Article Text

Abstract
Pancreatic ductal adenocarcinoma (PDAC) constitutes one of the most aggressive malignancies with a 5-year survival rate of <7%. Due to growing incidence, late diagnosis and insufficient treatment options, PDAC is predicted to soon become one of the leading causes of cancer-related death. Although intensified cytostatic combinations, particularly gemcitabine plus nab-paclitaxel and the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) protocol, provide some improvement in efficacy and survival compared with gemcitabine alone, a breakthrough in the treatment of metastatic pancreatic cancer remains out of sight. Nevertheless, recent translational research activities propose that either modulation of the immune response or pharmacological targeting of epigenetic modifications alone, or in combination with chemotherapy, might open highly powerful therapeutic avenues in GI cancer entities, including pancreatic cancer. Deregulation of key epigenetic factors and chromatin-modifying proteins, particularly those responsible for the addition, removal or recognition of post-translational histone modifications, are frequently found in human pancreatic cancer and hence constitute particularly exciting treatment opportunities. This review summarises both current clinical trial activities and discovery programmes initiated throughout the biopharma landscape, and critically discusses the chances, hurdles and limitations of epigenetic-based therapy in future PDAC treatment.
- PANCREATIC CANCER
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a dismal disease with a 5-year survival rate of <7%.1 Despite tremendous efforts in PDAC research and therapeutic development, no significant improvement in survival has been achieved within the last decades. Research efforts in PDAC have traditionally focused on genetic alterations underlying malignant transformation, carcinogenesis and tumour progression. Early studies on these genetic abnormalities defined common mechanisms of oncogenesis in PDAC, such as activating mutations of Kras or inactivation of the tumour suppressor genes TP53, DPC4 or CDKN2A.2 In addition to these well-characterised ‘driver mutations’, a plethora of diverse genetic events occurs in each pancreatic tumour, characterising PDAC as one of the most heterogeneous malignant diseases.1 ,3 These studies led to the testing of targeted therapies, which have been evaluated in preclinical and clinical settings as monotherapies or in combination with standard chemotherapy.4 ,5 While small subgroups of patients with PDAC show responsiveness towards selected therapeutic regimes, the majority of patients with PDAC is refractory to these treatments a priori or rapidly acquires therapeutic resistance.
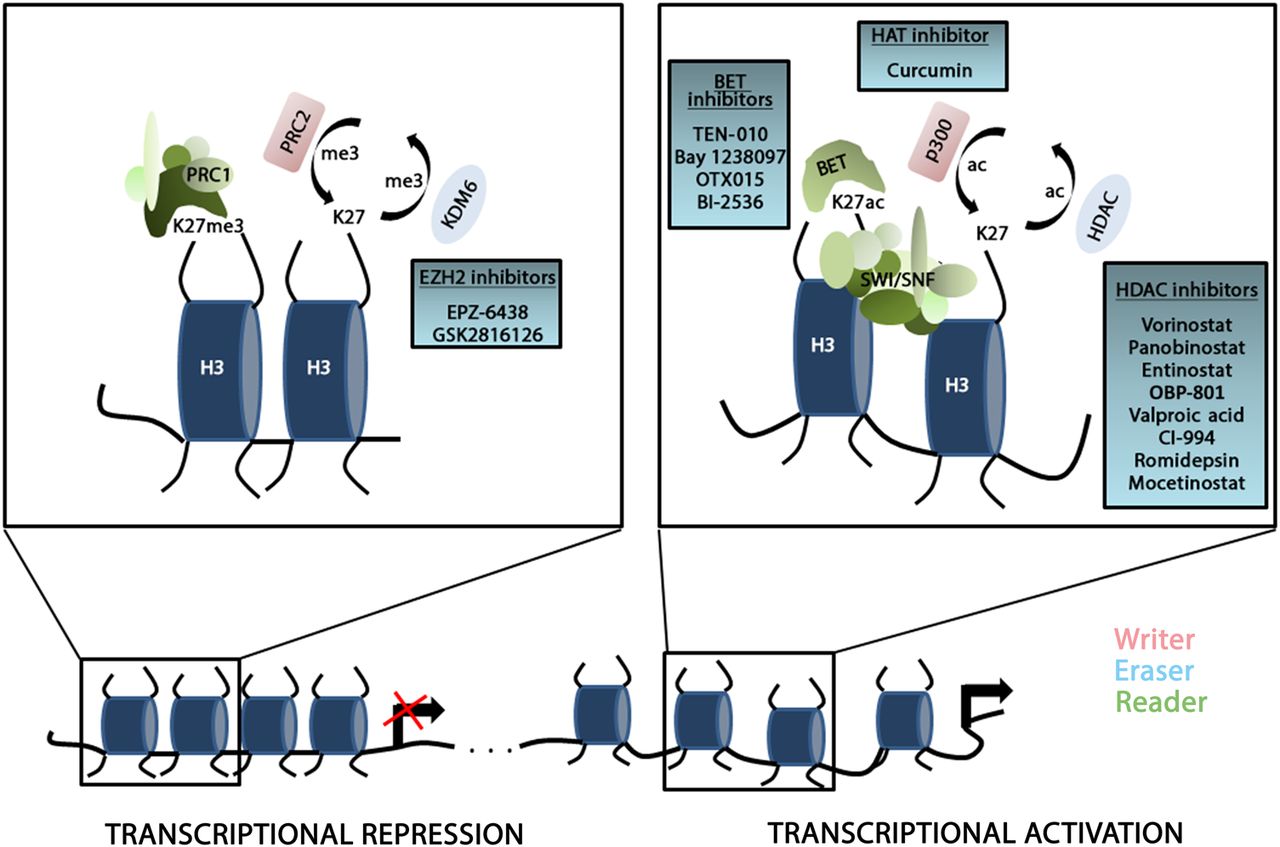
Drug resistance in PDAC is mediated by pronounced plasticity enabling PDAC cells to switch between phenotypic states and to select for cellular clones that eventually evade therapy.6 Importantly, apart from the restrictive contribution of genetic alterations, the acquisition of a drug-tolerant phenotype is frequently largely reversible. Indeed, the dynamic character of cell plasticity and drug resistance suggests the involvement of epigenetic regulation in controlling phenotypic heterogeneity in PDAC.7 The term ‘epigenetics’ describes processes that comprise non-genetic, heritable information through alterations that do not change the DNA sequence8 and refers to mechanisms involving DNA-methylation, post-transcriptional control of gene expression via non-coding RNAs as