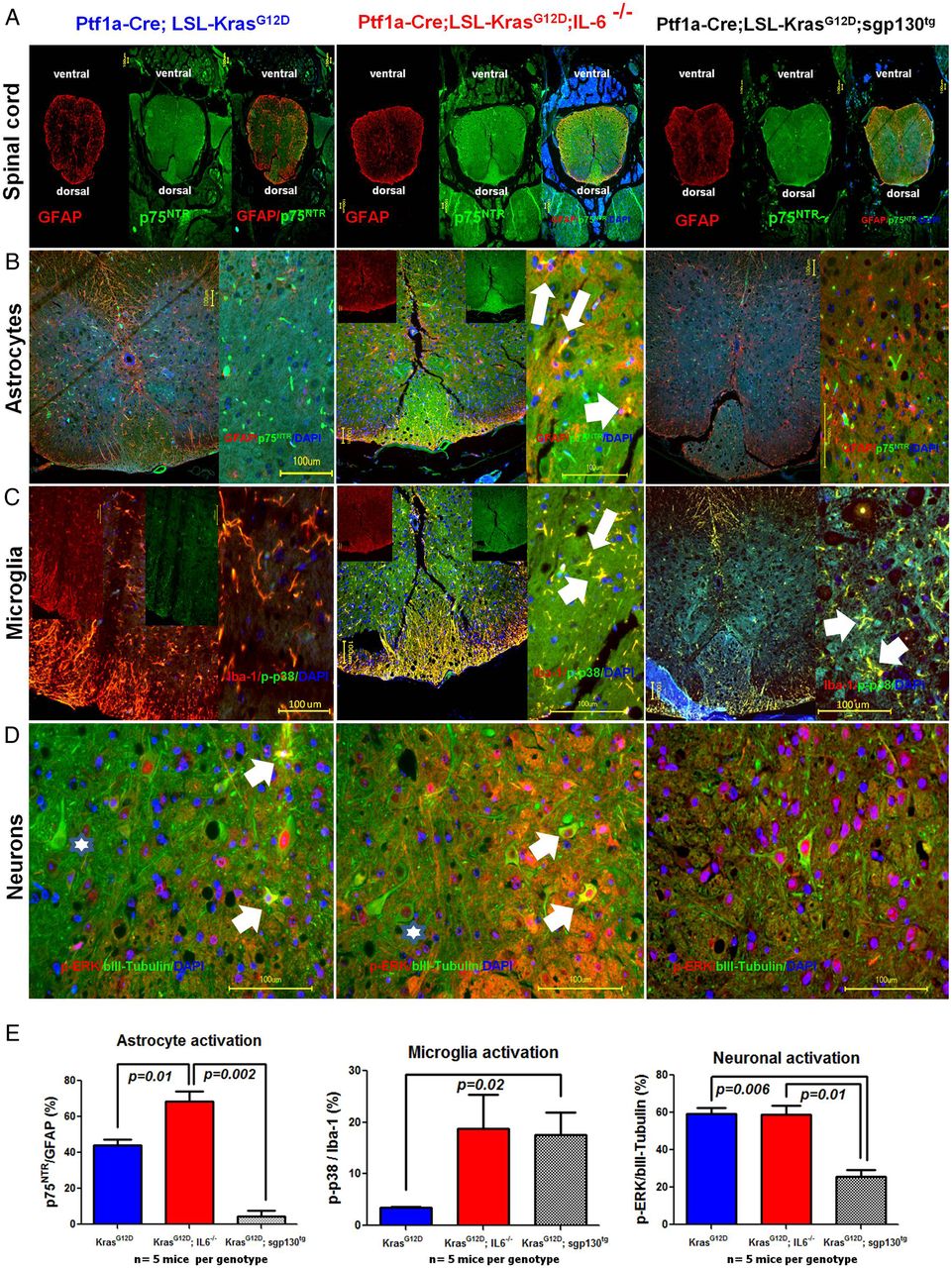
To understand the mechanism behind these indicators of decreased pain in these PCa mouse models with SC activation, we analysed the activation state of the spinal actors of nociception, that is, astroglia, microglia and spinal neurons (figure 7). Here, we observed prominently greater proportions of activated astroglia (figure 7B) and microglia (figure 7C) in the spinal dorsal horns of KC;IL6−/− mice (astroglia: 70.2%±5.4%; microglia: 18.8%±6.6%; figure 7B–E) compared with KC mice (astroglia: 45.3%±3.0%; microglia: 3.5%±0.1%; figure 7B–E). In KC;sgp130tg mice with isolated blockade of IL-6 transsignaling, there was a selective and potent suppression of spinal astrocyte activity (astroglia: 4.6%±3.0%, p=0.002 vs KC;IL6−/− and p=0.01 vs KC mice), whereas there was a potent microglial activation (17.63%±4.3%, p=0.02). Interestingly, there was no difference in the average portion of activated spinal neurons in the dorsal half of the spinal cord of KC versus KC;IL6−/− mice, as assessed via the proportion of p-ERK-containing neurons (KC mice: 59.3%±3.1%; KC;IL6−/−: 58.7%±4.7%; figure 7D,E). In contrast, the spinal neuronal activity at these thoracic segments of KC;sgp130tg mice was diminished (25.8%±3.6%, p=0.01 vs KC;IL6−/− mice and p=0.006 vs KC mice). Therefore, while there seems to be a potent parallel astroglia and microglia activity in KC;IL6−/− mice with complete abolishment of IL-6 signalling, the selective blockade of IL-6-transsignaling in KC;sgp130tg mice seems to maintain microglial activation and yet to block astroglia. Therefore, intact classical IL-6 signalling seems to suppress both types of spinal glia, whereas IL-6-transsignaling seems to selectively suppress astroglia and to maintain microglial activation, which may explain the high mechanosensitivity of KC;sgp130tg mice.

