Article Text

Abstract
Objective An inadequate host response to the intestinal microbiota likely contributes to the manifestation and progression of human inflammatory bowel disease (IBD). However, molecular approaches to unravelling the nature of the defective crosstalk and its consequences for intestinal metabolic and immunological networks are lacking. We assessed the mucosal transcript levels, splicing architecture and mucosa-attached microbial communities of patients with IBD to obtain a comprehensive view of the underlying, hitherto poorly characterised interactions, and how these are altered in IBD.
Design Mucosal biopsies from Crohn's disease and patients with UC, disease controls and healthy individuals (n=63) were subjected to microbiome, transcriptome and splicing analysis, employing next-generation sequencing. The three data levels were integrated by different bioinformatic approaches, including systems biology-inspired network and pathway analysis.
Results Microbiota, host transcript levels and host splicing patterns were influenced most strongly by tissue differences, followed by the effect of inflammation. Both factors point towards a substantial disease-related alteration of metabolic processes. We also observed a strong enrichment of splicing events in inflamed tissues, accompanied by an alteration of the mucosa-attached bacterial taxa. Finally, we noted a striking uncoupling of the three molecular entities when moving from healthy individuals via disease controls to patients with IBD.
Conclusions Our results provide strong evidence that the interplay between microbiome and host transcriptome, which normally characterises a state of intestinal homeostasis, is drastically perturbed in Crohn's disease and UC. Consequently, integrating multiple OMICs levels appears to be a promising approach to further disentangle the complexity of IBD.
- INFLAMMATORY BOWEL DISEASE
- GENE EXPRESSION
- INTESTINAL GENE REGULATION
- CROHN'S DISEASE
- BACTERIAL INTERACTIONS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- INFLAMMATORY BOWEL DISEASE
- GENE EXPRESSION
- INTESTINAL GENE REGULATION
- CROHN'S DISEASE
- BACTERIAL INTERACTIONS
Significance of this study
What is already known on this subject?
IBD is a complex disease that is characterised by chronic inflammation of the intestinal mucosa.
Inadequate host responses to intestinal microbiota, changes of the host transcriptome and altered host splicing patterns are known to be associated with IBD.
If and how the above factors interact to influence disease manifestation and progression is only poorly understood.
What are the new findings?
For the first time, we illustrate a close disease-relevant interaction between microbiota, host gene regulation and splicing of the host mucosal transcriptome.
A structured relationship between the three factors is most evident in healthy individuals and partially reduced in acute inflammation of the gut, but strikingly perturbed in patients with IBD.
Only a few inflammation-associated biological processes appear to drive the observed disease-relevant host-microbiome interaction.
How might it impact on clinical practice in the foreseeable future?
Our results emphasise that the microbiota and the host mucosa represent a joint intestinal ‘meta-organism’ that must be viewed as a single entity to attain a better understanding of the aetiology of IBD.
Introduction
Human chronic inflammatory bowel disease (IBD) (OMIM #266600) is an archetypical chronic immune-mediated disease, characterised by excessive T-lymphocyte-driven immune responses. IBD comprises a group of disorders with two main subentities, namely Crohn's disease (CD, OMIM #266600) and UC (OMIM #191390). Recently, advances have been made towards the identification of genetic IBD risk factors (by genome-wide association studies) and the molecular dissection of IBD pathophysiology (eg, by the detection of aberrant inflammatory signal transduction).1–4 However, why IBD preferentially manifests in a specific time window, spanning only a few decades in life and how the disease becomes chronic over time in a given individual is still poorly understood.
The human host together with their gut microbes forms a functional unit, called ‘meta-organism’.5 The commensal microbiota is essential for shaping the immune system and is involved in many host physiological functions including, among others, the digestion of nutrients.6 Host and microbiota are usually connected tightly in a resilient state believed to be important for the survival of the organism as a whole, thus qualifying as bona fide ‘symbiosis’. In contrast to the fixed repertoire of germline genetic variants inherited by a given individual, however, this symbiosis is influenced by a number of environmental factors as well, including lifestyle and hygiene. We may further assume that the interplay of gene and environment is much more complex than hitherto described by simple models7–9 even though defective host-microbiota crosstalk evidently contributes to the manifestation and progression of IBD.
The transcriptome of patients with IBD has been studied extensively since genome-wide approaches became feasible and popular, unravelling gene expression patterns of IBD in general, or CD or UC in particular.10–14 Various efforts have been made to characterise the transcriptional changes associated with IBD in more detail, from array-based studies to more recent RNAseq experiments. This work laid the foundation for a first map of mRNA expression changes characterising the disease.10–13 ,15 However, only a few studies so far have addressed the interaction between the human mucosal transcriptome and the gut microbiota16–19 in the context of intestinal inflammation. The gut microbiota has been shown to potentially interact with genetic IBD risk factors,20 the latter potentially influencing the response of the mucosa to the resident bacteria. Thus, it has been demonstrated that the NOD2 genotype likely influences21 and reshapes the microbiota in a disease-dependent fashion,22–25 a result further supported by observations of impaired mRNA expression in mouse models of colitis and colitis-associated carcinoma.