well as modifications and remodelling of chromatin. In addition to its functions to package DNA into a cell, its crucial involvement in mitosis and prevention of DNA damage, chromatin significantly impacts on the transcriptional activity of a gene. The nucleosome constitutes the functional subunit of chromatin and consists of approximately 147 bp of DNA wrapped around an octameric structure composed of two histones each of H2A, H2B, H3 and H4.9 Regulation of chromatin conformation is facilitated through DNA methylation, chromatin remodelling or histone modifications, such as acetylation, methylation, ubiquitination and phosphorylation.8 These post-translational modifications alter chromatin architecture by non-covalent interactions within and between nucleosomes, resulting in changes of nucleosome structure and accessibility of the transcription machinery.8 The major effectors of post-translational histone modifications are chromatin-modifying enzymes that either add (‘writers’) or remove (‘erasers’) modifications or factors that recognise specific histone modifications or combinations of modifications (‘readers’) (figure 1). Defects in these chromatin modulating proteins can have profound effects on vital cellular processes and hence contribute to development and progression of numerous diseases including cancer.10 Consequently, several recent preclinical studies investigated the impact of epigenetic alterations in diverse cancer models and emphasised the significance of epigenetics in malignant transformation and tumour progression in haematological malignancies and solid tumours including PDAC.1 ,11 Importantly, in contrast to genetic defects, epigenetic alterations are reversible and therefore represent bona fide targets for novel cancer therapies. Not surprisingly, first clinical trials aiming at altered epigenetic signalling in PDAC are underway. Furthermore, epigenetic alterations are increasingly being recognised for their predictive value in molecular tumour stratification in many cancer entities and thus extend the scientific and translational relevance of epigenetics in this particular tumour entity.

Schematic illustration of chromatin-associated regulation of gene transcription. The chromatin conformation significantly determines the accessibility of the transcription machinery to the DNA. The switch between condensed and transcriptional inactive heterochromatin (left panel) and open, accessible euchromatin (right panel) is controlled by chromatin regulators that establish (‘writers’), maintain (‘readers’) or remove (‘erasers’) post-translational modifications on the lysine residues of histone tails. Exemplary chromatin regulators are illustrated to show their impact on chromatin conformation and gene transcription. All inhibitors depicted in the blue boxes are evaluated in clinical trials in pancreatic ductal adenocarcinoma (PDAC). ac, acetylation; BET, family of bromodomain and external terminal proteins; HDAC, histone deacetylase; H3, histone 3; KDM6, lysine demethylase 6; K27, lysine 27; me3, 3 methyl groups; PRC1/2, polycomb repressor complex 1/2; SWI/SNF, SWItch/sucrose non-fermentable chromatin complex.
In this review, we summarise recent advances in our understanding of altered chromatin regulation in PDAC development and progression, and critically discuss ongoing initiatives to tackle epigenetic dysregulation in PDAC disease. Moreover, we highlight the potential of epigenetic approaches in therapeutic PDAC stratification strategies with a particular focus on chromatin-associated mechanisms (box 1).