Analysis of spinal neuron, astroglia and microglia activity in murine pancreatic cancer (PCa). Mice (KC: n=3; KC;IL6−/−: n=4, KC;sgp130tg: n=4) that were evaluated for mechanical hypersensitivity and pain-related behaviour were subsequently analysed for the activity of spinal cells. Double immunolabeling analysis of murine spinal cord sections were performed to colorimetrically quantify the proportion of explicitly activated cells (ie, for astroglia: p75NTR, for microglia: phospho-p38/p-p38, for neurons: phosho-ERK/p-ERK) to the total number of each cell subclass (glial fibrillary acidic protein (GFAP) as marker of astroglia, Iba-1 as marker of microglia, and beta-III-tubulin as marker of neurons) in both dorsal horns of the spinal cord at the level of the pancreas-innervating thoracic segments 8–11 (Th8-11). The white arrows denote examples of activated cells. The stars in the lower row (neurons) show examples of less active neurons that contain p-ERK (red) solely in the nucleus as opposed to activated neurons (shown by white arrows) that additionally have cytoplasmic p-ERK. Unpaired t test.
To dissect the molecular alterations that may be related to the analgesic effects of activated SCs, we performed PCR array analysis of hypoxic and normoxic hSC mRNA for different targets related to pain sensation (figure 8). Here, in addition to IL-6, we observed 18.1-fold upregulation of the anti-inflammatory IL-10 and downregulation of the nociceptive purinergic receptors P2RX7 (−2.1-fold), P2RX3 (−8.1-fold) and of the nociception-related adenosine A1 receptor (ADORA1, −56.1-fold) within hypoxic hSC compared with under normoxia (figure 8). Hence, hypoxia as a major activator of hSC was capable of activating analgesic and even more suppressing nociceptive target expression.

