Article Text

Abstract
Objective Endoplasmic reticulum (ER) stress is involved in liver injury, but molecular determinants are largely unknown. This study investigated the role of pleckstrin homology-like domain, family A, member-3 (PHLDA3), in hepatocyte death caused by ER stress and the regulatory basis.
Design Hepatic PHLDA3 expression was assessed in HCV patients with hepatitis and in several animal models with ER stress. Immunoblottings, PCR, reporter gene, chromatin immunoprecipitation (ChIP) and mutation analyses were done to explore gene regulation. The functional effect of PHLDA3 on liver injury was validated using lentiviral delivery of shRNA.
Results PHLDA3 was overexpressed in relation to hepatocyte injury in patients with acute liver failure or liver cirrhosis or in toxicant-treated mice. In HCV patients with liver injury, PHLDA3 was upregulated in parallel with the induction of ER stress marker. Treatment of mice with tunicamycin (Tm) (an ER stress inducer) increased PHLDA3 expression in the liver. X box-binding protein-1 (Xbp1) was newly identified as a transcription factor responsible for PHLDA3 expression. Inositol-requiring enzyme 1 (IRE1) (an upstream regulator of Xbp1) was required for PHLDA3 induction by Tm, whereas other pathways (c-Jun N-terminal kinase (JNK), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6)) were not. PHLDA3 overexpression correlated with the severity of hepatocyte injury in animal or cell model of ER stress. In p53-deficient cells, ER stress inducers transactivated PHLDA3 with a decrease in cell viability. ER stress-induced hepatocyte death depended on serine/threonine protein kinase B (Akt) inhibition by PHLDA3. Lentiviral delivery of PHLDA3 shRNA to mice abrogated p-Akt inhibition in the liver by Tm, attenuating hepatocyte injury.
Conclusions ER stress in hepatocytes induces PHLDA3 via IRE1–Xbp1s pathway, which facilitates liver injury by inhibiting Akt.
- LIVER
- HEPATOCYTE
- APOPTOSIS
- GENE EXPRESSION
- CELL SIGNALLING
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Tight regulation of endoplasmic reticulum (ER) stress response is fundamental in cell fate decision and organism survival. ER homeostasis is important in hepatocytes, an ER-rich cell type.
Sustained or unresolved ER stress contributes to hepatocyte dysfunction and death during the course of liver diseases. Restoration of ER homeostasis provides a therapeutic rationale, but new molecular targets need to be identified.
Serine/threonine protein kinase B (Akt) serves as a key signalling node modulating cell proliferation and energy metabolism.
Pleckstrin homology-like domain, family A, member-3 (PHLDA3) is a p53-regulated suppressor of Akt and acts as a tumour suppressor in lung and pancreatic neuroendocrine cancers.
What are the new findings?
PHLDA3 expression is elevated in patients with different liver diseases accompanying hepatocyte injury and ER stress.
ER stress increases PHLDA3 expression in hepatocytes, which is mediated by inositol-requiring enzyme 1 (IRE1)–Xbp1s pathway, but not by c-Jun N-terminal kinase (JNK), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6) pathways.
X box-binding protein-1 (Xbp1) is a previously unrecognised unique transcriptional regulator of PHLDA3 gene.
PHLDA3 induces ER stress-induced hepatocyte death through Akt inhibition, which occurs even in p53-deficient condition.
Suppression of PHLDA3 in vivo ameliorates ER stress-mediated liver injury.
How might it impact on clinical practice in the foreseeable future?
Our findings provide a novel insight on PHLDA3 regulated by IRE1 pathway in fine-tuning the fate of hepatocytes in acute or chronic liver disease, and may provide a new and attractive therapeutic strategy for the treatment of ER stress-associated pathological conditions.
Introduction
The liver provides functions required to maintain plasma proteins and produce proteins essential for cholesterol biosynthesis and biotransformation of xenobiotics. Endoplasmic reticulum (ER) homeostasis is important in hepatocytes, a major cell type rich in ER content since the ER is an organelle for synthesis, folding and trafficking of proteins.1 ,2 The accumulation of misfolded proteins induces ER stress response under various conditions, including nutrient deprivation and oxidative stress.1–3 In clinical situations, the secretory function (eg, albumin) is diminished in patients with hepatitis or cirrhosis.4 Thus, sustained or unresolved ER stress may account for hepatocyte dysfunction and death during the course of acute or chronic liver disease.1–3
Unfolded protein response (UPR) is an early adaptive mechanism that maintains the functional capacity of ER. UPR process is mainly mediated by three canonical ER stress transducers: inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6), and plays a critical role in cell survival.1 ,2 Once it is activated, signalling pathways lead to the recovery of homeostasis or can alternatively activate cascade events that ultimately cause cell death.1 ,2 However, the transducers that determine the execution of cell death following ER stress had not been fully elucidated. Moreover, the molecules responsible for the fate of hepatocytes accompanying liver injury after ER stress are largely unknown.
Serine/threonine protein kinase B (Akt) serves as a node in diverse signalling cascades downstream of growth factor receptor kinases and controls cell proliferation and energy metabolism.5 The pleckstrin homology-like domain, family A, member 3 (PHLDA3) was identified as a PH domain-only protein that inhibits Akt.6 PHLDA3 may limit cell proliferation and acts as a tumour suppressor in lung and pancreatic cancers.6 ,7 Although such a discovery indicates the antagonistic effect of PHLDA3 against Akt, the cellular and molecular determinants involved in this event are unclear. Moreover, it had not been explored whether PHLDA3 has a detrimental effect on acute or chronic liver diseases in association with ER stress. In addition, the signalling pathway by which the PHLDA3 gene expression is controlled remains largely unknown.
The evidence and lack of an understanding on the regulatory network of PHLDA3 encouraged us to hypothesise that ER stress induces PHLDA3 and which facilitates hepatocyte death in liver disease. Here, we provide the evidence that PHLDA3 expression is augmented in the liver of patients with acute liver failure, HCV infection and liver cirrhosis and of animal models with hepatitis. Our data also indicate that X box-binding protein-1 (Xbp1) serves as a transcription factor to induce PHLDA3 gene, which depends on IRE1 but not c-Jun N-terminal kinase (JNK), leading to hepatocyte death by inhibiting Akt. To unmask this, we carried out bioinformatic approaches and analysed human samples for the understanding of its role in human liver pathobiology and employed several chronic or acute animal models with liver disease. In addition, our results obtained from loss-of-function or gain-of-function experiments using hepatocytes and lentiviral delivery system of shRNA directed against PHLDA3 in vivo unravelled the new functional role of PHLDA3 in the event of hepatocyte death elicited by ER stress.
Materials and methods
Integrative network analysis
Gene expression data were obtained from the Gene Expression Omnibus (GSE25097). The procedures for analysis are described in the online supplement.
HCV patient samples
Liver samples from HCV patients without hepatitis (lower than the upper limit of normal in alanine transaminase (ALT) (ULN, 40 U/L), N=3) or those with hepatitis (>4 times ULN, N=3) were used. Studies using human samples were reviewed and approved by the institutional review boards.
Animal treatments
Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Use and Care Committee at Seoul National University. Male C57BL/6 mice at 6 weeks of age were intraperitoneally (i.p.) injected with a single dose of tunicamycin (Tm; 2 mg/kg) and were subjected to analyses at 24–72 h. For in vivo knockdown experiment, mice were injected with lentiviruses that express control shRNA or PHLDA3 shRNA through tail vein (2×107 viruses in 200 μL phosphate buffered saline (PBS) per mouse). Seven days after the injection, the mice were treated with vehicle or Tm (2 mg/kg, i.p.) and were killed 72 h afterwards.
RNA isolation and real-time RT-PCR assays
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, California, USA) and was reverse-transcribed to obtain cDNA. Quantitative reverse transcription-PCR was performed using the Light Cycler 1.5 (Roche, Mannheim, Germany).
Cell culture
HepG2 (human hepatocyte-derived cell line) and AML12 (mouse hepatocyte-derived cell line) were purchased from American Type Culture Collection (ATCC) (Rockville, Maryland, USA). The isolation of primary hepatocytes from rats is described in the online supplement.