Box 1 Inventory of epigenetic therapy in pancreatic cancer treatment
A plethora of phase I/II clinical trials evaluate epigenetic therapies in pancreatic ductal adenocarcinoma (PDAC)
Targeting chromatin dysregulation in PDAC can overcome cell plasticity and primary or secondary therapeutic resistance
A hitherto underestimated vulnerability towards inhibitors of chromatin regulatory proteins exists in PDAC
Monotherapies using epigenetic drugs have been shown to be not beneficial, but the pharmaceutical targeting of epigenetic modifications can significantly alter the susceptibility of PDAC towards standard chemotherapy
Combination of drugs targeting diverse chromatin regulators (eg, combined inhibitors of BET proteins/histone deacetylase inhibitors) to date represents the most promising concept for epigenetic therapies in PDAC
Box 2 Essential aspects that need to be considered to increase the potential of epigenetics-based therapeutic approaches in PDAC
Molecular stratification is inevitable to perform therapeutic response prediction in PDAC and thus to alleviate therapy decision making in PDAC
An intensified investigation of synthetic lethal interactions is essential to understand how epigenetic alterations influence each other and how they interfere with standard therapy
Alterations in the epigenetic landscape can be used to define certain PDAC subtypes with distinct response towards standard therapy
The elucidation of context-specific dependencies that facilitate maximal exhaustion of the therapeutic and predictive potential of epigenetic-based mechanisms in PDAC will significantly determine the success of the epigenetic translational PDAC research
Translational approaches to tackle epigenetic dysregulation in pancreatic cancer
Histone acetylation
Acetylation and deacetylation of lysine residues within histone tails represents a crucial mechanism within a plethora of epigenetic regulatory mechanisms controlling gene expression.10 While histone acetylation mediated by histone acetyltransferases (HATs), for example, CREB-binding protein (CBP), p300 and pCAF is associated with accessible chromatin and transcriptional activation, deacetylation by different groups of histone deacetylases (HDACs) is responsible for repression of gene transcription (figure 1). Histone acetylation and deacetylation levels are tightly controlled by antagonistic activities of HATs and HDACs and hence, unbalanced enzyme activation in one way or the other can foster malignant transformation and tumour progression.12
HATs control gene expression by catalysing acetylation of histones and non-histone proteins. Specifically, acetylation neutralises the positive charge on the amino group of specific lysine residues, thus weakening the DNA-chromatin complex and creating an open chromatin configuration.13 The p300 protein belongs to the best-studied HATs and represents a ubiquitously expressed global transcriptional coactivator with critical involvement in a wide variety of cellular mechanisms.14 Compared with HDACs, the contribution of HATs to pancreatic cancer formation and progression is multifaceted and highly dependent on the cellular context and the selection of regulated target genes.12 While certain studies designate a tumour-promoting function of p300 in PDAC, for example, by transcriptional activation of the c-Myc promoter,15 other reports describing p300 as a metastasis repressive protein16 as well as the frequent occurrence of loss of function mutations in the EP300 gene in PDAC cell lines14 argue for tumour-suppressive HATs functions in PDAC. Due to the contradictory preclinical findings as well as the unavailability of highly specific HAT inhibitors, little progress has been made in evaluating the potential utility of HAT inhibition for the treatment of patients with PDAC.
The natural turmeric-derived polyphenol compound curcumin represents a potent inhibitor of p300 HAT activity.14 A number of preclinical data have demonstrated antitumourigenic effects of curcumin in PDAC using in vitro and in vivo systems.17–19 These findings combined with a minimal toxicity profile led to the initiation of a few clinical trials to investigate the safety and efficacy of curcumin in PDAC therapy. The first-in-patient study performed with the natural compound tested the efficiency and feasibility of curcumin application in combination with gemcitabine in chemotherapy-naïve patients with advanced PDAC (NCT00192842) (compare tables 2 and 3).20 In contrast to the following trials, the daily oral dose of 8 g caused severe and intractable abdominal pain, indicating an increased GI toxicity of the drug when applied together with gemcitabine.21 Dhillon et al conducted a subsequent monotherapy trial with curcumin in patients with PDAC (NCT00094445). In a phase II setting, pretreated or untreated patients received 8 g curcumin daily, which was well tolerated and, despite its limited bioavailability, showed biological activity in some patients with PDAC with stable disease and a brief, but remarkable response (73% reduction of liver metastasis size) as the best outcome.21–23 Another group performed two clinical trials with curcumin and a nanoparticle-based curcumin (Theracurmin). In a phase I/II study, patients who became resistant to gemcitabine-based chemotherapy were treated with a combined curcumin/gemcitabine regime.23 No cumulative toxicity from curcumin was observed, but unfortunately, no patient experienced a complete or partial response.23 The described improvement of quality-of-life scores following Theracurmin administration needs to be confirmed in a randomised placebo-controlled trial.21
Overall, the clinical trials evaluating curcumin in PDAC therapy reflect the conflicting results obtained in preclinical studies. Although high p300 specificity of curcumin has been reported in vitro,14 it remains unclear whether the activity of the drug is limited to acetyltransferase inhibition. Careful preclinical investigations are required to understand the involvement of p300 in tumour-promoting functions in PDAC. If further evidence were to support a definitive function of HATs as oncogenic drivers in subgroups of PDAC, more specific and potent inhibitors would need to be developed to increase the chances of antitumourigenic activity of HAT blockade.
HDAC proteins counteract HAT activity and have been extensively described as significant drivers of transformation and tumour progression in multiple tissues, including the pancreas. Hyperactivity of HDAC proteins can foster proliferation and impair cell death regulation in pancreatic cancer24–26 and HDAC-mediated transcriptional control is associated with the regulation of epithelial-mesenchymal transition (EMT) programmes in PDAC cells (table 1),27–30 thereby contributing to PDAC invasion and metastasis. Based on their homology with yeast deacetylases, their subcellular localisation and diverse functions, HDAC proteins are grouped into three different classes.31 As a result of the frequently observed dysregulation of HDAC family members in cancer,32 ,33 and prompted by the finding that HDACs control various key oncogenic features of cancer cells,34 inhibition of HDAC activity has been evaluated as a therapeutic strategy to restore the balance of histone acetylation and to subsequently interfere with HDAC target gene expression. Several natural as well as synthetic compounds that inhibit HDAC activity are now available.35 ,36 HDAC inhibitors (HDACi) have been identified which either target all HDAC family members (pan-HDACi) or selectively interfere with subgroups of HDAC isoforms, thus facilitating a more specific manipulation of particular oncogenic HDAC functions (figure 2).12 ,31 Several HDACi have been investigated in clinical trials, where they have shown beneficial effects in selected haematological malignancies and solid tumour entities.34 Three HDACi, vorinostat (Zolinza), romidepsin (Istodax) and panobinostat (Farydak) have reached approval by the US Food and Drug Administration (FDA) or by the European Medicines Agency for the treatment of cutaneous T-cell lymphoma (vorinostat and romidepsin), peripheral T-cell lymphoma (romidepsin) and multiple myeloma (vorinostat and panobinostat).37
Exemplary HDAC targets in cancer

{kind=link}
{kind=link}
Histone deacetylase (HDAC) classes and their inhibitors. According to their structural and functional qualities, HDAC proteins are grouped into three classes. Class III deacetylases (silent mating type information regulation two (SIRT) proteins) are not depicted here. Asterisks indicate inhibitors that are investigated as therapeutic options in clinical trials.