Supplemental material
Although the relationship between variation of the human transcriptome and the human genome is increasingly better understood,26 to the best of our knowledge, all previous studies on inflammatory diseases have neglected the complexity of the transcriptome (eg, sense/antisense transcription, alternative splicing). Currently, gene expression changes are mostly assessed at a gene-condensed level, as recently shown in paediatric IBD.19 ,27 We hypothesise that studies focusing on the inference of functional changes from genetic data alone are inadequate in that they disregard the important layer of transcriptome information. In fact, a seminal publication recently emphasised that a variety of diseases can be caused by mutations that disrupt the splicing code,28 yet studies assessing splicing alterations globally for a given biological state, such as inflammation, are still rare.
To highlight one specific functional aspect of IBD pathophysiology, we jointly analysed comprehensive host transcriptome data and mucosa-attached microbial community profiles from a cohort of patients with IBD, healthy and non-IBD intestinal inflammation controls. We employed next-generation sequencing (RNAseq and 16S rRNA amplicon sequencing) and systems biological-inspired data integration to unravel previously unrecognised, global metabolism-connected and immunity-connected expression patterns in the host mucosa and related these to IBD. Following this approach, we were able for the first time to identify the molecular elements of the host-microbiome ‘meta-organism’ that are likely affected by IBD and to demonstrate a rather specific disconnection of the human transcriptome from the microbiota in patients with IBD.
Methods
Patient recruitment and biopsy selection
Biopsies from four groups of individuals were retrieved from the local hospital biobank at Kiel: healthy individuals without significant pathological findings, patients with acute non-IBD intestinal inflammation, patients with CD and UC. The samples were taken either from the terminal ileum or the sigmoidal colon. In the disease groups, both inflamed and non-inflamed material was collected. Overall, 63 biopsies from 41 individuals were available for further study (see tables 1, 2 and online supplement for further details). Patient identity was taken properly into account to allow for dependent samples in the statistical analyses. The study protocol was approved by the Ethics Committee of the Medical Faculty of Kiel University (reference #B231/98-1/13). All study participants gave written informed consent prior to sampling and data collection.
Characteristics of biopsies
Individual medication overview
RNA extraction and sample preparation
Biopsies were obtained endoscopically during routine diagnosis and stored in liquid nitrogen. Total RNA was extracted using RNeasy (Qiagen, Germany) according to the manufacturer's protocols, including repeated tissue crushing using a Teflon head (Omnilab, Bremen, Germany) under liquid nitrogen to augment bacterial cell wall disruption. RNA was prepared as previously described,29 with RNA integrity assessed using an Agilent Bioanalyzer following the manufacturer's instructions.
Mucosa-attached microbiota analysis
Transcriptionally active mucosa-attached bacterial profiles were generated from mucosal biopsy RNA (converted to cDNA using random hexamers, Qiagen). The 16S rRNA variable region V3-V4 was amplified using bacterial 16S rRNA gene-specific composite primers (319F and 806R) as described.30 Pooled amplicon libraries were sequenced employing an Illumina MiSeq platform (2×300 bp) and processed further as described in the online supplementary methods section. Unprocessed 16S rRNA reads (fastq) have been submitted in the European Nucleotide Archive under accession number PRJEB14288.
Host transcriptome analysis
Host transcriptome analysis was performed by RNA sequencing of the primary samples described above. Strand-specific sequencing libraries were prepared with the TruSeq stranded Total RNA kit (Illumina) from 1 µg total RNA of each sample and sequenced on an Illumina HiSeq2000 (100-nucleotide paired-end reads). Reads were quality-controlled, aligned and processed further using Bioconductor package DeSeq2. The Kyoto Encyclopedia of Genes and Genomes-GenomeNet (KEGG) enrichment and transcription factor binding site analyses were carried out using the Innate DB database.31 For further details, see online supplementary methods.
Analysis of splicing
RNA splicing products were identified and quantified using vast-tools.32 The level of alternative product inclusion was defined as either the percentage spliced in (PSI), for exons, the percentage intron retention (PIR), for introns, or the percentage splice site usage (PSU) for alternative splice site acceptor choice (ALTA) and alternative splice site donor choice (ALTD), as appropriate. A splicing event was defined by fulfilling the following criteria: (1) it had an alternative product inclusion level ≥10 and <100 and (2) it had sufficient read coverage in at least 90% of all the samples.32 For further details, see online supplementary methods.
Analysis of host-microbiome crosstalk
To obtain a quantitative measure of host-microbe crosstalk, the Spearman's rank correlation coefficient was calculated, for all differentially expressed genes and all operational taxonomical units (OTUs) present in at least 50% of all samples, between the respective gene expression level and the respective OTU abundance. To determine the statistical significance of individual correlation coefficients, the false discovery rate (FDR) was estimated employing a modified Westfall and Young permutation approach.33 Group-wise differences in Spearman's rank correlation coefficient were assessed for statistical significance employing a Mann-Whitney U test.
Reconstruction of context-specific metabolic models and analysis of metabolic network coherence
To create context-specific metabolic models of transcriptional activity, the gene expression data were mapped to the human metabolic model Recon 2.0434 using the intensity modulated arc therapy (iMAT)-implementation of the constraint-based reconstruction and analysis (COBRA)-toolbox.35 Metabolic network coherence was then computed based upon dichotomised gene expression data as described36 with modifications facilitating an application to human data.37 For details, see online supplementary methods.
Results
The IBD-associated mucosal transcriptome
We used deep RNA sequencing to identify transcriptome alterations that were associated with IBD. Filtering and trimming of RNA sequencing data from 63 mucosal biopsies excluded between 1% and 4% of reads (adapter contamination and low quality), leaving 89–167 million read pairs for subsequent mapping. The proportion of reads mapping to the human reference genome varied between 89% and 98% over biopsies. The raw read counts are provided in online supplementary table S1.