Transient transfection and reporter gene assays
The sources of the vectors and procedures used in this study for transfection and reporter gene assays are provided in the online supplement.
Details for in vitro and in vivo studies are given in the online supplementary materials and methods.
Results
PHLDA3 overexpression in patients with liver disease
To unravel the biological significance of PHLDA3 on liver disease progression in clinical situations, we analysed the human GEO database (GSE25097) available in public domain. In the patients with liver cirrhosis, PHLDA3 and Akt were closely linked to changes in the levels of molecules involved in an apoptosis-associated gene network compared with healthy individuals (figure 1A). Consistently, PHLDA3 mRNA levels were significantly elevated in cirrhotic liver, as were those of p53 upregulated modulator of apoptosis (PUMA, a gene related to cellular injury caused by ER stress)8 (figure 1B): a strong correlation existed between PUMA and PHLDA3 mRNA levels, suggestive of the role of PHLDA3 as a potential molecular marker of hepatocyte death. Moreover, the molecules enhanced by ER stress such as Bcl-2-associated X protein (BAX) and PUMA are directly or indirectly linked to PHLDA3 in the gene network. In the analysis of another set of GEO database (GSE38941) derived from the patients with acute liver failure, PHLDA3 levels were elevated as the degree of hepatocyte necrosis increased (figure 1C).
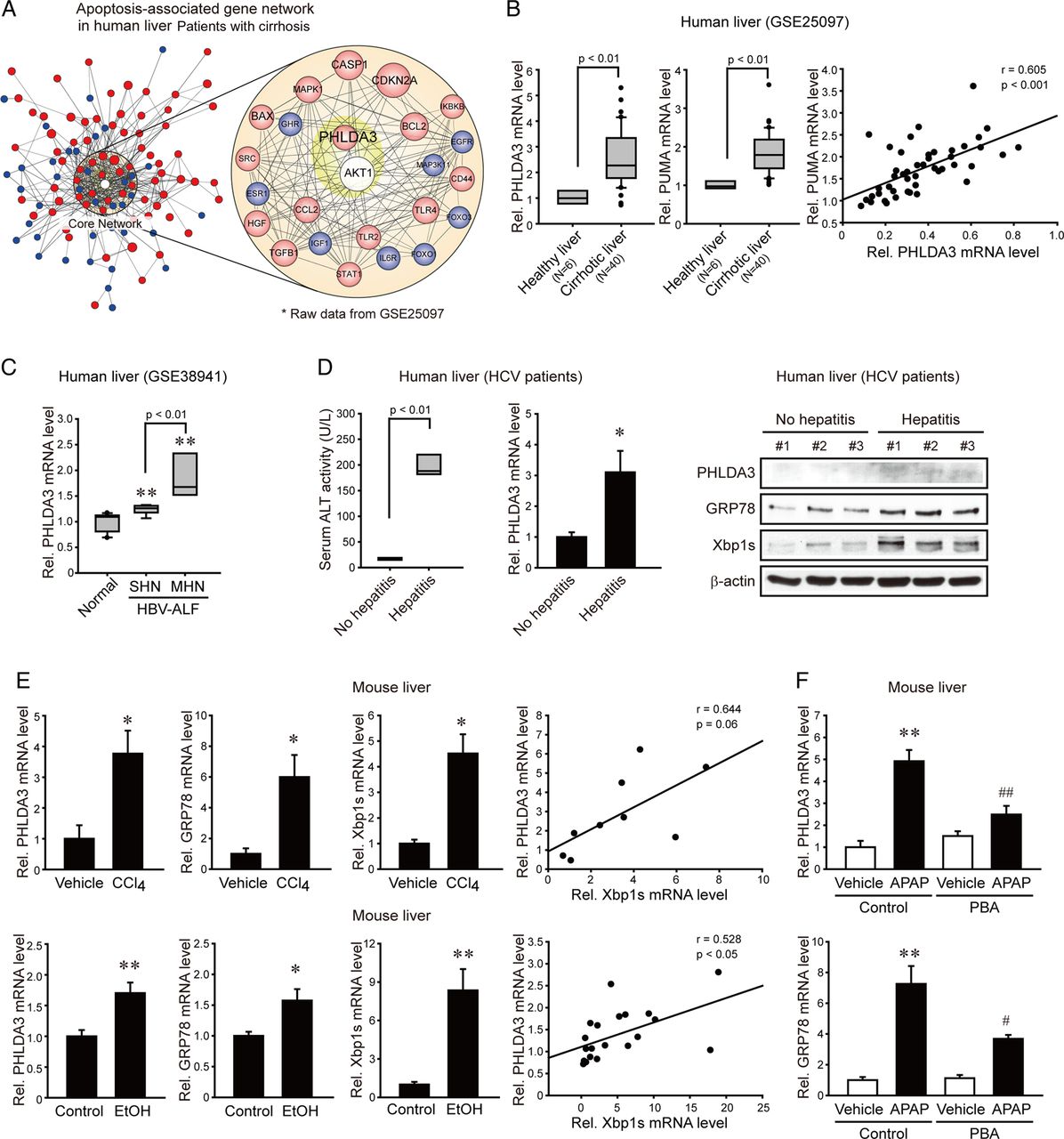
Pleckstrin homology-like domain, family A, member-3 (PHLDA3) overexpression in patients with different liver diseases. (A) Association of PHLDA3 and serine/threonine protein kinase B (Akt) in apoptosis-related gene network. Filled colours indicate the genes upregulated (red) or downregulated (blue) in the liver of patients with cirrhosis compared with healthy subjects. (B–C) PHLDA3 expression in patients with liver cirrhosis or liver failure. For B, hepatic PHLDA3 or p53 upregulated modulator of apoptosis (PUMA) mRNA levels were analysed in healthy individuals or patients with cirrhosis (GSE25097). For C, PHLDA3 mRNA levels were analysed in healthy individuals or patients with HBV-associated acute liver failure (GSE38941). SHN, submassive hepatic necrosis; MHN, massive hepatic necrosis. (D) PHLDA3 expression in HCV patients with hepatitis. Quantitative reverse transcription-PCR (qRT-PCR) assays for PHLDA3 or immunoblottings for PHLDA3, 78 kDa glucose-regulated protein (GRP78) and Xbp1s were done on HCV patient liver samples. No hepatitis, subjects with lower than upper limit of normal (ULN) in ALT; and hepatitis, subjects with >4 times ULN in ALT. (E) Increases of PHLDA3 or endoplasmic reticulum (ER) stress marker mRNAs after liver injury. Mice were injected with vehicle or carbon tetrachloride (CCl4) (0.6 mL/kg, i.p., 24 h) (N=3 or 6) or fed on a control diet or alcohol Lieber–DeCarli liquid diet for 5 weeks (N=9 or 11). (F) The effect of chemical chaperone on acetaminophen (APAP)-induced liver injury. Mice were injected with vehicle or phenyl butyric acid (PBA) (100 mg/kg, i.p.) 2 h prior to the treatment with vehicle or APAP (500 mg/kg, i.p., 6 h) (N=5 each). For B–F, data represent the mean±SE. Statistical significance of the differences between healthy individuals and patients with cirrhosis (B) or patients with liver failure (C) (**p<0.01) or HCV patient with hepatitis (D) or between each treatment group and the control (E) (*p<0.05, **p<0.01) or APAP (F) (#p<0.05, ##p<0.01) was determined.
It has been shown that patients with HCV infection exhibit ER stress in the liver.9 We found that PHLDA3 mRNA levels were significantly greater in HCV patients having hepatitis (>4 times ULN in ALT) than those with no hepatitis (lower than ULN) (figure 1D left and middle). Consistently, PHLDA3 protein levels were elevated in HCV patients having hepatitis along with the increases of 78 kDa glucose-regulated protein (GRP78) and Xbp1s (markers for ER stress) (figure 1D right). We next asked whether PHLDA3 was upregulated in animals by other pathological conditions inducing ER stress. Either a single injection of carbon tetrachloride (CCl4) to mice (24 h) or 5% ethanol diet feeding for 5 weeks significantly enhanced PHLDA3, GRP78 and Xbp1s transcript levels in the liver (figure 1E left); a positive correlation existed between PHLDA3 and Xbp1s mRNA levels (figure 1E right). In a separate animal experiment, we used acetaminophen as a different liver toxicant with or without chemical chaperone treatment, and found that acetaminophen treatment enhanced PHLDA3 and GRP78 transcript levels in the liver, which was significantly attenuated by phenyl butyric acid (PBA) (figure 1F). These findings directed our attention to the potential role of PHLDA3 in ER stress-mediated liver injury.