Based on the oncogenic activity of HDACs in PDAC, several clinical trials were initiated to evaluate the safety and efficacy of HDACi in patients with PDAC (tables 2 and 3). While HDACi monotherapy showed clinical activity in haematological malignancies, results have largely been disappointing in most solid tumours including PDAC.34 Consequently, recent clinical trials investigating HDACi in PDAC focus on combined approaches of HDACi with small-molecule inhibitors or chemotherapeutic agents. In fact, three clinical trials are currently evaluating HDAC inhibition in combination with gemcitabine (NCT00379639, NCT00372437, NCT00004861). While monotherapy with mocetinostat (MGCD0103) stabilised tumour disease at best in some cases,38 it was significantly more active in combination with gemcitabine, as described in a recent phase I/II study on advanced solid tumours (NCT00372437). Among 14 evaluable phase I patients, 2 out of 5 patients with PDAC showed partial response with tumour shrinking and 1 patient had stable disease (American Society of Clinical Oncology Meeting 2008, abstract 4625). In contrast to these results that suggest synergism of the gemcitabine/HDACi combination in patients with PDAC,39 the majority of studies revealed restricted effects in a limited number of patients with distinct histological PDAC subgroups40 ,41 or even characterised combination with HDACi to be inferior to gemcitabine monotherapy in this tumour entity (NCT00004861).42
Inhibitors of epigenetic regulators validated in clinical trials in pancreatic cancer (terminated, completed trials and active, but not recruiting clinical trails)
Inhibitors of epigenetic regulators validated in clinical trials in pancreatic cancer (recruiting trials)
As an alternative approach to chemotherapeutic agents, several trials have been conducted to evaluate the combination of HDACi and targeted therapies in PDAC treatment. Unfortunately, many HDACi combinatory treatment regimens with strong and promising mechanistic and functional synergism in preclinical PDAC models failed in first-in-patient studies, thus reflecting the difficulties of translating these findings into the clinical setting (eg, NCT00667082, NCT01056601).43 Nevertheless, a plethora of preclinical data on synthetic lethal interactions exist in the context of HDACi that can be used to optimise the potential of HDAC inhibition in PDAC treatment. For instance, a combinatory treatment of HDACi with LY294002-mediated phosphoinositide 3-kinase (PI3-kinase) inhibition has recently been reported to induce apoptosis in renal cancer cells44 and in a xenograft model of endometrial cancer.45 These findings might be of particular interest for PDAC therapy, as dysregulation of PI3-kinase activity occurs in subgroups of PDAC46 and might therefore represent an interesting target for synthetic-lethal approaches in the context of HDAC inhibition. Moreover, one of the neoadjuvant clinical trials investigating the efficiency of epigenetic drugs as a therapeutic option in PDAC treatment combines vorinostat-mediated HDACi with standard therapy (gemcitabine plus nab-paclitaxel or gemcitabine plus radiation) and sorafenib in patients with stage I-III PDAC (NCT02349867). The combination of HDACi with sorafenib-mediated tyrosine-proteinase inhibition has been reported to show additive effects in preclinical hepatocellular carcinoma (HCC) models.47 Until today, the mechanism of the reported synergistic or additive effects of combined HDAC inhibition and tyrosine protein kinase inhibition remains elusive. Nevertheless, the fact that erlotinib-resistant glioblastoma cells could be resensitised for tyrosine kinase inhibition upon blockade of HDAC activity48 and preclinical data on a vorinostat-driven overcoming of erlotinib resistance in PDAC cells,49 suggests that HDACi might represent a promising treatment option in combination with erlotinib-mediated inhibition of epidermal growth factor receptor (EGFR) signalling in patients with PDAC. However, clinical trials investigating combined EGFR inhibition/HDAC inhibition are currently limited to glioblastoma multiforme (NCT01110876), lung cancer (eg, NCT00251589) and head and neck tumours (NCT00738751).
Overall, preclinical and clinical data on HDACi as a therapeutic strategy in PDAC treatment are disappointing. To improve the potential of HDACi in PDAC treatment, preclinical studies and clinical trials need to (1) systematically dissect the impact of the different HDAC proteins on PDAC progression in order to clarify which HDAC classes represent preferable targets for HDACi and (2) should concentrate on the identification of predictive markers that allow therapeutic stratification of patients with PDAC that might benefit from HDAC inhibition. One such surrogate is the multiubiquitin chain receptor protein RAD23B, which has been recently characterised as a determinant for HDACi-induced apoptosis and is highly expressed in cutaneous T-cell lymphoma, a tumour which responds favourably towards HDACi-based therapy.50 These results suggest that therapy response prediction to HDACi is feasible, thus potentially opening new avenues to better translate HDAC inhibition strategies into the clinic. Moreover, future translational studies on HDACi need to extend the application of HDACi in combination with established and novel drugs in order to increase the potential of HDAC blockade in PDAC treatment. As indicated in the following sections of this review, the antitumourigenic potential of HDACi is highly dependent on the molecular context of the tumour and can be significantly enhanced when combined with the right therapeutic agents (box 2).
