Article Text

Abstract
Objective Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor expressed in tissues with high oxidative activity that plays a central role in metabolism. In this work, we investigated the effect of hepatocyte PPARα on non-alcoholic fatty liver disease (NAFLD).
Design We constructed a novel hepatocyte-specific PPARα knockout (Pparαhep−/−) mouse model. Using this novel model, we performed transcriptomic analysis following fenofibrate treatment. Next, we investigated which physiological challenges impact on PPARα. Moreover, we measured the contribution of hepatocytic PPARα activity to whole-body metabolism and fibroblast growth factor 21 production during fasting. Finally, we determined the influence of hepatocyte-specific PPARα deficiency in different models of steatosis and during ageing.
Results Hepatocyte PPARα deletion impaired fatty acid catabolism, resulting in hepatic lipid accumulation during fasting and in two preclinical models of steatosis. Fasting mice showed acute PPARα-dependent hepatocyte activity during early night, with correspondingly increased circulating free fatty acids, which could be further stimulated by adipocyte lipolysis. Fasting led to mild hypoglycaemia and hypothermia in Pparαhep−/− mice when compared with Pparα−/− mice implying a role of PPARα activity in non-hepatic tissues. In agreement with this observation, Pparα−/− mice became overweight during ageing while Pparαhep−/− remained lean. However, like Pparα−/− mice, Pparαhep−/− fed a standard diet developed hepatic steatosis in ageing.
Conclusions Altogether, these findings underscore the potential of hepatocyte PPARα as a drug target for NAFLD.
- GENE EXPRESSION
- NONALCOHOLIC STEATOHEPATITIS
- LIPID METABOLISM
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor expressed in many tissues and is responsible for several important metabolic controls, especially during fasting.
PPARα is a target for the hypolipidemic drugs of the fibrate family.
PPARα is less expressed in the liver of patients with non-alcoholic fatty liver diseases (NAFLD).
Several PPAR-targeting molecules, including dual agonists, are currently under investigation for NAFLD treatment.
What are the new findings?
Hepatocyte-restricted PPARα deletion impairs liver and whole-body fatty acid homeostasis.
Hepatic PPARα responds to acute and chronic adipose tissue lipolysis.
Hepatic PPARα regulates circadian fibroblast growth factor 21 (FGF21) and fasting-induced FGF21, and is partially responsible for the FGF21 increase in steatohepatitis.
Hepatocyte-restricted PPARα deletion is sufficient to promote NAFLD and hypercholesterolaemia during ageing, but does not lead mice to become overweight.
How might it impact on clinical practice in the foreseeable future?
This work emphasises the relevance and potential of hepatic PPARα as a drug target for NAFLD.
Introduction
Precise control of fatty acid metabolism is essential. Defective fatty acid homeostasis regulation may induce lipotoxic tissue damage, including hepatic steatosis.1 Peroxisome proliferator-activated receptors (PPARs) are transcription factors that serve as fatty acid receptors and help regulate gene expression in response to fatty acid-derived stimuli.2 PPARs act as ligand-activated receptors, controlling target gene transcription. The three PPAR isotypes, PPARα, PPARβ/δ and PPARγ, display specific tissue expression patterns and control different biological functions,3 but all bind lipids and control lipid homeostasis in different tissues, including the liver.2
A healthy liver does not accumulate lipids, but it plays central roles in fatty acid anabolism and export to peripheral organs, including white adipose tissue for energy storage.4 During dietary restriction, hepatic fatty acid catabolism is also critical for using free fatty acids (FFAs) released from white adipose tissues. PPARα is the most abundant isotype in hepatocytes and is involved in many aspects of lipid metabolism,5 ,6 including fatty acid degradation, synthesis, transport, storage, lipoprotein metabolism and ketogenesis during fasting.7–9 In addition, PPARα controls glycerol use for gluconeogenesis9 as well as autophagy10 in response to fasting. Moreover, PPARα regulates the expression of the fibroblast growth factor 21 (FGF21) during starvation.11 ,12 In turn, FGF21 acts as an endocrine hormone targeting various functions including metabolic control.13 Finally, PPARα helps repress the acute-phase response and inflammation in the liver.14
Obesity can lead to organ and vascular complications.15 Non-alcoholic fatty liver disease (NAFLD), which are considered the hepatic manifestation of metabolic syndrome, range from benign steatosis to severe non-alcoholic steatohepatitis (NASH), potentially further damaging organs.16 Sustained elevation of neutral lipid accumulation (mostly triglycerides in hepatocyte lipid droplets) initiates early pathological stages. Different fatty acid sources contribute to fatty liver development, including dietary lipid intake, de novo lipogenesis and adipose tissue lipolysis.4 In NAFLD, 60% of fatty acids accumulated in steatotic liver are adipose-derived.17
Preclinical18–21 and clinical22 studies highlight that PPARα influences NAFLD and NASH. Mice lacking PPARα develop steatosis during fasting,7 ,8 suggesting the importance of PPARα activity for using FFA released from adipocytes. However, PPARα is expressed and active in many tissues, including skeletal muscles,23 adipose tissues,24 ,25 intestines,26 kidneys27 and heart,28 which all contribute to fatty acid homeostasis. Therefore, it remains unknown whether the increased steatosis susceptibility in mice lacking PPARα depends on PPARα activity only in hepatocytes or also in other organs.
Here we investigated consequences of hepatocyte-specific Pparα deletion, focusing on effects on fatty acid metabolism in NAFLD, ranging from steatosis to steatohepatitis. We report the first evidence that adipocyte lipolysis correlates with and stimulates NAFLD when hepatocytes are lacking PPARα. Our data establish that hepatocyte-restricted Pparα deletion is sufficient to promote steatosis, emphasising this receptor's relevance as a drug target in NAFLD.
Materials and methods
Animals
Generation of floxed-Pparα mice and of Pparα hepatocyte-specific knockout (Pparαhep−/−) animals is described in online supplementary file 1.
In vivo experiments
In vivo studies followed the European Union guidelines for laboratory animal use and care, and were approved by an independent ethics committee.
Detailed experimental protocols are provided in online supplementary file 1.
Plasma analysis
Plasma FGF21 and insulin, respectively, were assayed using the rat/mouse FGF21 ELISA kit (EMD Millipore) and the ultrasensitive mouse insulin ELISA kit (Crystal Chem) following the manufacturer's instructions. Aspartate transaminase, alanine transaminase (ALT), total cholesterol, LDL cholesterol and HDL cholesterol were determined using a COBAS-MIRA+ biochemical analyser (Anexplo facility).