PHLDA3 induction in hepatocytes by ER stress
Next, we sought to identify the effects of ER stress on PHLDA3 expression in the liver and hepatocytes. Treatment of mice with a single dose of Tm gradually and substantially elevated PHLDA3 mRNA levels in the liver (figure 2A). Consistently, PHLDA3 protein levels were distinctly increased, as were GRP78. Moreover, immunohistochemistry showed PHLDA3 overexpression in the surrounding area of veins at day 3 after Tm treatment. Since AML12 is a non-transformed hepatocyte cell line and has typical hepatocyte features,10 we primarily used the cell line to study the effect of ER stress on hepatocytes. Treatment of AML12 cells with Tm increased PHLDA3 levels in a time-dependent manner (figure 2B). A similar increase was observed 24 h after thapsigargin (Tg) treatment. The ability of Tm to increase PHLDA3 expression was verified in HepG2 cells or rat primary hepatocytes (figure 2C). To validate the role of ER stress in the induction of PHLDA3, we examined the effect of chemical chaperones to reduce ER stress. As expected, treatment of AML12 cells with tauroursodeoxycholic acid (TUDCA) or PBA abrogated the ability of Tm to induce PHLDA3 (figure 2D). Our results demonstrate the effect of ER stress on the induction of PHLDA3 in hepatocytes, suggesting the role of PHLDA3 in hepatocyte injury caused by ER stress.
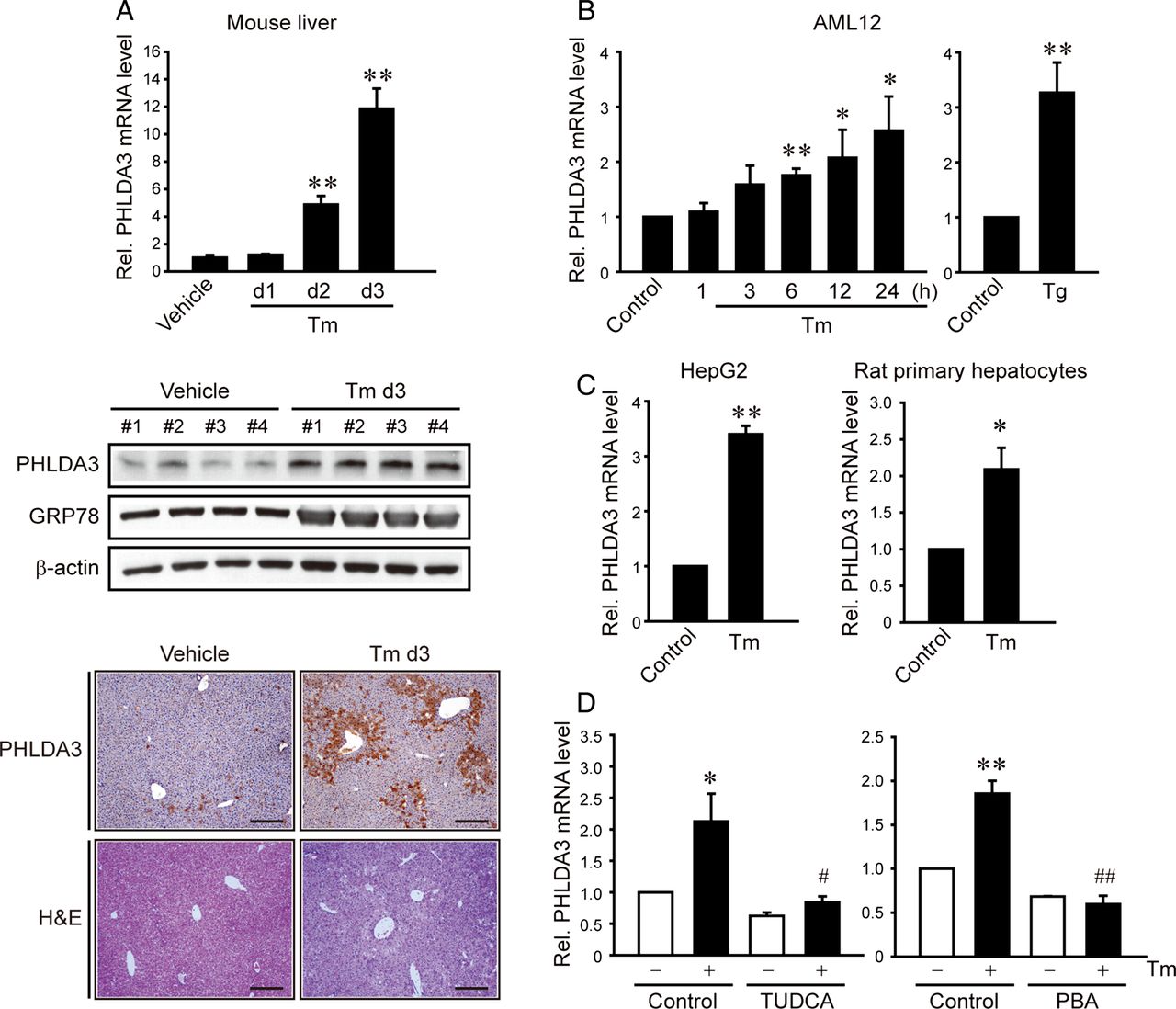
Increase in pleckstrin homology-like domain, family A, member-3 (PHLDA3) expression by endoplasmic reticulum (ER) stress. (A) PHLDA3 induction by ER stress in the liver. Quantitative reverse transcription-PCR (qRT-PCR), immunoblottings or immunohistochemistry for PHLDA3 was done on the liver of mice injected with vehicle or tunicamycin (Tm) (N=4 each). Scale bar: 200 μm. Data represent the mean±SE (significantly different compared with vehicle, **p<0.01). (B–C) PHLDA3 induction by ER stress in hepatocytes. AML12 cells were treated with Tm (2 μg/mL) for the indicated time period or thapsigargin (Tg) (0.3 μM) for 24 h. HepG2 and rat primary hepatocytes were treated with 5 and 2 μg/mL Tm, respectively. (D) The effects of chemical chaperones on PHLDA3 induction. HepG2 cells were pretreated with 100 μM tauroursodeoxycholic acid (TUDCA) or 1 mM 4-phenyl butyric acid (PBA) for 1 h and continuously incubated with Tm. For B–D, data represent the mean±SE of three separate experiments. Statistical significance of differences between each treatment group and the control (*p<0.05, **p<0.01) or Tm (#p<0.05, ##p<0.01) was determined.