BET proteins
While acetylation levels are regulated by HATs (‘writers’) and HDACs (‘erasers’), acetylation marks are recognised by bromodomains, which can be found in chromatin-associated and transcription-associated proteins that drive the formation of protein complexes that mediate active transcription.13 The bromodomain and extraterminal (BET) domain family of proteins (BRD2, BRD3, BRD4 and BRDT) constitutes the probably best characterised group of chromatin ‘reader’ proteins in cancer.51 By binding to acetylated chromatin via their tandem-bromodomains, BET proteins regulate the transcription of specific subsets of genes, including those that promote cell-cycle progression and the evasion of apoptosis.52 Furthermore, BET proteins function as critical mediators of transcriptional elongation by promoting the recruitment and activation of the positive transcription elongation factor-b complex (P-TEFb).53 Based on the importance of BET proteins in controlling important numerous cancer-relevant genes such as c-Myc, Bcl2, FosL154 and others, as well as the activity of the EMT-related transcription factor Twist1,55 several potent and selective inhibitors of BET proteins (BETi) have been developed.13 The apparent selectivity of BETi for tumour cells appears to originate from a particular dependence of many tumour-relevant genes (notably c-Myc) on the recruitment of BRD4 to larger, composite distal regulatory regions frequently referred to as ‘super enhancers’. This requirement is gene-specific and context-specific, where even for c-Myc different tumour types are dependent upon different BRD4-dependent enhancer regions.56 ,57 One of the founding BETi molecules was JQ1, which was initially described to be a potent suppressor of NUT midline carcinoma,53 B-cell lineage malignancies58 and other tumour entities.59 In a subcutaneous xenograft model of PDAC, Garcia et al60 observed a remarkable decrease of tumour size upon JQ1 administration, indicating antitumourigenic activity in this tumour entity. Moreover, administration of JQ1 blocked acinar-to-ductal metaplasia in the pancreas, a key event in PDAC initiation, decreased formation of PanIN lesions as well as PDAC cell proliferation61 and attenuated the progenitor phenotype of dedifferentiated PDAC in favour of enhanced cellular maturity.62 A more recent study confirmed a beneficial effect of BETi in mouse PDAC xenograft studies and suggested a specific importance of BET proteins in controlling the activity of Gli transcription factors downstream of oncogenic Hedgehog signalling.63 These data strongly support a role of the BET proteins as important drivers of both PDAC development and progression and clinical trials using different BETi agents have been initiated with great hope and expectation (NCT01987362, NCT02259114, NCT02369029). However, first results of clinical studies testing monotherapeutic applications of BETi in PDAC are discouraging: a randomised phase II trial in patients with PDAC with unresectable tumours using BI-2536, an inhibitor of the Polo-like kinase that has been shown to block BRD4 activity in vitro (NCT00710710), yielded poor response rates and the second stage of the study was not even initiated.13 Since it is unclear whether the dose of BI-2536 used in this study is sufficient to adequately inhibit BET protein activity in vivo, this result does not exclude the biological activity of BETi in PDAC. However, a recent preclinical approach in a genetically engineered mouse model of PDAC combining JQ1-mediated BET inhibition with gemcitabine also did not show a benefit in survival, when compared with single applications of the inhibitors,61 suggesting that neither BETi monotherapies nor BETi/chemotherapy combinations may represent beneficial therapeutic strategies in PDAC treatment.
In contrast to chemotherapy-based combinatory anticancer therapies, BET inhibitors applied together with non-chemotherapeutic agents have recently attracted much attention in various preclinical tumour models. Surprisingly, despite apparent opposing mechanistic effects, BET inhibitors seem to synergise with HDACi in defined tumour entities. For instance, Myc-induced murine lymphoma mouse models are highly responsive to BET inhibition, while BETi application sensitised Myc-overexpressing lymphoma cells to HDAC inhibition.52 Similar results have been reported in acute myelogenous leukaemia (AML), where panobinostat-mediated HDAC inhibition synergised with JQ1 to induce apoptosis in human AML cells.64 Most importantly, this mechanism of synergism has recently been confirmed in PDAC, where therapeutic co-application of JQ1 and the FDA-approved HDACi vorinostat potently suppressed tumour growth in advanced PDAC.61 Strikingly, PDAC bearing KrasG12D;p53ko mice, an established genetically engineered mouse model of PDAC,65 displayed a significantly reduced tumour volume upon combined JQ1 and vorinostat treatment. Importantly, mice that received the combination of both epigenetic drugs did not show any signs of tumour relapse, such as regularly seen with other therapies and died as a consequence of neurological symptoms rather than tumour burden.61 This strongly indicates that BETi/vorinostat cotreatment may be sufficient to overcome therapeutic resistance in PDAC. A similar effect could be observed in a second transgenic PDAC model that is driven by deletion of the CDKN2A gene locus in combination with oncogenic Kras activation, a genetic event that regularly occurs in PDAC1 and in experimental lung cancer models, arguing that the antitumourigenic activity of BETi/HDACi treatment is an attractive strategy in otherwise highly resistant Ras-driven cancers.61
Several mechanisms have been proposed to explain the antitumour activity of BETi in general and of combined BETi/HDACi efficiency in particular. While several reports suggest transcriptional downregulation of oncogenic c-Myc upon BET inhibition as the crucial mechanism of antitumour activity (reviewed in),66 other studies strongly support the notion that BETi activity is frequently independent of effects on c-Myc expression.52 ,57 Recently, JQ1 has been demonstrated to suppress tumour cell growth specifically in colon cancers that are characterised by a CpG island methylator phenotype (CIMP), one of the main subtypes of colorectal cancer.57 Genome-wide analyses led to identification of a specific BRD4-bound ‘super enhancer’ in CIMP colon cancers, which serves to promote the expression of the long non-coding RNA colon cancer-associated transcript 1 (CCAT1) and can be utilized as a marker for sensitivity to BETi. These data characterise CCAT1 as a potential clinical marker that predicts BETi responsiveness and might have a strong impact on the selection of patients who could benefit from BETi.57 Recent studies have also uncovered resistance mechanisms against BETi,67 demonstrating that their effective clinical application requires additional information about their precise, context-specific molecular mechanisms and biomarkers both predictive for and indicative of their biological activity.