Circulating glucose and ketone bodies
Blood glucose was measured using an Accu-Chek Go glucometer (Roche Diagnostics). β-Hydroxybutyrate content was measured using Optium β-ketone test strips with Optium Xceed sensors (Abbott Diabetes Care).
Histology
Paraformaldehyde-fixed, paraffin-embedded liver tissue was sliced into 5 μm sections and H&E stained. Visualisation was performed using a Leica DFC300 camera.
Liver lipids analysis
Detailed experimental protocols are provided in online supplementary file 1.
Gene expression studies
Total RNA was extracted with TRIzol reagent (Invitrogen). Transcriptomic profiles were obtained using Agilent Whole Mouse Genome microarrays (4×44k). Microarray data and experimental details are available in the Gene Expression Omnibus (GEO) database (accession number GSE73298 and GSE73299). For real-time quantitative PCR (qPCR), 2 µg RNA samples were reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Online supplementary file 2 presents the SYBR Green assay primers. Amplifications were performed using an ABI Prism 7300 Real-Time PCR System (Applied Biosystems). qPCR data were normalised to TATA-box-binding protein mRNA levels, and analysed with LinRegPCR.v2015.3.
Transcriptomic data analysis
Data were analysed using R (http://www.r-project.org). Microarray data were processed using Bioconductor packages (http://www.bioconductor.org, v 2.12)29 as described in GEO entry GSE26728. Further details are provided in online supplementary file 1.
Statistical analysis
Data were analysed using R (http://www.r-project.org). Microarray data were processed using bioconductor packages (http://www.bioconductor.org) as described in GEO entry GSE38083. Genes with a q value of <0.001 were considered differentially expressed between genotypes. Gene Ontology (GO) Biological Process enrichment was evaluated using conditional hypergeometric tests (GOstats package). For non-microarray data, differential effects were analysed by analysis of variance followed by Student's t-tests with a pooled variance estimate. A p value <0.05 was considered significant.
Results
Generation of hepatocyte-specific PPARα knockout mice
Progeny carrying the Pparαflox/flox alleles (figure 1A), referred to as floxed, were backcrossed in the C57Bl/6J background, and then crossed with albumin-Cre mice in the same genetic background, generating a hepatocyte-specific PPARα knockout (Pparαflox/floxalbumin-Cre+/−) referred to as Pparαhep−/− (figure 1B). PPARα mRNA was not detected in livers from Pparαhep−/− mice when compared with floxed and C57Bl6/J mice (figure 1C), suggesting that most hepatic PPARα expression is from hepatocytes. PPARα absence in hepatocytes did not alter mRNA expression of other PPAR isotypes (figure 1C).
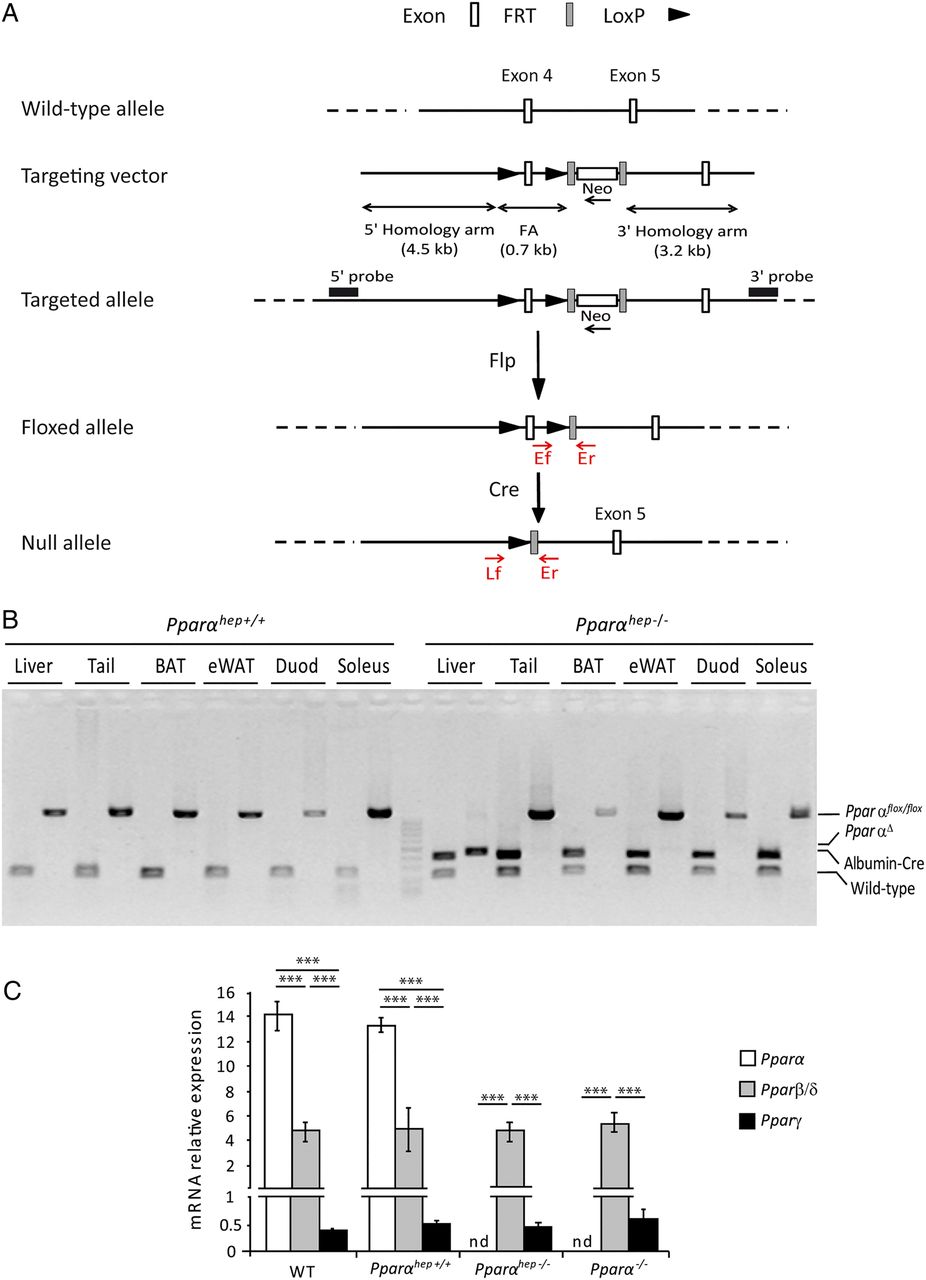
Characterisation of the hepatocyte-specific peroxisome proliferator-activated receptor α (PPARα) knockout mouse model. (A) Schematic of the targeting strategy to disrupt hepatic Pparα expression. (B) PCR analysis of Pparα floxed (Pparαhep+/+) and Albumin-Cre (Albumin-Cre+/−) genes from mice that are liver wild-type (WT), (Pparαhep+/+) or liver knockout (Pparαhep−/−) for Pparα using DNA extracted from different organs. (C) Relative mRNA expression levels of Pparα, Pparβ/δ and Pparγ from liver samples of WT, liver WT (Pparαhep+/+), Pparα liver knockout (Pparαhep−/−) and Pparα knockout (Pparα−/−) mice (n=8 mice per group). Data represent mean±SEM. ***p≤0.005. FA, floxed allele; Flp, flippase; FRT, flippase recognition target; LoxP, locus of X-overP1; nd, not detected; PparαΔ, Pparα deletion; WT, the Albumin-Cre−/− allele.