Xbp1s-mediated transcriptional induction of PHLDA3
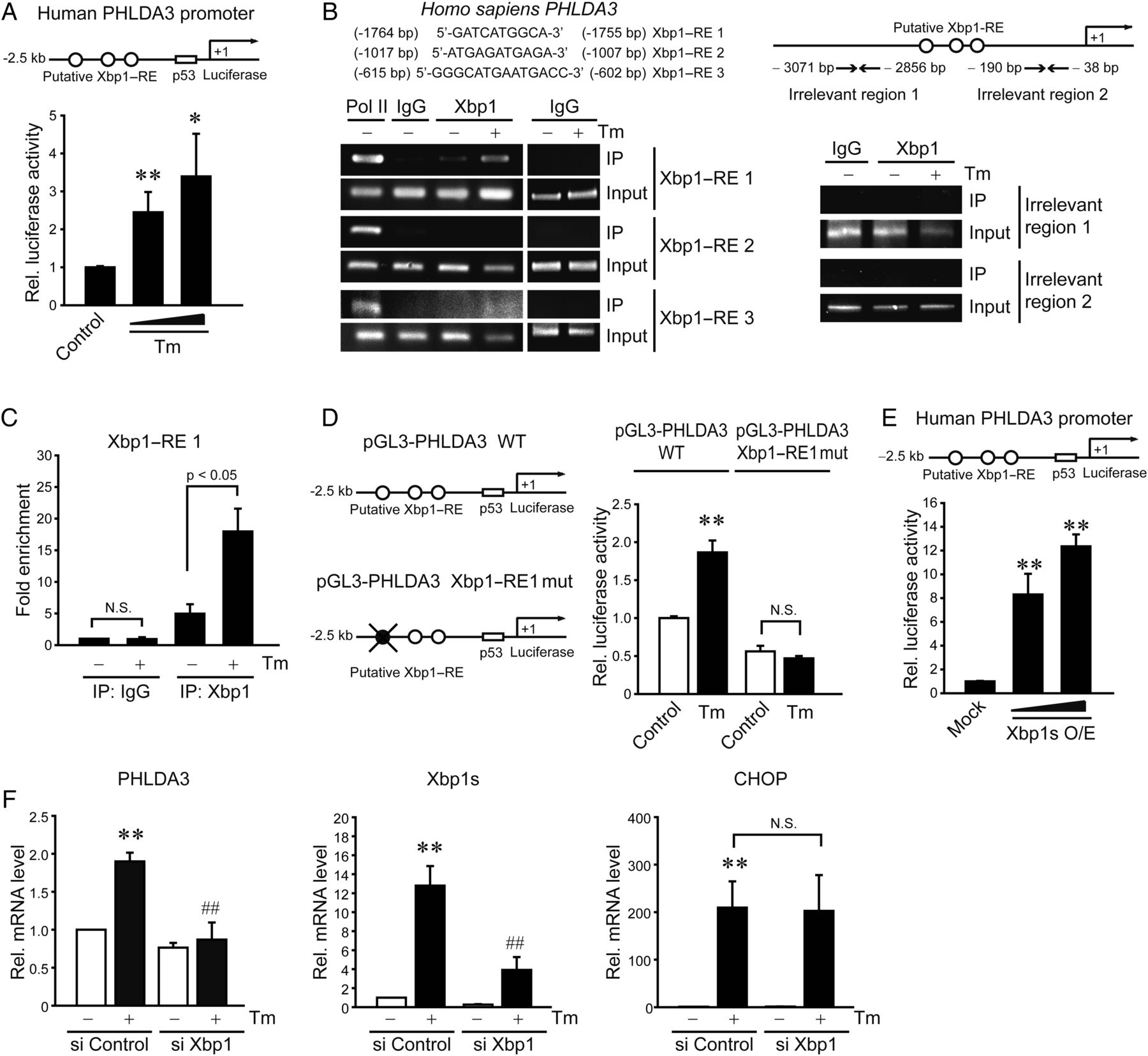
To elucidate the underlying basis of PHLDA3 induction by ER stress, we hypothesised that the transcriptional factor(s) activated by ER stress contributed to the gene transactivation. We postulated that should this transcriptional activation occur in hepatocytes, putative DNA-binding sites would be present in the gene promoter. We found three putative Xbp1-binding sites in PHLDA3 gene promoter. In addition, prediction using the PROMO analysis allowed us to extract transcription factors that potentially increase PHLDA3 expression, of which Xbp1 is the only protein involved in UPR pathways. Treatment of AML12 cells with Tm significantly promoted luciferase expression from a reporter construct containing −2.5 kb human PHLDA3 gene (figure 3A). Tm exposure to mice increased the Xbp1s mRNA levels (an active form as a spliced Xbp1) in the liver (see online supplementary figure S1A). Consistently, ER-localised DnaJ type II homologue 4 (ERdj4), a target gene controlled by Xbp1, was upregulated, and the time course was comparable with that of PHLDA3 mRNA. A time-course study also verified ER stress induction of Xbp1 mRNA and protein in AML12 cells (see online supplementary figure S1B and S1C): Xbp1s mRNA maximally increased 3–6 h after Tm treatment and remained elevated at 12–24 h, which matched with gradual increases in Xbp1s protein level at 3–24 h (ie, Xbp1 mRNA increase preceded the protein induction). The kinetics of Xbp1s protein paralleled that of PHLDA3 mRNA.
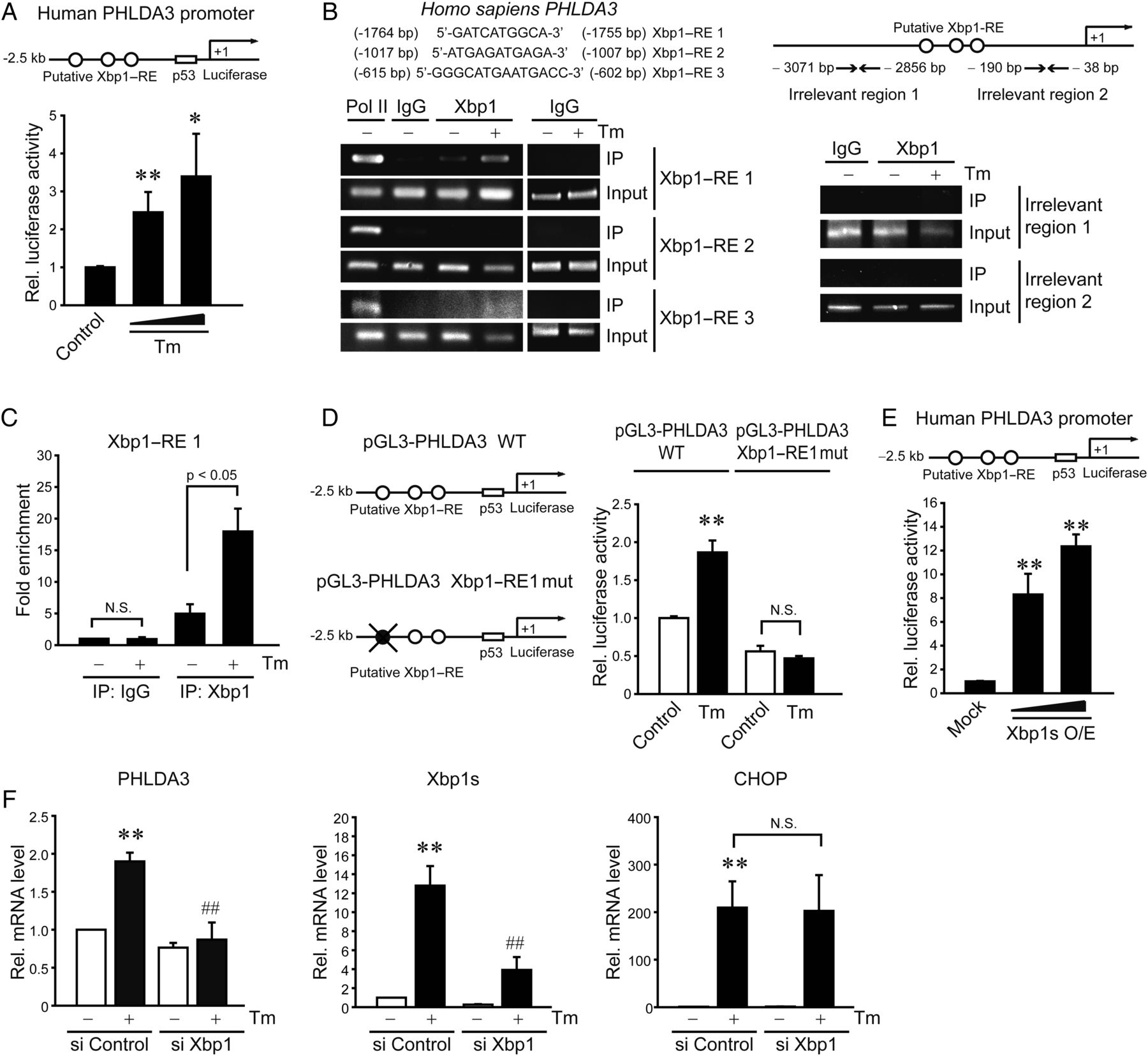
PHLDA3 gene induction by Xbp1s upon endoplasmic reticulum (ER) stress. (A) Transactivation of PHLDA3 gene by ER stress. Pleckstrin homology-like domain, family A, member-3 (PHLDA3) promoter reporter activity was measured in AML12 cells treated with vehicle or tunicamycin (Tm) (0.2 or 2 μg/mL) for 12 h following transfection with pGL3-PHLDA3 construct (0.5 μg, for 12 h). (B) X box-binding protein-1 (Xbp1) binding to PHLDA3 promoter. ChIP assays were done on the lysates of HepG2 cells treated with 5 μg/mL Tm. DNA–protein complexes were precipitated with anti-Xbp1 antibody and were subjected to PCR amplifications using the flanking primers for the Xbp1–response elements (Xbp1–REs). IgG immunoprecipitation represents negative control. The specificity of Xbp1 binding was verified using primers targeting irrelevant regions in the promoter. One-tenth of cross-linked lysates served as the input control. (C) qPCR assays for Xbp1 and Xbp1–RE1 binding. (D) The effect of Xbp1–RE1 mutation on PHLDA3 transactivation. (E) PHLDA3 transactivation by Xbp1s overexpression. AML12 cells were transfected with the PHLDA3 luciferase construct in combination with Mock or Xbp1s-overexpressing construct (as an active form of Xbp1, 0.5 or 1 μg). (F) The effect of Xbp1 knockdown on PHLDA3 induction. AML12 cells were treated with Tm after transfection with control siRNA or Xbp1 siRNA for 48 h. For A and C–F, data represent the mean±SE of three separate experiments. Statistical significance of differences between each treatment group and the control (*p<0.05, **p<0.01) or Tm (##p<0.01) was determined.