The unexpected synergism of HDACi and BETi in cancer therapy prompted several groups to dissect the mechanisms that underlie this phenomenon. Mazur et al61 proposed that de-repression of p57 upon combined JQ1 and vorinostat treatment is responsible for cell death induction upon treatment. Further mechanistic evidence comprises synergistic attenuation of c-Myc and BCL-2 expression.64 In their lymphoma model, Bhadury et al identified gene sets that are similarly induced upon isolated BET or HDAC inhibition. To explain this phenomenon, the authors used a model which has been proposed for BETi in HIV, inflammation and arteriosclerosis. Specifically, they propose that P-TEFb, which is inactivated by a complex containing HEXIM1 and the 7SK snRNP in untreated cells, is transiently released from this complex following HDAC or BET inhibition, thus enabling P-TEFb recruitment to and subsequent transcriptional induction of a defined set of alternative target genes.52 A more recent study demonstrated that HDACi treatment blocks transcriptional elongation and results in a re-distribution of BRD4 across the genome.68 Based on these findings, it is conceivable that BETi synergise with HDACi by targeting non-redundant regulatory mechanisms controlling transcriptional elongation of a specific subset of genes relevant for cancer progression. However, further studies will be necessary to definitively address the complex mechanisms by which BET and HDAC inhibition synergise in cancer treatment.
A BETi-similar effect on the transcriptional activity in cancer cells might be achieved by the cyclin-dependent kinase (CDK) inhibitor Dinaciclib, which also displays high potency against CDK9. BET bromodomains recruit CDK9, the catalytic subunit of P-TEFb, which catalyses the phosphorylation of serine 2 in the heptapeptide repeat sequence within the C-terminal domain of RNA polymerase II, a post-translational modification associated with transcriptional elongation.66 Importantly, Dinaciclib has been reported to abrogate PDAC growth and progression in vitro and in vivo69 ,70 and is currently being evaluated for its therapeutic potential in patients with PDAC in combination with the poly [ADP-ribose] polymerase (PARP) inhibitor veliparib (NCT01434316) and MK2206-mediated Akt inhibition (NCT01783171). Interestingly, CDK9 and BET inhibitors were also shown to function synergistically.71 Thus, it will be necessary to analyse overlapping and diverse functions of CDK9 inhibition and BET inhibition in cancer to determine in which context the combination of different therapeutic strategies may optimally work to have synergistic or additive effects in PDAC treatment.
Altogether, combined epigenetic targeting appears to be beneficial in certain tumour entities. Prospective clinical trials need to consider the promising effects of combined HDAC and BET targeting in preclinical PDAC models and have to validate whether this therapeutic strategy can be translated into the clinic.
Polycomb proteins
Monomethylation, dimethylation and trimethylation of histones elicit distinct changes in chromatin conformation and transcriptional activity dependent on the particular location and context of the modified lysine residues.10 For example, trimethylation of histone 3 lysine 4 by Trithorax group proteins is strongly associated with active gene expression, while the trimethylation of lysine 27 (H3K27me3) induces gene silencing.72 The histone mark H3K27me3 is catalysed and maintained by the polycomb repressor complex-2 (PRC2).10 The PRC2 constitutes an approximately 600 kDa complex with four subunits: Suz12, embryonic ectoderm development (EED), RaAp46/48 and the catalytic component enhancer of zeste homologue-2 (EZH2), which mediates H3K27 methylation.73–75 Physiologically, the polycomb proteins (PcG) function as crucial epigenetic repressors of differentiation and are responsible for the maintenance of stem cell capacities.76 Importantly, PcG proteins in general and EZH2 in particular are frequently overexpressed in cancer.77 High expression levels of the histone methyltransferase regularly correlate with tumour stage and poor prognosis, while depletion of EZH2 can restrict proliferation and tumour progression.78 The role of PRC2 proteins as epigenetic regulators of tumour progression has only recently become a focus in PDAC.79 Ougolkov et al80 showed nuclear overexpression of EZH2 in PDAC cell lines and in 68% of investigated human PDAC samples, while the nuclear accumulation of the enzyme was more prevalent in dedifferentiated PDAC (91%). Accordingly, an inverse expression profile of EZH2 and the epithelial marker protein E-cadherin81 and a robust EZH2-dependent promotion of cancer stem cell self-renewal76 have been demonstrated in PDAC, characterising EZH2 as a strong promoter of cancer cell plasticity in the pancreas. Consequently, EZH2 inactivation sensitised PDAC cells to chemotherapy in preclinical PDAC models.76 ,80 Reports demonstrating a significant longer survival of gemcitabine-treated patients with PDAC with low pancreatic EZH2 levels81 emphasise the oncogenic capacities of the histone methyltransferase.