Hepatocyte-autonomous effect of fenofibrate on PPARα activity
To determine whether PPARα response was hepatocyte-autonomous, we challenged wild-type (WT), floxed Pparαhep+/+, Pparα−/− and Pparαhep−/− mice with the PPARα agonist fenofibrate. We measured mRNA expressions of PPARα target genes, including Cyp4a10 (figure 2A) and Cyp4a14 (figure 2B). Their expressions were strongly induced by fenofibrate in WT and in floxed Pparαhep+/+ mice compared with Pparα−/− and Pparαhep−/− mice. These samples were also used for pangenomic expression profiling through microarray analysis (figure 2C). Differentially expressed gene (DEG) analysis was subjected to hierarchical clustering, highlighting similar expression profiles between WT and floxed Pparαhep+/+ mice within fenofibrate-treated or vehicle-treated groups. Whole-body Pparα−/− and Pparαhep−/− mice were unresponsive to fenofibrate, suggesting that fenofibrate-induced hepatic changes were mainly due to autonomous hepatocyte responses, not secondary to extrahepatic PPARα activation. GO biological function analysis revealed that fenofibrate upregulated lipid metabolism, and repressed immune and defence response, metabolic responses, and glycosylation and glycoprotein metabolism (figure 2C, groups 1, 2, 6 and 7). However, untreated Pparα−/− and Pparαhep−/− mice showed marked differences (figure 2C, groups 3, 4, 8 and 9). This implies that the absence of extrahepatic PPARα has a significant impact on the liver transcriptional profile and underscores the relevance of Pparαhep−/− mice to define the hepatocyte autonomous role of the receptor in the control of liver function.

Pharmacological peroxisome proliferator-activated receptor α (PPARα) activation using fenofibrate reveals hepatocyte-specific PPARα-dependent biological functions. Liver samples from wild-type (WT), PPARα knockout (Pparα−/−), liver WT (Pparαhep+/+) and Pparα hepatocyte knockout (Pparαhep−/−) mice treated with fenofibrate (Feno, +) or vehicle (−) by oral gavage for 14 days were collected. (A and B) The relative gene expression of two specific PPARα target genes Cyp4a10 (A) and Cyp4a14 (B) was measured by qRT-PCR. Data represent mean±SEM. **p≤0.01, ***p≤0.005. (C) Heat map representing data from a microarray experiment performed with liver samples. Hierarchical clustering is also shown, which allows the definition of nine gene clusters. Gene Ontology (GO) analysis of each cluster revealed significant biological functions (p≤0.05). nd, not detected.
Hepatocyte PPARα activity is context-specific
The Pparαhep−/− model was used to determine whether PPARα could drive hepatic regulations both in fasting-induced fatty acid catabolism as well as fatty acid anabolism during refeeding. The fasting–refeeding experimental design was validated by measuring glycaemia (figure 3A) and expression of fatty acid synthase (Fasn), which encodes the rate-limiting enzyme in lipogenesis (figure 3B). Both were low during fasting, intermediary in ad libitum-fed animals, and high in refed animals. Cyp4a14 (a well-known PPARα target) expression was low or undetectable in Pparαhep−/− animals, and strongly upregulated with fasting in WT mice (figure 3C).

Hepatocyte-specific peroxisome proliferator-activated receptor α (PPARα) function is dependent on nutritional status. Wild-type (WT) and PPARα liver knockout (Pparαhep−/−) male 8-week-old mice were fed ad libitum, fasted for 24 h, or fasted for 24 h and refed for 24 h. All mice were killed at ZT14, and sera and livers were collected. (A) Quantification of circulating glucose levels. (B, C) Relative mRNA expressions of Fasn (B) and Cyp4a14 (C) in liver samples quantified by qRT-PCR. Data represent mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005. (D) Heat map was performed based on average gene expression levels from WT (n=12 (6 WT and 6 Pparαhep+/+)) and from Pparαhep−/− (n=6). (E) Venn diagram and associated Gene Ontology (GO) function analysis (p≤0.05), GO categories corresponding to functions down in the absence of PPARα are in bold, GO categories corresponding to functions up in the absence of PPARα are in regular font.
Next we evaluated the hepatic transcriptome expression pattern using microarrays. We performed hierarchical clustering (figure 3D). Most PPARα-dependent changes were observed in fasted mouse livers. Venn diagrams were used to show nutritional status-related PPARα-dependent changes (figure 3E). Among the significant DEGs, 3048 were related to fasting, 390 to ad libitum-fed animals and 156 to refed mice, suggesting context-specific PPARα activity. The results further highlighted that fasting, rather than feeding or refeeding, triggered the broader PPARα-dependent hepatocytic response, with most upregulated genes related to metabolism (figure 3E). However, the expression of several genes was identified as PPARα dependent regardless of the nutritional condition tested (fasting, but also feeding and refeeding). These genes are mostly downregulated in the absence of PPARα and pathway analysis highlights their involvement in mitochondrial fatty acid catabolism (see online supplementary file 3).
Biological function analyses revealed that both transcriptional activation and repression were PPARα sensitive (figure 3E). The functions of PPARα-sensitive repressions (GO categories up in Pparαhep−/− mice) varied with context, and included GO categories not directly related to metabolism, including acute-phase response (fed), translation (refed) and protein glycosylation (fasted).