To validate the effect of Xbp1 on PHLDA3 induction, we sought to evaluate the Xbp1 binding to PHLDA3 gene promoter. Of the putative DNA-binding sites (Xbp1–RE1, RE2 and RE3), Xbp1 interacted with the site located in the distal region (RE1; −1764 to −1755 bp), as indicated by ChIP assays (figure 3B); the specificity of Xbp1 binding was confirmed using non-specific primers targeting irrelevant regions in the promoter. qPCR analysis verified Xbp1 and Xbp1–RE1 binding (figure 3C). Moreover, the mutation of Xbp1–RE1 abrogated the ability of Tm to transactivate PHLDA3 gene (figure 3D). This effect was verified in mouse primary hepatocytes (see online supplementary figure S1D). Consistently, Xbp1s overexpression notably facilitated PHLDA3 gene transactivation (figure 3E), whereas Xbp1 knockdown entirely inhibited PHLDA3 overexpression by Tm (figure 3F). In contrast, Xbp1 deficiency unaffected Tm induction of CCAAT-enhancer-binding protein homologous protein (CHOP), suggesting their mutual independence in the pathways. Together, these data demonstrate that Xbp1 induced by ER stress transactivates the PHLDA3 gene, which is distinct from ER stress-activated CHOP pathway.
Regulation of PHLDA3 induction by UPR pathway
The finding that Xbp1 splicing activation is required for the PHLDA3 induction prompted us to search for upstream UPR regulatory pathway. Because the splicing of Xbp1 mRNA to its active form depends on IRE1 endonuclease activity,11 we determined the role of IRE1 in the induction of PHLDA3 by ER stress. As expected, IRE1 knockdown diminished the increases in Xbp1s and PHLDA3 by Tm (figure 4A). Since IRE1 has a kinase activity that leads to JNK activation,12 we assessed whether the PHLDA3 induction by ER stress relies on JNK. JNKi treatment (an inhibitor of JNK) failed to affect the increase in PHLDA3 by Tm or Tg treatment (figure 4B). We additionally assessed the phosphorylation of signal transducer and activator of transcription 3 (STAT3) in this experiment. Tm treatment increased STAT3 tyrosine phosphorylation (an active form) and this effect was attenuated by JNKi (figure 4B) (Tg failed to increase p-STAT3 presumably because it has a different ER stress mechanism), indicative of the lack of STAT3 association with PHLDA3 induction by ER stress. Transfection with a dominant negative (DN)-PERK mutant or an ATF6 siRNA unaffected the PHLDA3 induction (figure 4C). Collectively, our results indicate that Xbp1s-mediated PHLDA3 induction by ER stress depends specifically on the endonuclease activity of IRE1 independently of its activation of JNK or other ER stress transducers.
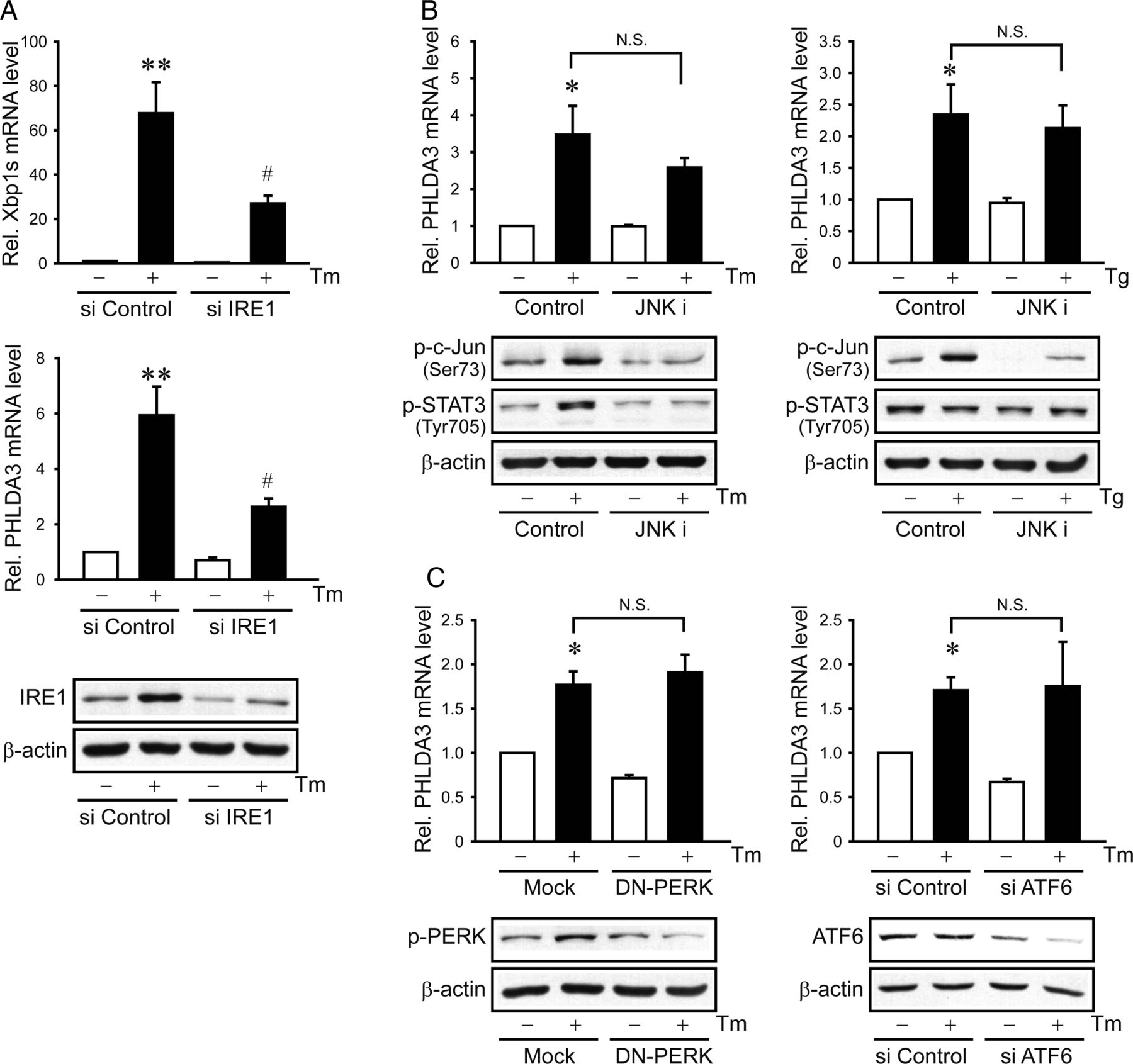
Inositol-requiring enzyme 1 (IRE1)-dependent induction of pleckstrin homology-like domain, family A, member-3 (PHLDA3). (A) The effect of IRE1 knockdown on PHLDA3 induction. (B) The effect of c-Jun N-terminal kinase (JNK) inhibition on PHLDA3 mRNA, p-c-Jun or p-signal transducer and activator of transcription 3 (STAT3) levels. AML12 cells were exposed to 20 μM JNKi for 1 h and were continuously treated with tunicamycin (Tm) or thapsigargin (Tg). (C) The effects of protein kinase RNA-like endoplasmic reticulum kinase (PERK) or activating transcription factor 6 (ATF6) inhibition. AML12 (left) and HepG2 (right) cells were treated with Tm after transfection with Mock or dominant negative (DN)-PERK construct or with control or ATF6 siRNA. Immunoblottings for p-PERK and ATF6 (90 kDa form) confirmed the effect of DN-PERK and ATF6 siRNA transfection, respectively. For A–C, data represent the mean±SE of three separate experiments. Statistical significance of differences between each treatment group and the control (*p<0.05, **p<0.01) or Tm (#p<0.05) was determined.