Regulation of pain-associated targets in human Schwann cells (hSCs) exposed to hypoxia. hSCs were treated with hypoxia for 24 h, and their mRNA was analysed for the expression of 84 different pain-related genes in a PCR array. Targets depicted in this graph exhibited a minimum twofold upregulation or downregulation. ACE, angiotensin I converting enzyme; ADORA1, adenosine A1 receptor; ALOX5, arachidonate 5-lipoxygenase; CCR2, c-c chemokine receptor type 2; CD200, cluster of differentiation 200/OX-2 membrane gylcoprotein; CNR2, cannabinoid receptor 2; CSF1, colony stimulating factor 1; EDN1, endothelin-1; GCH1, gtp cyclohydrolase I; IL10, interleukin-10; IL6, interleukin-6; ITGAM, integrin alpha m; MAOB, monoamine oxidase B; NTRK1, neurotrophic tyrosine kinase receptor type 1; P2RX3, purinergic receptor p2x ligand gated ion channel 3; P2RX7, p2x purinoceptor 7; PENK, proenkephalin; PTGER4, prostaglandin E receptor 4; PTGES3, prostaglandin e synthase 3; PTGS2, prostaglandin-endoperoxide synthase 2; SCN11A, sodium channel voltage gated, type xi alpha subunit; TACR1, tachykinin receptor 1; TLR4, toll-like receptor 4.
As summarised in figure 9, our observations collectively suggest that in PCa, hypoxia and IL-6 signalling induce reactive gliosis of SCs that exert an analgetic feature by suppressing spinal astroglia and microglia (figure 9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The proposed impact of activated Schwann cells on the course of pancreatic ductal adenocarcinoma (PDAC). Schwann cells in PDAC become activated under the influence of tissue hypoxia and of interleukin (IL)-6 secreted from cancer cells. These activated Schwann cells exhibit the same features of ‘reactive gliosis’ as known form astrocytes of the central nervous system. Increased Schwann cell activity in murine PDAC results in suppression of spinal astroglia and microglia activity and is associated with less pain, both in murine and human PDAC.
Discussion
The present study demonstrated that activated hSCs in PCa exhibit the same cardinal features of reactive glia (astrocytes) in the CNS, that is, upregulation of the intermediate filament GFAP, cellular hypertrophy (stellation) and increased secretion of pro-inflammatory cytokines. This intratumoral glial activation seems to be augmented within hypoxic nerves, at sites of neuroinflammation, to depend on IL-6 in vitro and on IL-6 signalling in vivo. Intriguingly, glial activation in the pancreas is shown to be linked to decreased pain sensation both among patients with PCa and in murine PCa models, owing to the suppression of spinal astroglia and/or microglia. Hence, cancer cell-induced SC activation seems to disguise a key symptom, that is, pain, during cancer progression.
Glial activation is a well-characterised trait of CNS and peripheral nervous system (PNS) during injury, ischaemia and infection.6 Similarly, enteric glia were previously shown to increasingly proliferate and upregulate GFAP in UC and Crohn's disease,27–29 to ensure mucosal integrity30 and to differentiate into neurons after enteric injury.31 ,32 However, to our knowledge, the present study is the first one to investigate the activation state of the glia cells of peripheral extrinsic nerves, that is, SCs, in a GI disorder and GI malignancy.
Hypoxia was detected to be one of the strongest activators of hSC in the current study. Although the reaction of CNS astrocytes to hypoxia is well known, it has not received the same extent of attention in studies of the PNS and SC.33 Tumour hypoxia is a key feature of solid desmoplastic tumours and especially of PCa,34 and a driver of pro-angiogenetic and pro-metastatic pathways.35 In the present study, we observed HIF-1α and CA-IX expression not only in PCC, but specifically also in intrapancreatic nerves within PCa. In similarity to hypoxia-induced GFAP upregulation in vitro, the amount of GFAP in nerves in PCa increased in parallel with upregulation of HIF-1α and CA-IX in these nerves. Therefore, one can conclude that nerves in PCa are subject to hypoxia and that glial activation in these nerves is accompanied by activation of hypoxia-associated signalling pathways.