Hepatocyte PPARα is required for liver and whole-body fatty acid homeostasis in fasting
We next used Pparαhep−/− mice to determine the contribution of hepatocyte PPARα, and compared it with Pparα−/− and WT mice. We measured FFA and β-hydroxybutyrate (ketonaemia) levels in fasted and non-fasted mice (figure 4A). Plasma FFA was elevated in fasting mice of all three genotypes, but was significantly higher in Pparαhep−/− and Pparα−/− mice compared with controls. Fasting strongly increased ketone body levels in WT mice and to a lesser degree in Pparαhep−/− and Pparα−/− mice. This suggests that hepatic PPARα is required for FFA disposal and for β-hydroxybutyrate production. Correspondingly, fasting Pparαhep−/− and Pparα−/− mice showed elevated hepatic triglycerides and cholesterol esters (figure 4B), and substantial centrilobular steatosis (figure 4C), confirming that hepatic PPARα expression is required for fasting-induced FFA catabolism. PPARα absence led to defective expressions of PPARα target genes (figure 4D), including those involved in fatty acid catabolism and processing in lipid droplets (figure 4E). As a consequence of PPARα deficiency in hepatocytes, Pparαhep−/− mice exhibit a distinct fasting-induced fatty acid profile with a significant increase in oleic acid (C18:1n–9) and linoleic acid (C18:2n–6) when compared with WT mice (see online supplementary file 4).

Fasting is the major inducer of hepatic peroxisome proliferator-activated receptor α (PPARα) activity. Wild-type (WT), hepatocyte-specific PPARα knockout (Pparαhep−/−) and total PPARα knockout (Pparα−/−) mice were fed ad libitum or fasted for 24 h and then killed. (A) Quantification of plasma free fatty acids (FFAs) and ketone bodies (ketonaemia). (B) Hepatic triglycerides and cholesterol esters hepatic levels. (C) Representative pictures of H&E staining of liver sections. Scale bars, 100 µm. (D) Relative mRNA expression levels of Pparα, Cyp4a14 and Vnn1 in liver samples determined by qRT-PCR. (E) Quantification of mRNA expression of Acox1, Hmgcs2, Acadl, Fsp27 and Plin5 by qRT-PCR. Data shown as mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005.
Hepatocyte-specific Pparα deletion impairs constitutive and fasting-induced FGF21 expression
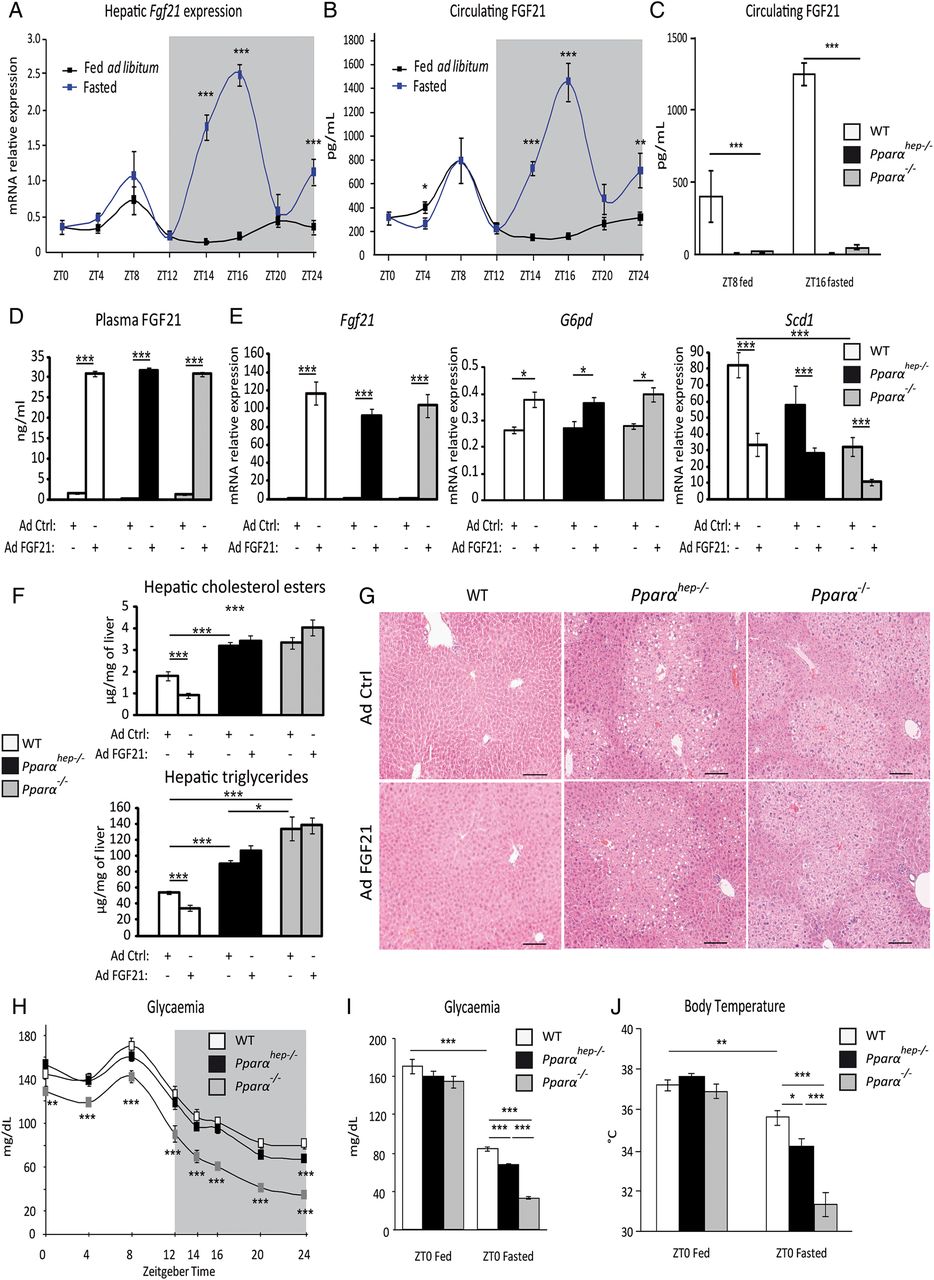
FGF21 is a hepatokine mainly produced by the liver. We examined liver Fgf21 mRNA expression (figure 5A) and plasma FGF21 levels (figure 5B) in fed and fasted animals. We identified a constitutive expression peak during the day (ZT8) in both groups, and a fasting-triggered night-time peak (ZT16). In Pparαhep−/− mice, we examined whether fasting-induced FGF21 expression/production was strictly dependent on PPARα hepatic activity. Pparα−/− and Pparαhep−/− mice showed very low plasma FGF21 protein at ZT8 or at ZT16 with fasting (figure 5C).