Effects of PHLDA3 on cell death caused by ER stress
Diverse pathophysiological situations cause cell death through ER stress activation. Serum deprivation causes energy imbalance, leading to ER stress in the cell.13 ,14 Thus, we examined serum starvation effect on PHLDA3 expression and cell viability. PHLDA3 mRNA was significantly increased by serum deprivation in AML12 cells, being consistent with the change in Xbp1s mRNA (see online supplementary figure S2A), which supports that Xbp1s increased by ER stress contributes to PHLDA3 overexpression. Serum starvation significantly decreased the cell viability, which was abolished by treatment with chemical chaperones (see online supplementary figure S2B), indicating that ER stress plays a role in the cell death elicited by serum deprivation (the increased PHLDA3 mRNA levels after 24 h serum starvation may be in part due to UPR signalling-independent, eg, p5315). These results support the contention that PHLDA3 overexpression by ER stress is associated with hepatocyte death.
To determine the role of PHLDA3 induction in hepatocyte injury by ER stress, we next evaluated the functional association between PHLDA3 and liver damage in mice. Administration of Tm to mice caused the increases not only in hepatic PHLDA3 levels as shown in figure 2A, but also in serum transaminase activities (ie, ALT and aspartate transaminase (AST)), and there existed a highly significant correlation (figure 5A). We confirmed a gradual increase in terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cell number in the liver, which also positively correlated with PHLDA3 expression levels. To validate the causal relationship between PHLDA3 and cell death, we examined the effect of PHLDA3 modulation on the cell viability decreased by ER stress. PHLDA3 knockdown prevented Tm from diminishing the cell viability (figure 5B). Consistently, PHLDA3 overexpression significantly decreased the cell viability and enhanced the cytotoxic effect of Tm. We verified the effect of ER stress regulators responsible for PHLDA3 expression on the cell viability by modulating Xbp1 or IRE1. Xbp1 knockdown had the similar effect as did that of PHLDA3 (figure 5C). Overexpression of Xbp1s tended to increase the effect of Tm but was statistically insignificant probably because the amount of Xbp1s activated by Tm might suffice PHLDA3 induction and/or Xbp1s may also be involved in the adaptive response for cell survival.16 ,17 In addition, IRE1 deficiency enabled AML12 cells to survive against ER stress, and this effect was abrogated by PHLDA3 overexpression (figure 5D). PHLDA3 overexpression decreased the cell proliferation rate (see online supplementary figure S3).
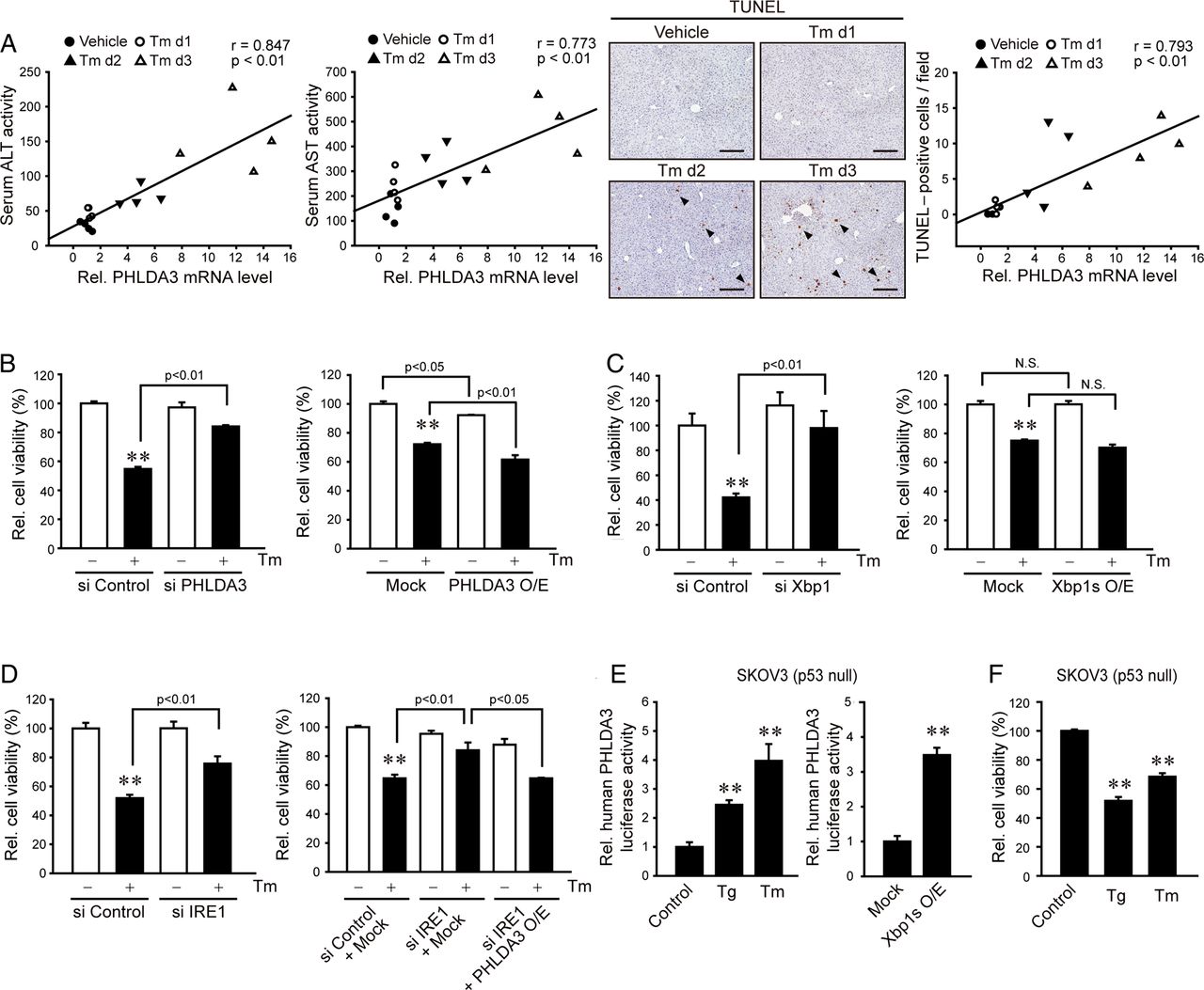
The causative role of pleckstrin homology-like domain, family A, member-3 (PHLDA3) in endoplasmic reticulum (ER) stress-induced cell death. (A) A positive correlation between PHLDA3 and serum transaminases or TUNEL positivity in the liver. Mice were treated with tunicamycin (Tm) as described in figure 2A. Four separate samples in each group were used for the analysis. Scale bar: 100 μm. (B) The effect of PHLDA3 modulation on ER stress-induced cell death. After the transfection of HepG2 (left) or AML12 (right) cells with control or PHLDA3 siRNA or with Mock or PHLDA3 construct, the cells were continuously incubated with Tm. (C) The effect of X box-binding protein-1 (Xbp1) modulation on ER stress-induced cell death. AML12 cells were transfected with control or Xbp1 siRNA or with Mock or Xbp1s construct and were treated with Tm. (D) Attenuation of ER stress-induced cell death by inositol-requiring enzyme 1 (IRE1) knockdown, and the reversal of this effect by PHLDA3 overexpression. (E) PHLDA3 induction in p53-null cells. SKOV3 cells were treated with thapsigargin (Tg) (1 μM) or Tm (2 μg/mL) for 24 h after transfection with PHLDA3 luciferase construct alone or in combination with Mock or Xbp1s construct. (F) The cytotoxic effects of ER stress inducers on SKOV3 cells. For B–F, data represent the mean±SE of at least three samples. Statistical significance of differences between each treatment group and the control (**p<0.01) was determined.