Supplementary table
Some 4576 genes were found to be significantly differentially expressed between cases and controls (see online supplementary table S2). Using global scaling methods, most of the variation in expression level was explained by tissue type, followed by inflammation status. In contrast, clinical diagnosis did not discriminate well between expression profiles, as was highlighted by principle component analysis (figure 1A–C). The first two dimensions explained 19% of the overall variance in expression level (figure 1D). Of the differentially expressed genes, 2466 were upregulated and 2110 were downregulated in patients with IBD (figure 2A: unique and shared features in differentially gene expression; figure 2B: top 200, ranked by fold change after applying a cutoff of p≤0.01). The top 50 genes (ranked by fold change after applying a p value cutoff of 0.01) were differentially regulated exclusively in either of the two IBD subtypes and are displayed in figure 2C (CD) and 2D (UC).

Principal component analysis (PCA) of mRNA expression levels. The first two components shown explain the largest part of the variation in mRNA expression. Individual insets are colour-coded by tissue (A), inflammation status (B) and diagnosis (C). (D) Variance explained by the first 20 PCA components.
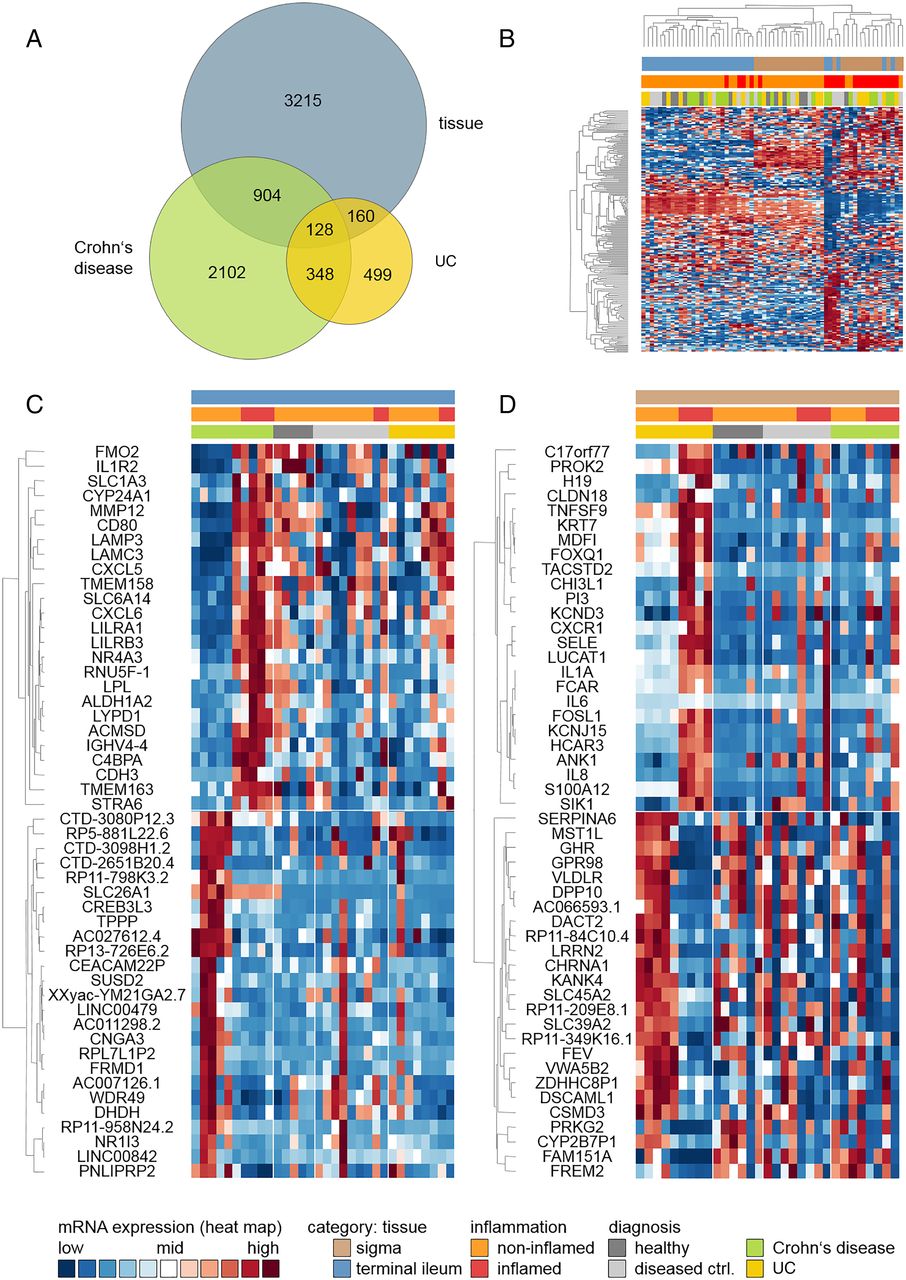
Hierarchical clustering of genes differentially expressed in IBD, Crohn's disease (CD) or UC. Hierarchical clustering was based upon relative expression levels. Each row corresponds to a single gene, whereas each column corresponds to a single sample. (A) Venn diagram illustrating overlaps and unique features identified in different pair-wise comparisons (tissue: terminal ileum vs sigmoidal colon; CD inflamed vs non-inflamed; UC inflamed vs non-inflamed); (B) Clustering of top 200 genes differentially expressed between tissue type, inflammation status or IBD subtype; (C) Clustering of top 50 genes differentially expressed in CD only and (D) clustering of top 50 genes differentially expressed in UC only. Genes were ranked by fold change after applying a p value cutoff of 0.01. Genes are labelled with gene symbols.