Hepatocyte and extrahepatocyte peroxisome proliferator-activated receptor α (PPARα) regulate fibroblast growth factor 21 (FGF21), glycaemia and body temperature during fasting. (A and B) Eleven-week-old male mice of the C57Bl/6J background were fed ad libitum or fasted for 24 h, and were killed around the clock from ZT0 to ZT24. (A) Fgf21 mRNA was quantified by qRT-PCR. (B) Quantification of circulating FGF21 levels by ELISA. (C) Twelve-week-old wild-type (WT), PPARα-hepatocyte knockout (Pparαhep−/−) and PPARα knockout (Pparα−/−) male mice were fed ad libitum or fasted for 16 h and blood was collected at ZT8 (ZT8 fed) or at ZT16 (ZT16 fasted). FGF21 plasma level was determined by ELISA. (D–G) Male mice of WT, Pparαhep−/− and Pparα−/− genotypes were infected with an adenoviral construct containing cDNA of Fgf21 or an empty vector. Mice were sacrificed after a 24 h fasting period at ZT14. (D) Quantification of circulating FGF21 levels by ELISA. (E) Fgf21, G6pd and Scd1 mRNAs were quantified by qRT-PCR. (F) Quantification of hepatic cholesterol esters and triglycerides. (G) Representative pictures of H&E staining of liver sections. Scale bars, 100 µm. (H) Plasma glucose level was monitored over a 24 h fasting period from ZT0 to ZT24 in WT, Pparαhep−/− and Pparα−/− mice. (I, J) Plasma glucose (I) and body temperature (J) were determined at ZT0 in fed mice or at ZT0 in mice fasted for 24 h. Data are shown as mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005.
Since FGF21 has been shown to reduce steatosis and lipotoxic lipids13 ,30 we questioned whether the absence of FGF21 determines fasting-induced steatosis observed in Pparαhep−/− and Pparα−/− mice. FGF21 expression was rescued by adenoviral delivery both in Pparαhep−/− and in Pparα−/− mice (figure 5D). Comparable expression of FGF21 (figure 5E) was obtained in liver of WT, Pparαhep−/− and in Pparα−/− mice. FGF21-sensitive genes such as G6pd and Scd1 showed significantly different expression in response to FGF21 overexpression (figure 5E). However, FGF21 only reduced hepatic triglycerides and cholesterol esters in WT mice, but not in Pparαhep−/− and in Pparα−/− mice (figure 5F, G). These results indicate that the fasting-induced steatosis occurring in Pparαhep−/− and in Pparα−/− mice does not depend on the lack of FGF21. This is in line with our observations that FGF21- and PPARα-sensitive target genes are different (see online supplementary file 5A). Moreover, it is also consistent with the observation that FGF21 overexpression does not rescue the expression of PPARα target genes and conversely that PPARα-sensitive regulations occur in Fgf21−/− mice (see online supplementary file 5B, C).
In addition to their defective fatty acid catabolism, Pparα−/− mice are hypoglycaemic and hypothermic during fasting.7 Because FGF21 is important for glucose homeostasis and for thermogenesis,13 we investigated the role of hepatocyte PPARα in controlling fasting glycaemia and body temperature. Both Pparαhep−/− and Pparα−/− mice were hypoglycaemic and hypothermic compared with WT mice during fasting. However, this phenotype was much stronger in fasted Pparα−/− mice compared with fasted Pparαhep−/− mice (figure 5H-J), indicating that extrahepatic PPARα strongly influenced whole-body glucose homeostasis and temperature independent of hepatocytic PPARα activity and FGF21 production.
Fasting-enhanced hepatocytic PPARα activity is time-restricted and sensitive to adipocyte lipolysis
We next tested the kinetics of other fasting-induced hepatic PPARα activity in vivo. We used several measures of PPARα activity, including Fgf21 (figure 5A) and Vanin1, Cyp4a10, Cyp4a14 and Fsp27 mRNAs (figure 6A), since these genes were most sensitive to fasting and to fenofibrate, and were strictly PPARα dependent (see online supplementary files 6–10A). Plasma FFA and glucose levels were also measured during fasting (figure 6B). FFA were markedly increased in the early night (ZT14–ZT16). The FFA pattern was correlated with the PPARα mRNA expression profile and expressions of Fgf21, Vanin1, Cyp4a10, Cyp4a14 and Fsp27 (figures 5A and 6A). This strongly suggested that FFA released from adipocytes during fasting-influenced hepatic PPARα expression and activity without inflammatory response since hepatic Tnfα mRNA expression was not sensitive to fasting. We further determined that acute treatment of fasted mice with the β3-adrenergic receptor agonist CL316243 enhanced circulating FFA levels in WT and Pparαhep−/− mice (figure 6C), and increased expressions of Fgf21, Cyp4a14, Vanin1, Cyp4a10 and Fsp27 in WT mice but not Pparαhep−/− mice (figure 6D) without inducing Tnf α in response to fasting or in response to CL316243 (see online supplementary file 10C and D). These data support a role for acute adipocyte lipolysis as a signal for hepatocyte PPARα activity during fasting.

Hepatocyte peroxisome proliferator-activated receptor α (PPARα) activity is induced by adipose tissue lipolysis. (A and B) Liver samples were collected from male wild-type (WT) C57Bl/6J mice that were fed ad libitum (black curve) or fasted (blue curve) over 24 h. (A) Hepatic mRNA expression levels of Pparα, Cyp4a14, Vnn1, Cyp4a10, Fsp27 and Tnfα were quantified by qRT-PCR. (B) Plasma glucose and free fatty acids (FFA) were measured. (C and D) WT and PPARα hepatocyte-specific knockout (Pparαhep−/−) mice were treated with the β3-adrenergic receptor agonist CL316243 at ZT6 and then killed at ZT14. (C) Quantification of plasma FFA. (D) Relative mRNA expression levels of Fgf21, Cyp4a14, Vnn1, Cyp4a10 and Fsp27 were measured by qRT-PCR. Data are shown as mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005.