Based on our observation that Xbp1 serves as the transcription factor of PHLDA3 gene and the report that p53 may transcriptionally activate it,6 we asked whether ER stress induces PHLDA3 and can execute cell death in the absence of p53. In SKOV3 cells deficient of p53,18 either Tg or Tm treatment increased PHLDA3 gene transactivation, as did Xbp1s overexpression (figure 5E). Consistently, treatment with either Tg or Tm significantly decreased the cell viability (figure 5F). Collectively, our results support that PHLDA3 overexpression by ER stress through IRE1–Xbp1s pathway contributes to cytotoxicity even without p53.
Role of Akt inhibition by PHLDA3 in ER stress-induced cell death
Given the link between PHLDA3 and Akt as core signalling nodes in the network of proapoptotic molecules (figure 1A) and the effect of PHLDA3 on Akt activity for anti-tumour effect,6 we hypothesised whether PHLDA3 inhibition of Akt mediates ER stress-induced cell death. Indeed, p-Akt levels were notably diminished in the livers of HCV patients with hepatitis compared with those without hepatitis (figure 6A), which matched with the induction of PHLDA3 (figure 1D). Additionally, we measured p-Akt levels in mice treated with Tm. Basal p-Akt levels in the liver were decreased by Tm, being inversely correlated with PHLDA3 levels (figure 6B). There was also an inverse correlation between TUNEL-positive cell numbers and p-Akt. Moreover, we found that the inhibitory effect of Tm on p-Akt was reversed by PHLDA3 knockdown, but was enhanced by PHLDA3 overexpression in hepatocyte cell models (figure 6C). Next, we assessed the effects of phosphatidylinositol 3-kinase (PI3-K) or specific Akt inhibitor alone or in combination with ER stress inducer on the cell viability. Treatment of AML12 cells with LY294002 (a PI3-K inhibitor) decreased viability of the cells by ∼30% and augmented Tm's cytotoxic effect (∼70%) (figure 6D). A443654 (a specific Akt inhibitor) treatment synergistically increased the Tm effect (∼90%), although A443654 alone had only a minor effect. Consistently, PHLDA3 overexpression fortified the cytotoxic effect of LY294002 or A443654. To address whether PHLDA3 participates in established mechanisms to impair Akt signalling, we measured p-IRS1 and p-JNK; PHLDA3 knockdown slightly enhanced IRS1 (Ser307) or JNK phosphorylation (figure 6E), which may reflect adaptive changes (PHLDA3 inhibits Akt recruitment to the plasma membrane,6 an event downstream from IRS1). So, PHLDA3 inhibition of Akt may not directly link to IRS1 phosphorylation or JNK signalling. Overall, our results indicate that PHLDA3 overexpression by ER stress inhibits Akt phosphorylation, which promotes hepatocyte death.

The effect of pleckstrin homology-like domain, family A, member-3 (PHLDA3)-mediated serine/threonine protein kinase B (Akt) inhibition on hepatocyte death. (A) Akt inhibition in the liver of HCV patients. Immunoblottings for p-Akt and total form of Akt were done on the liver homogenates as used in figure 1D. (B) An inverse relationship between PHLDA3 and p-Akt or between p-Akt and TUNEL positivity in the liver. Immunoblottings for p-Akt were done on the liver specimens. (C) The effect of PHLDA3 knockdown or overexpression on p-Akt and total Akt levels. (D) The effects of phosphatidylinositol 3-kinase (PI3-K) and Akt inhibitors on endoplasmic reticulum (ER) stress-induced cell death. AML12 cells were treated with LY294002 (20 μM) or A443654 (2 μM) in combination with vehicle or tunicamycin (Tm) for 24 h or with Mock or PHLDA3 transfection for 24 h. (E) The effect of PHLDA3 knockdown on p-IRS1 or p-c-Jun N-terminal kinase (JNK) levels. For B–D, data represent the mean±SE of at least three samples. Statistical significance of differences between each treatment group and the control (*p<0.05, **p<0.01) was determined.
Amelioration of liver injury by lentiviral delivery of PHLDA3 shRNA
As a continuing effort to verify the impact of PHLDA3 on hepatocyte injury, we employed lentiviral delivery system of shRNA interfering with PHLDA3 expression in a mouse model (figure 7A), and found that this effect prevented Tm's inhibition of p-Akt in the liver (figure 7B). PHLDA3 knockdown also prevented histological changes, including hepatocyte ballooning, degeneration, and necrosis, elicited by Tm (figure 7C). Consistently, TUNEL+-staining intensities were notably diminished (figure 7D). Likewise, a deficiency in PHLDA3 using the shRNA significantly lessened increases of ALT, AST and lactate dehydrogenase activities in serum (figure 7E). Our data support the contention that inhibition of PHLDA3 and the resultant recovery of Akt activity effectively protect the liver from injury against ER stress (figure 7F).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of lentivirus expressing pleckstrin homology-like domain, family A, member-3 (PHLDA3) shRNA on liver injury by endoplasmic reticulum (ER) stress. (A) PHLDA3 knockdown by lentiviral delivery of shRNA. Seven days after a tail vein injection with lentiviruses that express control or PHLDA3 shRNA, the mice were administered with vehicle or tunicamycin (Tm) (2 mg/kg, i.p.). Quantitative reverse transcription-PCR (qRT-PCR) assays for PHLDA3 were done on the liver after 72 h (N=3 or 8). (B) Immunoblottings for p-serine/threonine protein kinase B (Akt) and total Akt in the liver homogenates. p-Akt levels were assessed by scanning densitometry of the immunoblots. The values indicate the ratio of p-Akt to total Akt. (C–D) H&E and TUNEL stainings. Scale bar: 100 μm. (E) Blood biochemistry. (F) A proposed scheme illustrating the mechanism by which ER stress causes liver injury via PHLDA3 overexpression. For A and E, data represent the mean±SE. Statistical significance of differences between each treatment group and the control (*p<0.05, **p<0.01) was determined. LDH, lactate dehydrogenase.
Discussion
The tight and fine-tuning regulation of ER stress response is fundamental in cell fate decision since UPR process is an evolutionarily conserved pathway for organism survival. Here, our findings show that ER stress promotes PHLDA3 gene induction in hepatocytes, facilitating cell death during the course of liver diseases. An important finding of this study is the discovery of PHLDA3 as a molecule that triggers hepatocyte injury elicited by ER stress. This outcome was supported by the analyses of clinical GEO databases; PHLDA3 was overexpressed in the liver of patients with cirrhosis; there was a variation in PHLDA3 mRNA level in these patients, probably reflecting individual differences in the degree of hepatocyte death (Xbp1s mRNA was not statistically different in this database presumably because of wide variations and/or difference in the mRNA half-life). We could see the significantly greater induction of PHLDA3 in HBV-infected patients with massive necrosis than those with submassive necrosis. In our samples obtained from HCV-infected patients with hepatitis, PHLDA3 overexpression was observed along with ER stress. Clinically, we evaluated PHLDA3 expression in different liver diseases and/or different stages (ie, acute/chronic, HCV/HBV or hepatitis/cirrhosis). Although the aetiology and characteristics of the diseases vary, one common feature is the association of ER stress with disease occurrence (or progression).2 The positive correlation between PHLDA3 and ER stress markers was validated using several different animal models (CCl4, Lieber–DeCarli diet, APAP, ER stress inducer and chemical chaperone treatment). The functional impact of PHLDA3 on ER stress-induced liver damage was further corroborated by the results of experiment using lentiviral shRNA delivery system in mice. Overall, our results support the notion that PHLDA3 overexpression may contribute to hepatocyte death in different stages of liver diseases in association with ER stress.