The KEGG pathway analysis identified several functional categories comprising differentially expressed genes, including overexpression of adhesion molecules and of genes related to fatty acid biosynthesis and natural killer cell-mediated cytotoxicity (figure 3A) as well as underexpression of genes related to spliceosome assembly and nicotinate (NA)/nicotinamide (NAM) metabolism (figure 3B).
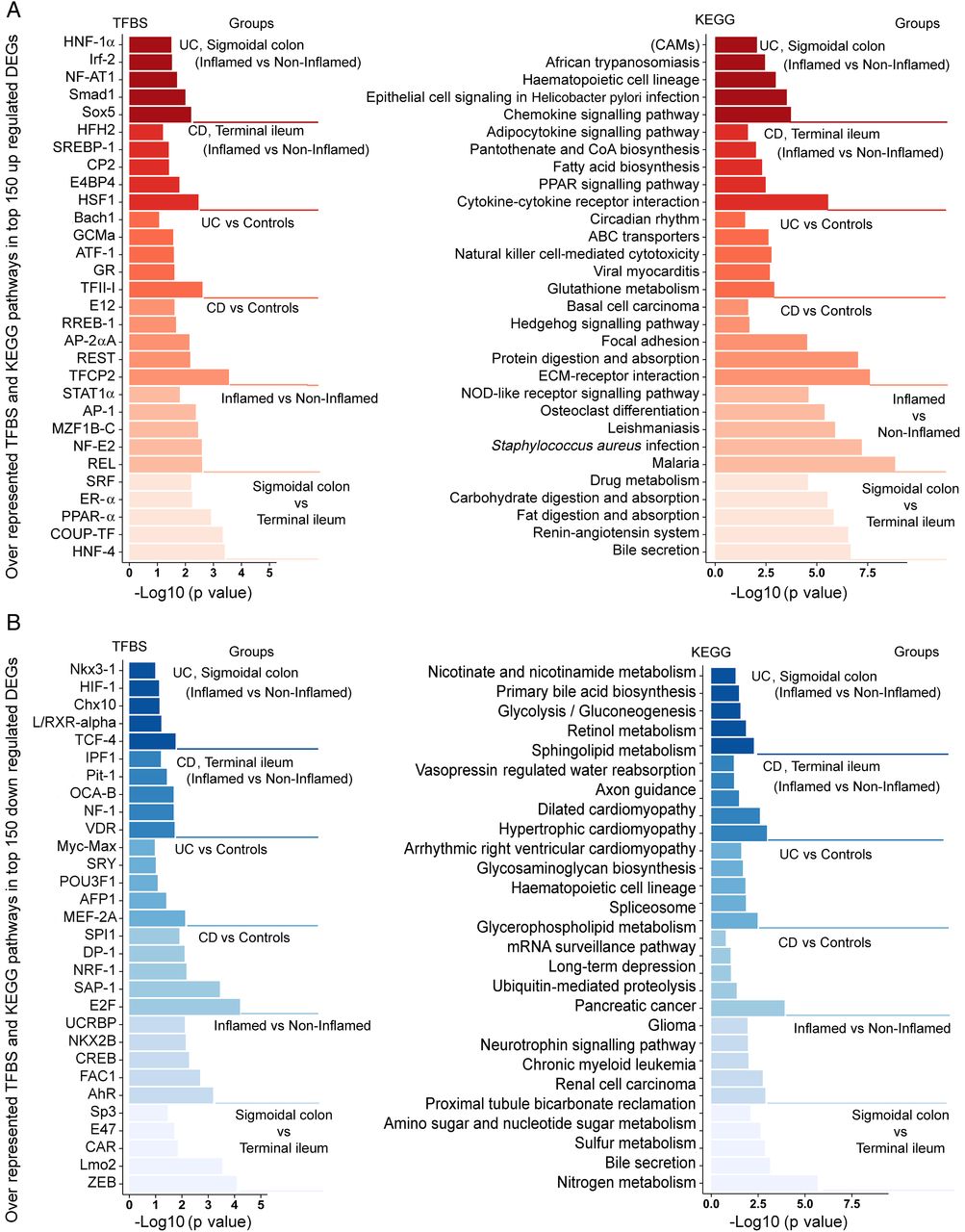
Transcription factor binding side and the KEGG pathway analysis of genes differentially expressed in IBD. Transcription factor binding sites (left panel) and the KEGG pathway categories (right panel) were identified for the top 150 genes upregulated (A) or downregulated (B) in IBD. The differentially expressed genes included origin from the pair-wise comparisons of the tissue type (sigmoidal colon vs terminal ileum), inflammatory stage (inflamed vs non-inflamed) and diagnostic status (Crohn's disease (CD) terminal ileum inflamed vs non-inflamed, UC sigmoidal colon inflamed vs non-inflamed).