Independently from hypoxia, cancer cells either alone or in co-culture with T-cells were sufficient to induce hSC activation in vitro, and this activation was reversible upon inhibition of IL-6, but not of IL-1β, within PCC supernatants. IL-6, IL-6R and IL-1β were previously reported to be upregulated in hSC subsequent to nerve injury36 and to be required for the induction of GFAP expression after nerve injury.37 ,38 In our study, cancer cell-derived IL-6 emerged as a potential molecular activator of hSC in vitro. The in vivo depletion of IL-6 in mice that progressively develop PCa from murine PanIN lesions resulted in prominently decreased numbers of GFAP-immunoreactive or Sox10-immunoreactive glial cells around PanIN lesions. In this model, the early appearance of SC around PanIN lesions is a typical finding that precedes the invasive cancer stage.7 Interestingly, a similar decrease was less pronounced when the IL6-soluble-IL6R complexing was depleted in sgp130-transgenic mice. These findings underline that IL-6 signalling mediates emergence of GFAP-immunoreactive and Sox10-immunoreactive cells around murine PanIN lesions.
Classically, glial activation is assumed to be linked to more pain sensation.6 While our findings certainly necessitate additional validation, we could show that the paradigm of ‘glial activation resulting in pain’ does not seem to hold true for SC activation. Rather, it seems to be the accompanying reaction of spinal/central microglia and astroglia that determines the pain sensation. In this regard, control of intrapancreatic neuroinflammation by activated SCs may be a potential mechanism that could explain the decreased pain sensation among patients with PCa with reactive gliosis. Correspondingly, in the expression array analysis, we detected prominently elevated levels of the anti-inflammatory IL-10 within hypoxic hSC, and downregulation of pro-nociceptive receptors like the purinergic P2RX3, P2RX7 and of the ADORA1. In the KC;sgp130tg mice, we observed a more complex pain-related behaviour as these animals contrastingly had both high mechanosensitivity and enhanced locomotion. These seemingly contrasting observations can though be attributable to potential selective modulatory roles in the PNS and CNS and thus to differential and selective effects of IL-6-transsignaling on spinal microglia and astroglia. In any case, blockade of the classical IL-6 or of the IL-6-transsignaling reduced the SC frequency around PanINs compared with KC mice, and both conditions were associated with higher abdominal mechanosensitivity than in KC mice. Furthermore, it seems that in animals with high abdominal mechanosensitivity, there seems to be a high spinal microglial activation, which goes in line with previous reports on neuropathic pain.39–41 Hence, our observations imply that activated SC downregulate, clear or suppress factors that would activate peripheral neurons in cancer and thereby inhibit the potential subsequent spinal astroglial and microglial activation. The peculiar peripheral–central glia interaction in the suppression of PCa-associated pain demonstrates a novel, ominous mechanism on how cancer cells can exploit the tissue response to carcinogenesis to escape from surveillance mechanisms.
In summary, the present study demonstrated that hypoxia, cancer cells and neuroinflammation constitute a network that activates hSC in the PCa microenvironment. Reactive hSC exhibit the same cardinal characteristics as activated astrocytes of the CNS and largely depend on IL-6 signalling in the acquisition of this phenotype. Activation of SC leads to suppression of astroglial and microglial activity in the spinal cord, which in turn decreases pain sensation (figure 9). Collectively, cancer cells seem to activate SC in PCa, which seems to result in a prolonged asymptomatic phase and in potentially delayed diagnosis.
Acknowledgments
The authors are grateful to Mr Bernhard Haller (Institute of Medical Statistics and Epidemiology at Technische Universität München) for the statistical review of the manuscript, to Ms Ulrike Bourquain for her excellent technical assistance and to Professor Stefan Rose-John for providing the sgp130 mice. This work is part of ET's MD thesis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table
Footnotes
Contributors IED, ET and SS contributed equally. IED, HF, ML, HA and GOC designed the study. ET, IED, SS, ÖCS, PLP, ST, KW, CW, MUK, SMW and VES performed the experiments. IED, ET, SS, ÖCS, PLP, ST, CW, VES and EC performed the data analysis. HF, HA, TK, EC and GOC supervised the experiments. All authors contributed to the draft of the manuscript and approved the final version.
Funding IED was supported by an institutional KKF (B10-10) grant of the Faculty of Medicine of the Technische Universität München, Munich, Germany.
Competing interests None declared.
Ethics approval Technische Universität München, 1926/07.
Provenance and peer review Not commissioned; externally peer reviewed.