Another intriguing finding of this study is the identification of Xbp1 as a transcriptional factor responsible for the PHLDA3 gene induction by ER stress. Of putative DNA-binding elements, the distal site located in the region between −1764 bp and −1755 bp was found as the active element in the human PHLDA3 gene. This idea was reinforced by the observation that a mutation of the site nullified PHLDA3 transactivation by Tm. Consistently, Xbp1s overexpression facilitated PHLDA3 induction, whereas Xbp1 knockdown had the opposite effect. Although the Xbp1-binding sequence in mouse PHLDA3 gene was not exactly identical to that in the human, it seems to be conserved across the species. In other studies, Xbp1-binding elements were reported as unfolded protein response element (UPRE), endoplasmic reticulum stress element (ERSE) or endoplasmic reticulum stress element II (ERSE-II).19 ,20 Xbp1 preferentially interacted with UPRE, whereas ATF6 bound to ERSE.19 Consistently, we found that the identified Xbp1–RE1 in the PHLDA3 gene promoter resembled UPRE core sequence (CGTGG). As the most conserved ER stress sensor, IRE1 mediates Xbp1 mRNA splicing and activation,11 contributing to adaptive UPR and ER homeostasis.16 ,17 ,21 We therefore hypothesised that of the canonical ER stress transducers, IRE1 regulates PHLDA3 gene via Xbp1s during the course of ER stress in hepatocytes, as supported by decrease of PHLDA3 after IRE1 knockdown.
Despite intensive studies on UPR regulators, the pathway involved in liver pathogenesis had not been entirely clarified. Xbp1 increases lipid synthesis in the liver.22 Our data shown here suggest the role of Xbp1 in ER stress-mediated cell death process. However, the Xbp1s overexpression alone did not notably cause cell death. Thus, it is likely that Xbp1 is necessary for the PHLDA3 induction and is controlled by a negative feedback pathway. In most of stress conditions, adaptive response and apoptotic signalling can be simultaneously activated because the removal of injured cells would be important for overall homeostasis of multicellular organisms if damages are not resolved. In previous studies, attention has been focused on the role of Xbp1 in the adaptive response. Our results demonstrated that Xbp1 also contributes to the apoptotic process. Thus, the IRE1–Xbp1 pathway may orchestrate the switch from the UPR to proapoptotic signalling. The fine-tuning between UPR and cell death pathway may rely on the amount of PHLDA3 along with other mediators (eg, p53 and PUMA). Our data that IRE1 knockdown attenuated ER stress-induced hepatocyte death imply that IRE1 may control a rheostat switch between prosurvival and apoptosis.
IRE1 has diverse effects and the activity may vary depending on cell type in a context-specific manner.23 In pancreatic beta cells, excessive activation of IRE1 by ER stress causes inflammation.24 JNK may be involved in BAX-mediated apoptosis,25 and c-Jun activated by JNK promotes cell survival.26 Taking the role of JNK in hepatocyte death and the function of IRE1 as a JNK-activating kinase into consideration,12 we evaluated the role of JNK in PHLDA3 induction; the lack of change in PHLDA3 expression by JNKi supports the notion that JNK pathway may not be linked to ER stress induction of PHLDA3. Of the other pathways, PERK phosphorylation and the consequent inactivation of eukaryotic initiation factor 2 attenuated general protein synthesis. Prolonged PERK signalling may enhance apoptosis, and such an effect possibly relies on CHOP induction.27 ATF6 also induces UPR target genes.28 In our study, neither DN-PERK overexpression nor ATF6 knockdown diminished PHLDA3 overexpression by ER stress in hepatocytes. These results support that IRE1 specifically regulates PHLDA3 expression, being independent of JNK pathway or other ER stress transducers.
Studies have shown the intrinsic pathway of apoptosis activated by ER stress, which may involve BH3 domain-containing proteins (ie, BAX, Bak and Bim).1 ,2 ,29 ,30 Our results demonstrate the role of PHLDA3, a PH domain-only protein, in the process of cell death activated by ER stress: it acts as an effector for the execution of apoptosis. Treatment of hepatocytes with chemical chaperones ameliorated the increase in PHLDA3, supporting the bona fide effect of ER stress on PHLDA3. Our findings that PHLDA3 deficiency abrogated ER stress-mediated hepatocyte death, whereas PHLDA3 overexpression exacerbates it strengthen the role of PHLDA3 in determining cell fate. This concept was reinforced by the in vivo experiment using lentivirus of PHLDA3 shRNA. Additionally, our recent study that PHLDA3 serves as an effector molecule for acute kidney injury supports its effect on cell death.31 Here, we expanded the knowledge on the new function of PHLDA3 as a determinant of pathological progress in hepatocytes. Moreover, PHLDA3 expression correlated with the degree of hepatocyte injury in mice exposed to ER stress (ie, serum transaminases and TUNEL positivity). Since PHLDA3 protein is excreted in the urine (Kim et al, unpublished data), it may serve as a non-invasive biomarker of ER stress and/or liver injury.
In a time-course study, PHLDA3 was gradually increased after ER stress in vivo and in vitro. At an early time after ER stress, the degree of PHLDA3 induction was weak or moderate. A large increase of PHLDA3 at later times would favour the process of hepatocyte death by triggering the threshold. The greater increase of PHLDA3 in the liver than the hepatocyte may be due to activation of inflammatory cells and/or different pharmacokinetics. PHLDA3 competes with Akt, as shown by our result and others.6 The data that PHLDA3 knockdown helps increase Akt phosphorylation in hepatocytes, whereas its overexpression had the opposite effect support this concept. The proposed hypothesis is in line with the finding that mild decrease of Akt signalling attenuates cell growth, whereas its severe decrease causes apoptosis.5 ,32 Additional inhibition of Akt by chemical means augmented ER stress-induced cytotoxicity. Consistently, PHLDA3 overexpression in addition with Akt inhibition fortified hepatocyte death, supporting the role of PHLDA3 inhibition of Akt in cell death (low concentrations of PI3K or Akt inhibitors were chosen in this experiment to show enhanced cytotoxicity). PHLDA3 may have effects on the molecules working with Akt, PI3K and/or adaptor proteins, which remains clarified in future. In our study, Xbp1-mediated PHLDA3 induction by ER stress occurred even without p53 in SKOV3 cells. In hepatocytes that have an appropriate p53 pathway, a decrease in p-Akt would enhance p53 activity since Akt inhibits p53. Collectively, a sufficient amount of PHLDA3 may trigger cell death presumably by reinforcing the positive feed-forward activation between PHLDA3 and p53, implying that PHLDA3 serves as a rheostat to determine the UPR or the cell death pathway.
Diverse stimuli generating reactive oxygen species facilitate protein aggregation and lipid peroxidation, leading to ER stress and liver injury. Progression of liver diseases, including viral, alcoholic or non-alcoholic steatohepatitis, cholestasis and ischaemia–reperfusion injury, to more severe conditions may be linked to ER stress.1–3 Thus, it is highly likely that PHLDA3 serves as a determinant to interrogate the fate of hepatocyte in liver disease progression. In addition, PHLDA3 inhibition of Akt from insulin receptor signalling may provoke insulin resistance, supporting the link between aberrant energy metabolism and the pathogenesis of liver disease.
Overall, our results shown here provide a novel insight on PHLDA3 regulated by IRE1 pathway in fine-tuning the fate of hepatocytes in acute or chronic liver disease, and may provide a potential new strategy for the protection of hepatocytes from ER stress-associated pathological conditions.
Acknowledgments
A443654 was kindly provided by Dr RF Novak (Shriners Hospitals for Children International, FL).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors CYH and SWL: study concept and design, acquisition of data, analysis and interpretation of data, statistical analysis, drafting of the manuscript. JHK: acquisition of data, analysis and interpretation of data. WK: study using human samples with administrative, technical and material support. SGK: study concept and design, analysis and interpretation of data, critical revision of the manuscript for important intellectual content, obtained funding, study supervision.
Funding This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2007-0056817).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethic committees of Seoul National University and Seoul Metropolitan Government Seoul National University Boramae Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.