Supplementary table
Transcription factor analysis inferred signal transducer and activator of transcription 1 (STAT1) (figure 3A) and nuclear factor-κB (NF-κB) reticuloendotheliosis proto-oncogene, NF-kB subunit (REL) binding motifs as major inflammation-associated sites, regardless of diagnosis, whereas heat shock transcription factor 1 (HSF1) and Sox5 binding motifs were the most prevalent sites for CD and UC, respectively. Binding sites for aryl hydrocarbon receptor (AhR) and fetal Alzheimer antige clone 1 (FAC1) were significantly depleted in genes downregulated by non-specific inflammation. For further information, see figure 3B.
Reconstruction of context-specific metabolic networks and host metabolic network coherence in IBD
Models comprising between 2235 and 2668 metabolic reactions were generated and analysed using classical data mining and a previously described metabolic network coherence approach.36 ,37 Comparing the number of reactions found in networks reconstructed from samples of non-inflamed versus inflamed tissues, we observed a significant decrease of inferred metabolic activity in the latter (FDR-corrected p value=4.0×10−2), with 8 of 12 altered pathways displaying a significant decrease of inferred activity (table 3). Furthermore, metabolic coherence values (M) measuring the connectedness of expression level alterations in a predefined network (Recon2.04) differed substantially between groups defined by tissue type, inflammation status and disease type (see online supplementary figure S1). The differential expressed genes originating from the comparison of inflamed and non-inflamed regions of the terminal ileum for CD, resulted in a positive metabolic network coherence (M=0.74). The effective network consisted of a single large and dense component in addition to several smaller network fragments (see online supplementary figure S1B), pointing towards a rather specific change in metabolic activity. Similarly, differentially expressed genes originating from the comparison of samples from inflamed and non-inflamed sigma regions in cases of UC yielded a high metabolic network coherence (M=1.44). Again, one cluster dominated the effective network (see online supplementary figure S1C). The contextualisation of these metabolic networks by way of gene ontology analysis highlighted the associated metabolic functions and processes (see online supplementary figure S2).
Metabolic pathways significantly differentially regulated in inflamed versus non-inflamed tissues
Supplementary figures
Alternative splicing in IBD
The proteomes of both healthy and diseased tissues attain an extra level of complexity by alternative splicing that may fall into one of four major categories (figure 4A): intron retention, exon skipping (ES), ALTA and ALTD. To reflect the distribution of splicing events, we further categorised them with respect to their relative occurrence in the corresponding sample groups (events occurring in either 100%, 90%, 50% or 10% of all samples, figure 4B). Only high-quality splicing events that occurred in at least 90% of all samples per experimental group were subjected to further analysis. Note that the sensitivity of alternative splicing event detection by way of RNA sequencing is heavily dependent upon the read coverage across samples.
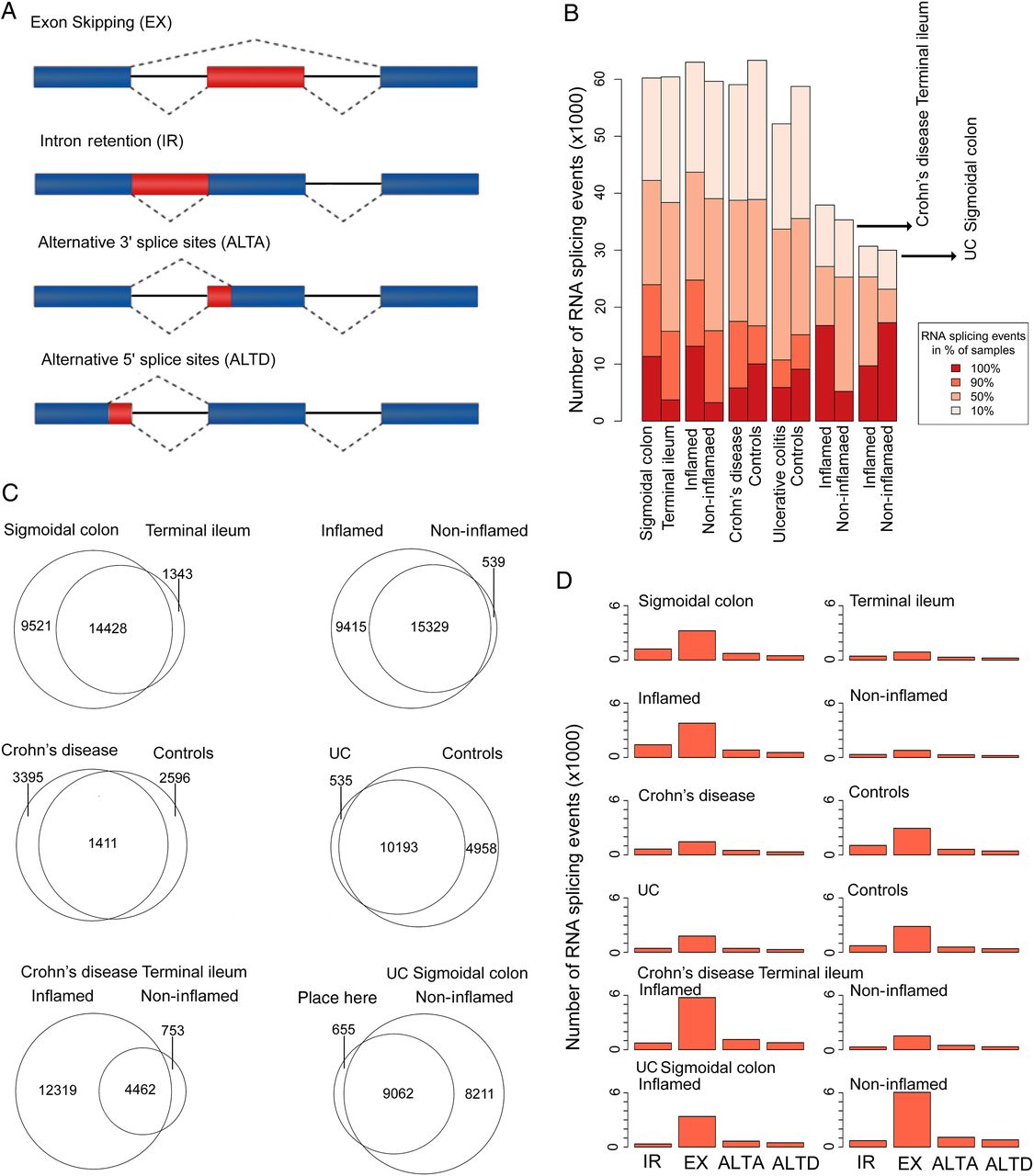
Alternative splicing in relation to tissue type, inflammation status and IBD subtype. (A) Types of alternative splicing events; (B) distribution of alternative splicing events of different prevalence, stratified by group-wise comparison type (tissue, diagnostic status, inflammation); (C) Venn diagram of alternative splicing events shared or unique in different group-wise comparisons (only high-quality events detected in at least 90% of IBD biopsies included). (D) Distribution by alternative splicing type detected in different group-wise comparisons (only high-quality events that occurred in at least 90% of all samples included). IR, intron retention; EX, exon skipped; ALTA/ALTD, alternative splice-site acceptor/donor.