Hepatocyte PPARα is required for protection in steatohepatitis
We next examined whether the hepatocytic PPARα response to chronic lipolysis occurred during methionine-deficient and choline-deficient diet (MCD)-induced weight loss. In rodents, this diet rapidly promotes lipolysis in adipocytes, resulting in steatohepatitis. On the MCD diet, mice of each genotype showed weight loss (figure 7A), steatosis (figure 7B), and increased hepatic triglycerides, cholesterol esters (figure 7C) and plasma ALT (figure 7D). Compared with WT, Pparαhep−/− and Pparα−/− mice showed greater steatosis and liver damage, suggesting a more severe MCD diet-induced phenotype without hepatocyte PPARα. MCD also induced increased expressions of Cyp4a14 and Vanin1 in WT mice, but not Pparαhep−/− or Pparα−/− mice (figure 7E). Fgf21 mRNA (figure 7E) and circulating FGF21 (figure 7F) were increased through a mechanism that is partly dependent on hepatic PPARα. Overall, hepatocyte-specific Pparα deletion aggravated MCD diet-induced liver damage, correlating with defective PPARα-dependent pathway upregulation in response to chronic lipolysis.

Liver peroxisome proliferator-activated receptor α (PPARα) deficiency aggravates non-alcoholic steatohepatitis in response to a methionine-deficient and choline-deficient diet (MCD). Wild-type (WT), PPARα hepatocyte knockout (Pparαhep−/−) and PPARα knockout (Pparα−/−) mice were fed a MCD or a control diet for 2 weeks and were killed at ZT8. (A) Body weight gain was measured over 2 weeks. (B) Representative pictures of H&E staining on liver sections. Scale bar, 100 µm. (C) Quantification of hepatic triglycerides and cholesterol esters. (D) Alanine transaminase activity level in plasma. (E) Hepatic mRNA expression levels of Cyp4a14, Vnn1 and Fgf21. (F) Plasma levels of fibroblast growth factor 21 (FGF21). Data are shown as mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005.
Additionally, we questioned whether hepatocyte PPARα may also be required for the protection of the liver during early hits in steatosis such as those occurring in response to short-term exposure to a high-fat diet (HFD). Over 2 weeks of HFD, mouse liver accumulated hepatic triglycerides and cholesterol esters. Importantly, this steatosis was twice higher in Pparαhep−/− mice than in WT mice, and was further elevated in Pparα−/− mice (see online supplementary file 11). Altogether, these data suggest that hepatic PPARα is essential in hepatoprotection.
Hepatocyte PPARα deficiency leads to steatosis and hypercholesterolaemia but not excess weight gain in ageing mice
Lastly, we questioned the long-term consequences of hepatocyte-specific Pparα deletion during ageing. More specifically, since PPARα is broadly expressed in metabolic tissues, we aimed at clarifying whether the steatosis that develops in aged whole-body Pparα−/− mice is due to the hepatocytic defect in PPARα activity. WT, Pparαhep−/− and Pparα−/− mice were fed a standard diet over 1 year. Pparα−/− mice, but not Pparαhep−/− mice, grew overweight with ageing (figure 8A–C). Both Pparαhep−/− and Pparα−/− mice showed spontaneous centrilobular steatosis (figure 8D), elevated hepatic triglycerides and hepatic cholesterol esters (figure 8E), as well as hypercholesterolaemia (see figure 8F online supplementary file 12) without hyperglycaemia (figure 8G). Overall, hepatocyte-specific PPARα deficiency was sufficient to induce spontaneous steatosis and disrupt whole-body fatty acid as well as cholesterol homeostasis, but did not affect weight gain and diabetes during ageing.
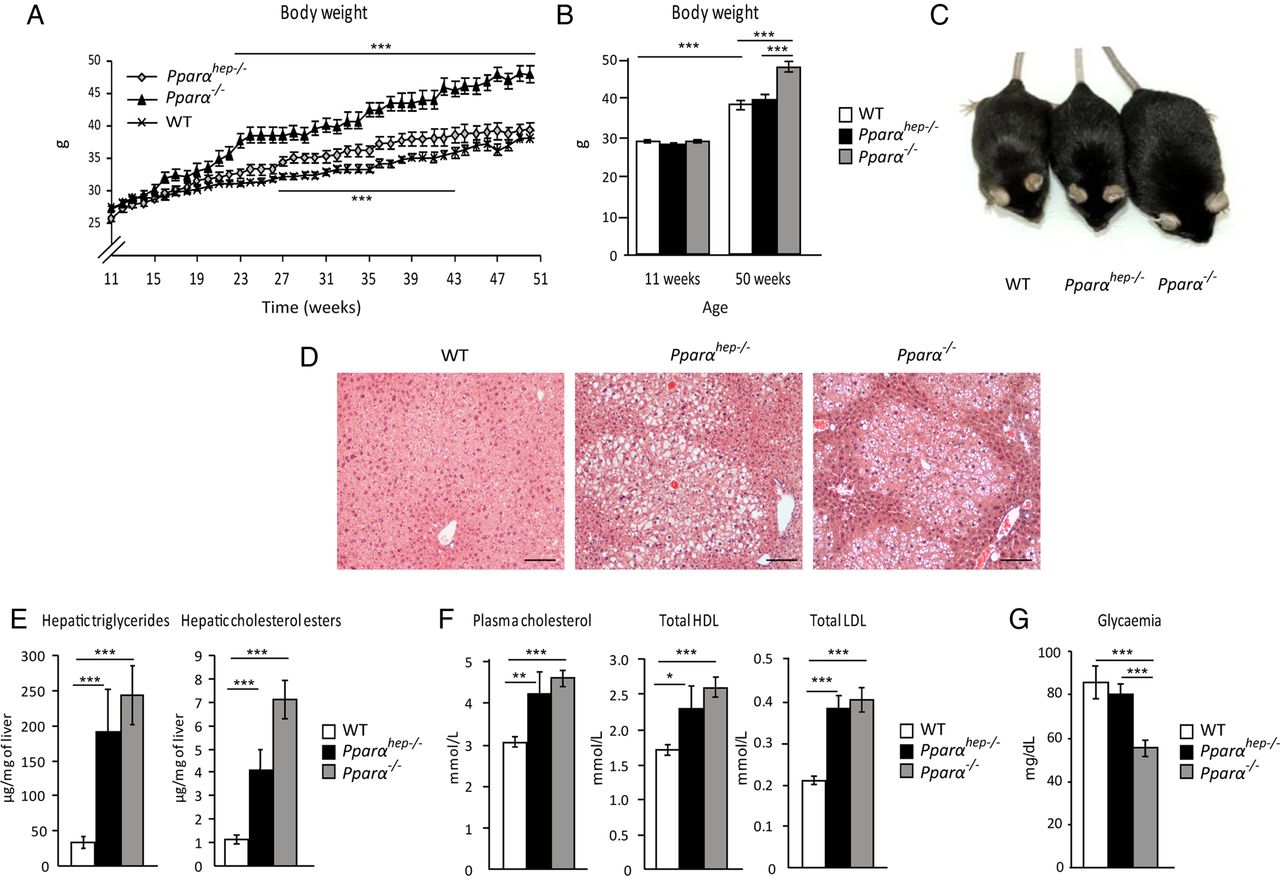
Mice deficient in hepatic peroxisome proliferator-activated receptor α (PPARα) develop spontaneous hepatic steatosis during ageing. Wild-type (WT), PPARα hepatocyte knockout (Pparαhep−/−) and PPARα knockout (Pparα−/−) mice were fed a chow diet for 51 weeks. All mice were killed at ZT16 in a non-fasted state. (A) Body weight gain was followed over time. (B) Comparison of body weight between weeks 11 and 50. (C) Representative pictures of 52-week-old mice of the three genotypes. (D) Representative images of H&E staining of liver sections. Scale bar, 100 µm. (E) Quantification of hepatic triglycerides and cholesterol esters. (F) Measurement of plasma total cholesterol, HDL cholesterol and LDL cholesterol. (G) Fasting glycaemia. Data are shown as mean±SEM. *p≤0.05, **p≤0.01, ***p≤0.005.