Its involvement in a wide range of oncogenic functions in several cancer entities has prompted the development of specific inhibitors of EZH2 activity. With tazemetostat (EPZ-6438), CPI-1205 and GSK2816126 three different EZH2 inhibitors are being assessed, for their tolerability and efficiency in clinical trials for haematological and solid malignancies. The potent inhibitor of EZH2 methyltransferase activity tazemetostat82 is currently being evaluated in a multicentre, open-label phase I/II study (NCT01897571). Patients with haematological and solid tumours including PDAC are enrolled in the phase I arm, while the phase II portion is restricted to lymphoma patients. Preliminary results for a tazemetostat trial investigating dose escalation and expansion of the drug in B-cell lymphoma and refractory or relapsed solid tumour patients (NCT02601950) have been presented at the European Society for Medical Oncology (ESMO)'s European Cancer Congress in Vienna, Austria in September 2015. Response assessment was conducted after 8 weeks of application. Importantly, tazemetostat monotherapy induced a 55% disease control rate in solid tumours, including rhabdoid tumours and epitheloid sarcoma. It is worth mentioning that the subgroup of responding tumours exhibited inactivating mutations of either SMARCB1 (also known as INI1 or SNF5) or SMARCA4 (also designated as BRG1). Both proteins belong to the SWItch/sucrose non-fermentable chromatin complex (SWI/SNF) chromatin remodelling complex and have been characterised for their tumour-suppressive potential. Intriguingly, mutations of the SWI/SNF complex can be found in approximately 20% of all human cancers.83 The dependency of antitumour activity of EZH2 inhibition on SWI/SNF mutations has recently been described in a preclinical approach. In a detailed mechanistic study, Kim et al demonstrate that genetic depletion of EZH2 sufficiently blocked proliferation of a wide panel of SWI/SNF mutation containing cancer cells, while loss of EZH2 had no impact on cells harbouring wild-type expression of SWI/SNF proteins. Surprisingly, the dependency of SWI/SNF-mutated cells on PRC2 expression did not necessarily always require EZH2 enzymatic activity, but was rather based on the stabilisation of the PRC2 complex in the context of Ras or Raf mutation.78 These data suggest the existence of methyltransferase-independent oncogenic EZH2 functions and indicate that EZH2 inhibitors may not fully repress the oncogenic activity of the enzyme under some contexts, unless they are able to intervene with EZH2 expression or its interaction with the PRC2 complex. Whether this observation holds true for pancreatic cancer is currently unclear. Although patients with PDAC are eligible for the EZH2 inhibitor trials, no data are currently available on the enrolment of patients with PDAC into the trials or the efficiency of EZH2 blockade in PDAC. Nevertheless, since recent whole genome sequencing approaches reported frequent occurrence of SWI/SNF mutations (of complex members such as ARID1A, ARID1B, SMARCA2 and SMARCA4) in PDAC,1 ,3 beneficial effects of EZH2 inhibition in this subgroup of patients with PDAC are worth further investigation.
Synthetic lethal interactions between EZH2 inhibition and the disturbance of other epigenetic mechanisms or signalling pathways are not limited to the SWI/SNF complex, but have been described in other contexts as well. For example, EZH2 inhibition sensitised colorectal cancer cells to EGFR inhibition by the induction of autophagy84 and blockade of EZH2 activity increases the susceptibility of small cell lung cancer with EGFR gain-of-function mutations towards topoisomerase II inhibition.85 Moreover, an interesting connection between PRC2 integrity and rat sarcoma (RAS) activity has been recently revealed in malignant peripheral nerve sheath tumours. Herein, deficiency of the non-catalytic PRC2 subunit Suz12 or EED has been shown to amplify RAS-driven transcription.86 As attenuation of the H3K27me3 mark upon Suz12 or EED ablation resulted in an increase of H3K27 acetylation and subsequent recruitment of bromodomain proteins, the authors hypothesised that Suz12-deficient tumours might respond to pharmacological bromodomain blockade. Indeed, JQ1 treatment counteracted activation of RAS signalling upon Suz12 deficiency by suppressing RAS signature genes.86 These preclinical studies indicate that the functional consequences of PRC2 inactivation are diverse across tumour entities and underscore that methyltransferase-independent functions of EZH2 should be considered to exhaust the therapeutic potential of PRC2 disruption in PDAC treatment. Moreover, the experience and knowledge gained from preclinical and clinical studies in the context of EZH2 highlight the utmost importance of molecular stratification approaches as a central component of epigenetically based cancer therapy.