The nature of the alternative splicing events detected in individual sample groups is summarised in figure 4C, highlighting unique and shared events for all pair-wise group comparisons. Briefly, tissue difference accounted for 25 292 alternative splicing events, while 25 283 events were attributable to general inflammation status. At the individual disease level, 17 534 alternative splicing events were associated with inflammation in CD and 17 928 events occurred in UC. Across all samples, ES was found to be the most prevalent event (figure 4D). Simultaneous consideration of tissue type, inflammation stage and diagnosis identified 320 shared alternative splicing events (see figure 5A and online supplementary table S3). The KEGG pathway analysis of genes linked to these 320 events revealed a significant enrichment of a number of different categories (figure 5B), including bacterial invasion of epithelial cells. Spearman's correlation analysis of PSI/PIR/PSU values and the respective gene expression levels revealed only weak relationships, suggesting that splicing occurs largely independent of gene regulation (figure 5C).

Differentially regulated splicing events between tissue types, inflammatory stages and IBD subtypes. (A) Distribution of differentially regulated splicing events found in different comparisons (tissue type: sigmoidal colon vs terminal ileum; inflammatory stage: inflamed vs not-inflamed; diagnosis status: Crohn's disease, terminal ileum inflamed vs non-inflamed and UC, sigmoidal colon inflamed vs non-inflamed) in IBD biopsy samples (differentially regulated splicing events were considered to have Δ|(PSI/PIR/PSU)|≥10 between two conditions in each pair-wise comparison). (B) The KEGG pathway enrichment categories for all genes linked to splicing events in (A) that were observed to be differentially upregulated/downregulated in IBD biopsy samples. The x-axis indicates their associated enrichment p value for each category. (C) The distribution of Spearman's rank correlation coefficients between PSI/PIR/PSU of differentially regulated splicing events and their corresponding gene expression values that are upregulated/downregulated or not regulated across IBD biopsy samples. p Values were generated employing a Wilcoxon rank-sum test. PSI, percentage spliced in; PIR, percentage intron retention; PSU, percentage splice site usage; IR, intron retention; EX, exon skipped; ALTA/ALTD, alternative splice-site acceptor/donor.
Supplementary table
Subtle alteration of the mucosa-attached microbiome associated with IBD
Analysis of mucosa-attached active bacterial communities resulted in a total of 1 918 235 16S rRNA amplicon reads, with an average of 32 517 sequence reads per sample. For quality reasons, three samples were excluded from further analysis. Some 19 phyla and 3120 OTUs were detected, including the most dominant phyla Firmicutes (62.23%), Bacteroidetes (26.62%), Proteobacteria (8.71%) and Actinobacteria (1.51%) (see online supplementary figure S3; for the full dataset and online supplementary table S4). Similar to other studies at the RNA level, bacterial community richness and diversity did not differ significantly between patients with CD or UC, respectively, and controls. Only a small number of OTUs and low Shannon diversity were observed in patients with CD (see online supplementary figure S4A: terminal ileum and online supplementary figure S4B: sigmoidal colon), while multivariate analysis Permutational Multivariate Analysis of Variance (PERMANOVA) of abundance-based (Bray-Curtis) and non-abundance-based (Jaccard) distance matrices revealed significant differences between patient and control microbial communities. Regardless of tissue type (terminal ileum or sigma), inflammation status was significantly associated with altered microbiota. Principal coordinate analysis of the two distance matrices showed seemingly distinct clusters, based upon diagnosis, while clustering based upon tissue or inflammation status was not evident (see online supplementary figure S5). These observations support the view that active mucosa-attached microbiota are different in healthy individuals and patients with IBD, and that they are also significantly altered by inflammation status.