Discussion
NAFLD are a spectrum of diseases presenting a major public health concern that is strongly linked with obesity. Most accumulated hepatic fatty acids in NAFLD come from increased non-esterified FFA in the fasting state.17 Thus, it is essential to define the mechanisms by which the liver adapts to this influx. FFA processing largely involves the fatty acid oxidative pathway, coupled to ketogenesis allowing the liver to use lipids,31 which is critical during fasting and requires transcriptional regulation of rate-limiting enzymes.32
Whole-body Pparα−/− mice show impaired coping with prolonged fasting, resulting in defective fatty acid oxidation and steatosis, hypoglycaemia and hypothermia. However, PPARα also contributes to metabolic homeostasis through expression in other tissues. Here we investigated the impact of hepatocyte-specific PPARα deletion on liver physiology and lipid metabolism in vivo. To our knowledge, this is the first report that selective PPARα deletion in hepatocytes (Pparαhep−/−) was sufficient to promote hepatic steatosis.
PPARα is targeted by several fibrate drugs,33 and by pan-agonists for PPAR isotypes21 that are currently in clinical trials for NASH treatment. Using Pparαhep−/− mice, we demonstrated an autonomous transcriptional response of hepatocytes to fenofibrate, indicating that fibrates’ effects on the liver gene expression are largely independent from those in extrahepatic tissues. Moreover, liver gene expression profiles markedly differed between untreated Pparα−/− and Pparαhep−/− mice, suggesting that extrahepatocytic PPARα activity substantially influenced the hepatic transcriptome.
Food restriction induces PPARα activity, and endogenous PPARα ligand production requires hepatic lipogenesis, which increases upon feeding.34 ,35 Thus, PPARα may be important during fasting-induced lipid catabolism and in the response to anabolic fatty acid-derived signals. Our data revealed the context dependency of PPARα hepatocytic activity defined by DEGs. This activity was clearly the highest during fasting. During fasting, hepatocyte-specific PPARα deletion resulted in steatosis, increased plasma FFA and impaired ketone bodies. This supports the concept that FFA released from adipose stores during fasting may activate PPARα for hepatic use. Accordingly, we found that Pparαhep−/− mice accumulate high oleic and linolenic acids in the liver during fasting (see online supplementary file 4), which is in agreement with the fact that both of them are the main fatty acids stored in the white adipose tissues of mice fed a chow diet.36 Importantly, we found a high correlation between the kinetics of circulating FFA increase and expression of PPARα and several of its target genes. Moreover, treatment with a β3-adrenergic receptor agonist further enhanced this response in vivo through PPARα but did not induce detrimental FFA-sensitive response driven by toll-like receptor 4 (TLR4). This is likely due to the mixture of FFA released from the adipose stores. Indeed, fatty acids that accumulated in the liver of Pparαhep−/− mice during fasting were mostly oleic (C18:1n–9) and linoleic acids (C18:2n–6), and not only saturated fatty acids such as palmitic acid (C16:0). Interestingly, it has been shown that palmitic acid cannot activate TLR4 in the presence of unsaturated FFA.37
Overall, our data highlight hepatic PPARα activity regulation by fatty acids released from adipocytes. This contrasts with the previous evidence that PPARβ/δ rather than PPARα may act as a FFA sensor.38 However, our data support the possibility that this adipose-derived signal is time-restricted and specifically efficient in early night. Moreover, other pathways likely influence PPARα activity by providing ligands.34 ,35 ,39 ,40 Several insulin-sensitive signalling mechanisms influence hepatic PPARα, and adipocyte lipolysis is insulin sensitive.41 Thus, insulin may coordinate hepatic PPARα, both through cell-autonomous mechanisms and adipocyte lipolysis inducing interorgan communication mediated by FFA release. Our findings also correspond with the recent evidence that adipocyte lipolysis may regulate hepatic Fgf21.42 Circulating FGF21 was strictly dependent on hepatocytic PPARα activation during fasting. Most circulating FGF21 is liver-derived43 and Pparα−/− mice show very little FGF21.11 ,12 Other transcription factors can also regulate hepatic Fgf21 expression44–48 and PPARα is also expressed in extrahepatic tissues.13 Our findings in Pparαhep−/− mice showed very little FGF21 without hepatic PPARα in both fed and fasted states. Pparα−/− mice are hypoglycaemic and hypothermic during fasting7 and FGF21 is known for its endocrine effect on glucose homeostasis and thermogenesis.13 However, compared with fasted Pparα−/− mice, fasted Pparαhep−/− mice showed reduced hypoglycaemia and hypothermia while FGF21 was equally absent in both models. This indicates that extrahepatocytic PPARα strongly influenced whole-body glucose homeostasis and temperature independently of hepatocyte PPARα and FGF21 production during fasting. In addition, while FGF21 prevents steatosis in different mouse models13 ,30 and FGF21 reduces hepatic lipids in WT mice, its overexpression is not sufficient to protect from lipid accumulation in Pparαhep−/− and in Pparα−/− mice. Therefore, the absence of FGF21 is not the primary cause for the steatosis observed in Pparαhep−/− mice.