Conclusion and perspective
Epigenetic mechanisms in general, and chromatin alterations in particular, are involved in all aspects of cancer development and progression. A number of elegantly conducted translational studies have provided significant evidence that dysregulation of chromatin organisation strongly contributes to the aggressive behaviour of PDAC, for instance, by promoting cellular plasticity and therapeutic resistance. Consequently, several clinical trials have been initiated to determine the safety and efficiency of epigenetic drugs in patients with PDAC. The small number of predominantly non-randomised studies on epigenetic targeting in this tumour entity and restrictive enrolment of patients with PDAC in some of the studies both limit the informative value of these trials. Nevertheless, the following conclusions can be drawn: (1) similar to chemotherapy, monotherapeutic targeting of epigenetic alterations in PDAC has thus far not proven to be beneficial; (2) efficiency of epigenetic drugs could be reported in selected patients with PDAC, especially following targeting of HDAC or BET proteins; (3) the mechanistic basis of the tumour cell selectivity of some epigenetic inhibitors remains largely unclear.
As demonstrated in diverse preclinical studies, a striking vulnerability of PDAC cells exists towards epigenetic-based therapy in this disease. However, targeting chromatin alterations in cancer to date is limited to a subset of histone writers, readers and erasers, while the potential targetability of other epigenetic regulators in the therapeutic landscape of cancer treatment is unknown. Whether this is a consequence of less academic and pharmaceutical research interest or a result of reduced vulnerability compared with the established targets of epigenetic drugs is unclear. For instance, it is conceivable that inhibition of selective histone demethylase activities may be effective in the treatment of tumours displaying mutations in histone methyltransferases such as MLL2, MLL3 and SETD2,1 ,3 while tumours with mutations in KDM6A might be expected to respond well to EZH2 inhibitors. It remains a tremendous hurdle to identify the most beneficial therapeutic targets of epigenetic cancer therapy and to detect those subpopulations of patients that will profit from targeting of a particular epigenetic alteration in PDAC. While initial indications suggest that SMARCB1 and/or SMARCA4 mutations may be predicative of tumour responsiveness to EZH2 inhibitors in rhabdoid tumours, it remains to be determined whether pancreatic tumours displaying mutations in KDM6A, ARID1A, ARID2 or other BAF complex components also depend upon EZH2 activity. Significant preclinical and clinical efforts are required to develop a paradigm of molecular stratification to address this challenge. First advances in the prediction of responses towards epigenetic therapies57 indicate that this is a difficult, but feasible task. Recently developed high-throughput techniques can be used to identify molecular signatures that allow therapeutic response prediction to certain epigenetic drugs and alleviate therapy decision making in PDAC. This strategy is extremely important to enrol patients into the right clinical trials and to learn about the determinants of epigenetic drug responses. Intriguingly, considering molecular stratification approaches in PDAC, chromatin deregulation contributes to the development of epigenetic targeting strategies, and also influences therapy decision making in standard care, as alterations in the epigenetic landscape might characterise certain subtypes of PDAC, which are susceptible to defined (non-epigenetic) therapies. Therefore, epigenetic dysregulation in PDAC can function as both a therapeutic target and therapy-response predictor.
As a result of its tremendous cellular plasticity, monotherapeutic strategies usually fail in PDAC. Therefore, the identification of the right drug combinations should significantly impact the therapeutic response of patients with PDAC. Combinations of epigenetic drugs with standard chemotherapy or targeted therapies or even combinations of different epigenetic drugs with one another are conceivable approaches that need to be validated in preclinical and clinical settings. Due to the dynamic character of epigenetic modifications, their pharmaceutical targeting can significantly alter the susceptibility of the tumour towards standard chemotherapy, when applied in the right combination and sequence.
Intriguingly, a rising number of preclinical and clinical studies in the past years emphasise the relevance and chances of therapeutic strategies that combine epigenetic inhibitors with each other to combat cancer. The increasing understanding of the mechanistic and functional consequences of targeting central chromatin regulatory proteins in PDAC cells and the subsequent implications of these therapeutic strategies on the epigenetic balance of a tumour reveals a hitherto underestimated vulnerability of these cancer cells towards inhibitors of other chromatin-modifying or remodelling proteins with similar or inverse functions. In this context, the identification and characterisation of synthetic lethal interactions represents a crucial component to understanding how epigenetic mechanisms interact with each other and how they function within the genetic background of a tumour cell. Unfortunately, in contrast to the good tolerability of epigenetic monotherapies, combinatory applications of epigenetic drugs with other antitumourigenic agents frequently show additive and dose-limiting toxicities (table 4).28 Considering both the documented chances of epigenetic combination therapies in PDAC treatment as well as their toxicity-related limitations, extensions of synthetic lethality studies and their translation to clinical settings might significantly expand the therapeutic potential and the feasibility of epigenetic therapeutic approaches in PDAC treatment. Furthermore, the utilisation of epigenetic drugs at or near the maximum tolerated dose may not be the best approach. Given their mechanisms of action, which function primarily through gene expression changes rather than cytotoxic effects, the definition of an ‘effective dose’ based on biomarkers may significantly increase the potential utility of combinatorial approaches and reduce toxicity.
Side effects of exemplary clinical trials testing epigenetic inhibitors in cancer treatment
Without a doubt, the explosive scientific and pharmaceutical interest in epigenetic mechanisms in PDAC biology and therapy represents a significant advance in the field and contributes to our understanding of key mechanisms of PDAC biology. Finally, the success of the epigenetic era in translational PDAC research will be determined by the elucidation and creation of molecular and context-specific dependencies that guarantee maximal exhaustion of the therapeutic and predictive potential of epigenetic-based mechanisms in pancreatic cancer.
References
Footnotes
Contributors EH, SJ, JS and VE wrote the manuscript and designed the images for this review article.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.