Supplementary table
Co-abundance analysis of microbiome and host transcriptome
As a measure of host-microbiome crosstalk, we correlated the expression level of differentially expressed genes with the abundance of bacterial OTUs, revealing 5988 significantly correlated pairs (FDR≤0.05). These pairs resulted from six different correlation analyses, namely for terminal ileum samples of (i) healthy individuals, (ii) disease control individuals and (iii) patients with CD, and for sigmoidal colon samples of (iv) healthy individuals, (v) disease control individuals and (vi) patients with UC. Inspection of the top 500 correlations for each tissue type (ranked by a combination of Spearman's rank correlation coefficient and FDR) revealed a trend from healthy individuals (318 significantly correlated pairs) via disease controls (155 pairs) to patients with CD (terminal ileum only: 31 pairs). With the exception of sigmoidal colon from patients with UC and non-IBD controls (p=0.523), all other Spearman's rank correlation coefficients were found to differ statistically significantly between patient/tissue groups (p<10−5). The majority of the top 200 differentially expressed genes (ranked by fold change after applying a p value cutoff of 0.01) also showed a correlation to bacterial OTUs (186 of 200 in CD and 194 of 200 in UC). With both diseases, only three bacterial families accounted for over 70% of all disease-specific significant correlations between abundance and transcription level, namely Lachnospiraceae, Bacteroidaceae and Ruminococcaceae. Assigning the involved genes to functional categories showed that, in patients with CD, the majority of the correlations (12 of 31) were attributable to one of two categories: chemotaxis and inflammation. In patients with UC, a single bacterial OTU, which originated from the Bacteroidetes phyla, accounted for 40% of these correlations and paired with host transcripts from all functional categories (figure 6). Finally, functional classification of those genes that were involved in at least one significant correlation revealed that 208 of 1507 genes were functionally related to splicing and RNA processing. On the other hand, a total of 390 genes actually showed signs of alternative splicing. Similarly, for UC, 431 of 2286 categorised genes were functionally related to splicing and RNA processing, 617 genes were identified actually to be alternatively spliced. Taken together, these findings indicate a notable disease-associated loss of interaction between the host transcriptome and the mucosal microbiota.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Loss of host-microbiome interaction. Host-microbiome interaction was quantified by Spearman's rank correlation coefficient between relative host transcript amount (mRNA) and operational taxonomical unit (OTU) count. Only mRNA-OTU pairs with a significant correlation (false discovery rate (FDR)≤0.05) are shown. Spearman's rank correlation coefficients are colour-coded, whereas the circle size depicts the FDR. (A) Terminal ileum in healthy individuals; (B) terminal ileum in diseased controls; (C) terminal ileum in patients with Crohn's disease; (D) sigmoidal colon in healthy individuals; (E) sigmoidal colon in diseased controls; (F) sigmoidal colon in patients with UC. Host gene transcripts are grouped by functional category based upon gene ontology terms. Genes that are part of a metabolic network are labelled with by the corresponding gene symbol. OTUs were categorised by associated bacterial class.
Discussion
In our study, we jointly investigated three closely related OMICs layers, namely host gene expression level, host transcript splicing pattern and mucosa-associated active microbiota, in the context of IBD. We described for the first time how these three components interact quantitatively and how the interactions are altered in the presence of IBD. Our key finding was a significant uncoupling of host gene expression and microbial signature in patients with IBD that was accompanied by substantial changes in splicing patterns. These findings seem to be an IBD-specific phenomenon because, in the disease control group available to us (mainly infectious acute non-IBD intestinal inflammation), a much weaker uncoupling was observed than in patients with IBD.
Before alluding in more detail to the genome-wide aspects of our study, we will discuss several selected host gene sets in order to put our results in the context of previous findings. Of course, this highlighting of individual genes does not mean to suggest that the respective results are necessarily of higher relevance than the network-based findings discussed later.
Several of the genes found to be upregulated or downregulated in our samples from patients with IBD have been addressed in previous studies. Many of them were thus shown to be of pathophysiological importance, including interleukin-1 receptor type II, interleukin 1α, interleukin-6 (IL6) and interleukin-8, all of which are key regulators of inflammation in IBD.38 Similarly, we observed an upregulation of claudin 18 in UC samples, which corroborated similar findings obtained before irrespective of disease severity, thereby suggesting an epithelial defect underlying the disease.39 The KEGG pathway enrichment analyses pointed towards a functional role of differentially regulated genes associated with chemokine signalling, natural killer cell-mediated cytotoxicity, NOD-like receptor signalling, drug metabolism or spliceosome assembly, all of which are highly relevant in the context of microbiota-associated inflammation and the development of IBD. At the same time, we also observed many known IBD-relevant genes to be alternatively spliced in IBD. However, this does not mean that alternative splicing is causally related to changes in gene expression. In fact, in concordance with a previous study,40 we found only a weak correlation between alternative splicing and differential gene expression. Genes found in our study to be alternatively included autophagy-related 16-like 1, forkhead box P1, various interleukins (IL22RA1, IL10RA/B, IL17RA/B/C/E, IL1R1/2, IL2RG, IL4R, IL6R, IL6ST, IL18, IL32) and dual oxidase 2 (DUOX2). Interestingly, mutations in DUOX2 and in splicing elements of other NAM adenine dinucleotide phosphate oxidase genes have been linked to very early onset IBD before, suggesting a direct link between altered splicing of a risk gene and susceptibility to IBD.41 Transcription factors reflect key regulatory elements of transcriptional response. The inferred transcription factor binding sites we identified here point towards inflammation pathways that overlap between IBD subentities, for example, NF-κB, activator protein 1 and STAT1-α, and also invoke potentially relevant unique targets, for example, Nuclear factor of activated T-cells 1 (NF-AT1) and SMAD (small mothers against decapentaplegic) family member 1 (SMAD1) (enriched in upregulated transcripts in UC inflamed tissue only) or vitamin D receptor (uniquely enriched in downregulated transcripts in CD inflamed tissue).