Lack of hepatic PPARα impairs the liver's ability to use FFA from acute lipolysis, resulting in steatosis. MCD diet-induced weight loss49 ,50 also correlated with hepatic PPARα activity, suggesting that chronic lipolysis elevates hepatocytic PPARα activity in non-fasted mice. In agreement with the findings in whole-body PPARα-deficient mice,20 our data demonstrated that the absence of hepatocytic PPARα was sufficient to increase MCD diet-induced liver damage. FGF21 expression/circulating levels increased in steatohepatitis, supporting the possibility that elevated FGF21 may reflect liver stress without fasting. This MCD diet-induced FGF21 increase was not strictly PPARα-dependent, consistent with the findings that amino acid deprivation induces hepatic FGF21 expression through ATF4.44 PPARα presence led to greater FGF21 increase, and may contribute to hepatoprotection from lipotoxic lipid accumulation.30
MCD diet is widely used for preclinical NASH studies. However, it has many limitations, including the important weight loss that occurs in mice fed such diet. Therefore, we also tested the role of hepatocyte PPARα in lipid homeostasis in response to a short-term HFD feeding, which is sufficient to initiate early neutral lipid accumulation that may promote NAFLD. Pparαhep−/− mice showed marked increase in hepatic steatosis in response to 2 weeks of HFD feeding (see online supplementary file 11) suggesting that hepatocyte PPARα plays a dual role in exogenous (dietary) as well as in endogenous (released from adipocyte lipolysis) fatty acid homeostasis.
Previous studies have shown that Pparα−/− mice show a significant alteration of systemic lipid metabolism that leads to hepatic steatosis in ageing mice. Since PPARα is active in skeletal muscles,23 adipose tissues,24 ,25 intestines,26 kidneys27 and heart,28 which all contribute to fatty acid homeostasis, it is impossible to determine whether the spontaneous steatosis that occurs in ageing Pparα−/− mice originates from a defect in the hepatocytic PPARα activity. This led us to investigate ageing-related differences between Pparα−/− and Pparαhep−/− mice. During ageing, Pparα−/− mice became overweight and developed steatosis, while Pparαhep−/− mice only suffered steatosis. Therefore, neither obesity nor hyperglycaemia, which are both known to promote NAFLD,15 ,16 is responsible for the steatosis observed in mice with hepatocyte-specific PPARα deletion.
Furthermore, both Pparα−/− and Pparαhep−/− ageing mice were hypercholesterolaemic. This is likely due to the dysregulation of apolipoproteins gene expression as well as cholesterol transport (Abcg8) as revealed in microarray analysis (see online supplementary file 12A). It is also possible that the cholesterol biosynthesis pathway driven by SREBP-2 may be dysregulated in the absence of PPARα since some of the SREBP-2 genes are elevated in Pparα−/− and/or in Pparαhep−/− mice (see online supplementary file 12B). Therefore, this suggests that drugs that activate hepatocytic PPARα will likely influence whole-body fatty acid and cholesterol homeostasis.
Altogether, our extensive analysis performed in Pparαhep−/− mice has allowed us to extend the evidence for the central role of PPARα in hepatocyte fatty acid homeostasis (figure 9). PPARα is strikingly essential to many aspects of fatty acid homeostasis including degradation through oxidative pathways. Our work provides the first demonstration that hepatocyte-specific PPARα deletion impairs whole-body fatty acid homeostasis during fasting, MCD and HFD feeding as well as in ageing. These findings underscore the central role of PPARα in the clearance of dietary fatty acids and of FFA released from adipocytes, the major source of lipid accumulation in NAFLD. These data highlight the relevance of PPARα as a drug target for NAFLD treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Overview of hepatocyte-specific peroxisome proliferator-activated receptor α (PPARα)-regulated genes involved in fatty acid metabolism. This figure was designed based on transcriptome analysis of PPARα-dependent gene expression in hepatocytes. Genes listed in regular font are induced by fenofibrate and by fasting in wild-type (WT) but not in Pparαhep−/− mice. Genes in italics are repressed by fenofibrate and by fasting in WT but not in Pparαhep−/− mice. Genes referenced in bold are downregulated in Pparαhep−/− compared with WT mice, whatever the conditions.
Acknowledgments
We thank all members of the EZOP staff, particularly Colette Bétoulières for her careful and outstanding help from the early start of this project. We thank Aurore Dequesnes and Laurent Monbrun for their excellent work on plasma biochemistry. We thank Christine Salon and Florence Capilla for their excellent work on histology. We thank the staff from the Genotoul: Anexplo, Get-TriX and Metatoul-Lipidomic facilities. The authors wish to thank Professor Daniel Metzger, Professor Pierre Chambon (IGBMC, Illkirch, France) and the staff of the Mouse Clinical Institute (Illkirch, France) for their critical support in this project. We thank Professor Didier Trono (EPFL, Lausanne, Switzerland) for providing us with the Albumin-cre mice. We thank Professor David Mangelsdorf (Howard Hughes Medical Institute, Dallas, TX) and Professor Steven Kliewer (UT Southwestern, Dallas, TX) for providing us with the FGF21-deficient mice. We thank Alice Marmugi and Géraldine Michel for their technical assistance. We thank Professor Bertrand Cariou and Professor Bart Staels for constructive discussions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors AM initiated the project, designed experiments, performed experiments, analysed the data and wrote the paper. AP, EF, SD, YL, FL, MR, CL, FB and AI contributed to design experiments, perform experiments and to analyse the data. VB designed and performed a critical experiment. JB-M, TAS, PC and LL provided critical analysis and technical support. SL contributed to analyse the data. GM, FR and TP provided critical materials and contributed to design the project. NL, CP and DL critically contributed to design the project and supervised experiments. WW provided critical reagents, designed the project, analysed the data and wrote the paper. HG designed the project, performed experiments, analysed the data and wrote the paper.
Funding This work was funded by grants from the Human Frontier Science Program (HFSP) (WW), by Start-Up Grants from the Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore (to WW), by SFN (to HG), by ANRs ‘Crisalis’ (to CP and HG), by ‘Obelip’ (to DL, CP, AM and HG). DL is a member of the Institut Universitaire de France. AM, DL, WW and HG were supported by Région Midi-Pyrénées.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Gene expression array raw data are deposited in GEO as indicated in the manuscript.