We next tried to interpret the complex individual relationships between different data layers from a systems biology perspective by mapping our transcriptome data to a predefined model of cellular metabolism. Although a metabolic component of mucosal inflammation can be assumed to be present,42 metabolome-wide approaches to contextualise gene expression profiles in IBD are still missing. Importantly, the employed model contains information on the directionality of flux, which may be used to infer cause-effect relationships without knowing exact metabolite levels. The role of cellular metabolism in regulating the gut microbiome is receiving increasing attention and has recently been suggested to play a more important role in shaping the gut microbiome than the immune system itself.43 Coherence analysis showed a striking ‘connectedness’ (ie, high coherence) of transcriptome changes in IBD, particularly for subnetworks highlighting only downregulated genes. At the level of individual pathways, notable regulatory changes included a decreased biosynthesis of bile acids and an inferred downregulation of the short-chain fatty acid propanoate metabolism. Both metabolites are key components of host-microbiome crosstalk. The inferred decrease in propanoate metabolism indicates that this metabolite may be produced in smaller quantities by the microbiome, which could drive inflammation due to its anti-inflammatory effect.44 Bile acids, in turn, have previously been implied as key regulators of microbial composition.45 Hence, a reduction of their synthesis would point towards a potential causal mechanism underlying the breakdown of microbiota-host interactions as observed in our IBD samples. The upregulation of tryptophan metabolism represents another hallmark of inflammation.46 Our results indicate that the previously reported reduction of serum tryptophan levels in IBD47 may be due to an active upregulation of tryptophan degradation in the intestine during inflammation. A critical pathway downstream of tryptophan breakdown, which constitutes further processing of NAM/NA, is downregulated in the colonic mucosa of patients with UC and CD, which may explain the low level of NAM vitamin observed in the serum of patients with IBD.48 Indeed, countering tryptophan/NAM depletion by dietary supplementation of NAM has been found to reduce inflammation in a murine model of IBD.49
Finally, we made the striking observation that the vast majority (>90%) of genes differentially expressed in IBD and controls showed a significant correlation of their expression level to the abundance of microbial taxa only in the healthy state. The correlation was slightly lower in cases of non-IBD inflammation but was almost completely lacking in patients with IBD. This gradient may point towards an uncoupling of the host and intestinal microbiota, which has already been suggested before by a small study of monozygotic twins discordant for IBD.18 Likewise, the disease-associated gradient in crosstalk may also assign functional relevance to the correlated bacterial OTUs. The finding that three bacterial families accounted for over 70% of the residual host-microbiota correlations in patients with IBD, namely Lachnospiraceae, Bacteroidaceae and Ruminococcaceae, supports their potential role in disease pathogenesis.
It is clearly not possible to draw safe functional conclusions from our rather preliminary observations. Naturally, clinical samples exhibit a high degree of variation. In addition to inter-individual differences such as genotype and medication, altered cell compositions in biopsies can also contribute to this variation. We have tried to allow for these imponderabilities by appropriate sampling and a choice of robust statistical methods. However, it cannot be excluded, for example, that the proportion of one or the other type of immune cells differed between our inflamed IBD and non-IBD disease control biopsies or some signals were even lost due higher sample-to-sample variability introduced by the different medications. This notwithstanding, taken together, our results highlight a close interaction between microbiota, gene regulation and splicing architecture of the host mucosal transcriptome, which is only partially impeded in acute non-IBD inflammation, but significantly reduced in chronic IBD. Unravelling the precise nature of this interaction will be a challenge for future research, but the findings presented here emphasise that the intestinal meta-organism comprising microbiota and host mucosa has to be regarded as a joined entity to allow a better understanding of the aetiology of IBD.
Acknowledgments
The authors would like to thank all the participating patients for supporting this study by donating biomaterial. The expert technical assistance by Dorina Oelsner, Karina Greve as well as the support by Markus Schilhabel is greatly acknowledged.
References
Footnotes
RH and RS-T contributed equally. SS and PR share senior authorship. AS, MB and AR should be considered as equal second authors.
Contributors RH, RS-T and PR analysed the data, drafted and revised the manuscript; AS, MB, AR, DE, KA, CK, BB, SN, SaS, CK, AF and CF collected and analysed the data; WM, M-TH, MK, StS and PR contributed the conception of the work; all authors approved the final version of the manuscript.
Funding This work was funded by the BMBF as part of the e:Med framework (‘sysINFLAME’, grant 01ZX1306), the Cluster of Excellence ‘Inflammation at Interfaces’ (ExC 306), SysMedIBD EU FP7 under grant agreement n° 305564 and DEEP IHEC (TP2.3 and 5.2, BMBF).
Competing interests None declared.
Ethics approval The Ethics Committee of the Medical Faculty of Kiel University, Kiel, Germany.
Provenance and peer review Not commissioned; externally peer reviewed.