Article Text

Abstract
Objective Estimates of cancer risk and the effects of surveillance in Lynch syndrome have been subject to bias, partly through reliance on retrospective studies. We sought to establish more robust estimates in patients undergoing prospective cancer surveillance.
Design We undertook a multicentre study of patients carrying Lynch syndrome-associated mutations affecting MLH1, MSH2, MSH6 or PMS2. Standardised information on surveillance, cancers and outcomes were collated in an Oracle relational database and analysed by age, sex and mutated gene.
Results 1942 mutation carriers without previous cancer had follow-up including colonoscopic surveillance for 13 782 observation years. 314 patients developed cancer, mostly colorectal (n=151), endometrial (n=72) and ovarian (n=19). Cancers were detected from 25 years onwards in MLH1 and MSH2 mutation carriers, and from about 40 years in MSH6 and PMS2 carriers. Among first cancer detected in each patient the colorectal cancer cumulative incidences at 70 years by gene were 46%, 35%, 20% and 10% for MLH1, MSH2, MSH6 and PMS2 mutation carriers, respectively. The equivalent cumulative incidences for endometrial cancer were 34%, 51%, 49% and 24%; and for ovarian cancer 11%, 15%, 0% and 0%. Ten-year crude survival was 87% after any cancer, 91% if the first cancer was colorectal, 98% if endometrial and 89% if ovarian.
Conclusions The four Lynch syndrome-associated genes had different penetrance and expression. Colorectal cancer occurred frequently despite colonoscopic surveillance but resulted in few deaths. Using our data, a website has been established at http://LScarisk.org enabling calculation of cumulative cancer risks as an aid to genetic counselling in Lynch syndrome.
- CANCER GENETICS
- CANCER PREVENTION
- CANCER SYNDROMES
- COLONOSCOPY
- COLORECTAL CANCER
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Inherited colorectal cancer may be caused by mismatch repair gene mutations and is then commonly referred to as Lynch syndrome.
Lynch syndrome is under-recognised and results in about 0.1% of the population having a significantly increased risk of early onset colorectal, endometrial and ovarian cancer.
Endoscopic surveillance with removal of precursor adenomas is recommended to prevent colorectal cancer.
What are the new findings?
This is the first comprehensive prospective study to provide empirically observed data on colorectal cancer incidence and survival in Lynch syndrome.
Colorectal cancer occurred despite colonoscopic surveillance with removal of adenomas.
Colonoscopic surveillance with early detection and treatment of invasive colorectal cancer was associated with excellent survival. Survival after first endometrial or ovarian cancer was also excellent.
Revised estimates of the different penetrance and expression patterns in carriers of MLH1, MSH2, MSH6 and PMS2 mutations.
Introduction
Lynch syndrome (LS) is associated with a high probability of GI, gynaecological and other cancers. It is caused by inherited mutations affecting any of four DNA mismatch repair (MMR) genes, MSH2, MLH1, PMS2 or MSH6, or by a deletion in the EPCAM gene, which leads to methylation of the adjacent MSH2 promoter. It is an under-recognised condition accounting for about 1–3% of colorectal cancers (CRCs) in the population.1 To date, most LS patients have been identified following investigation because of their family or personal histories of multiple and/or early-onset cancers.
Carriers of pathogenic MLH1, MSH2, MSH6 or PMS2 mutations require reliable information about their future cancer risk so that they can be offered appropriately targeted surveillance, but published risk estimates are extremely variable. One obvious factor is reliance on retrospective data. Another is the impact of initial selection criteria for molecular testing. In clinical practice, these have included the Amsterdam I or Amsterdam II criteria, the Bethesda guidelines or simply age at cancer diagnosis. Previous estimates of the cumulative risk at 70 years for CRC in MLH1 or MSH2 mutation carriers range from 22% to 74%. Mutations in MSH6 and PMS2 genes have lower penetrance and different patterns of expression: MSH6 mutation carriers are thought to have a high risk of endometrial cancer, similar to that in MSH2 mutation carriers, but lower risks of CRC. For a comprehensive and updated overview of literature, see two recently published reviews by us and others.1 ,2
Colonoscopy enables the identification and removal of preinvasive neoplasia or early cancers in the absence of symptoms, and is the mainstay of secondary prevention in LS patients. Although adenoma removal is considered to represent a surrogate for the prevention of CRC and death, the evidence supporting this assumption in LS is controversial.1–11
The European ‘Majorca group’ (http://mallorca-group.eu) and colleagues in the International Society for Gastrointestinal Hereditary Tumours (http://insight-group.org) have developed a pooled prospective database of LS mutation carriers to better characterise their cancer risks and the effects of interventions. This first report focuses on the cumulative incidence of first cancers and associated survival.
Patients and methods
The LS prospective database
Data were stored and organised as an Oracle relational database adapting the core structure of CGEN.12 This structure has the capability for later addition of other information and classes of information to the patients already filed and for inclusion of new patients. Data were manipulated by TOAD inside Oracle and exported by TOAD to SYSTAT13 and Excel for further manipulations and statistical calculations.
The variables used for this first report were age at inclusion for follow-up, gender, age at most recent observation, age at death, mutated gene, age at diagnosis of any cancer, age at complete surgical removal of organs and which preventive modalities had been applied. Cancer diagnoses were scored by using the first three positions in the International Classification of Disease, ninth revision system. Ages were scored as integers.
Patients and interventions
Each centre had identified each patient to be at increased risk of CRC according to internationally recognised guidelines1 ,2 or local adaptations of these. Patients had then been subject to follow-up by colonoscopy and modalities for early detection of endometrial and ovarian cancer, and mutational analysis of the MMR genes. All patients in this study were proven or obligate carriers of pathogenic mutations as judged by the reporting centre in the MLH1, MSH2, MSH6 or PMS2 genes at the time of reporting. EPCAM mutations that lead to methylation of the adjacent MSH2 promoter were included and scored as MSH2 mutations. The mutations were assumed to be germline, regardless of when they were identified. All mutations reported in the 1942 patients were searched for in the Leiden open variant database (LOVD) database (http://chromium.lovd.nl/LOVD2/colon_cancer/) during October 2015: 1310 patients (67%) had pathogenic (class 5) mutations, 28 patients (1%) had probably pathogenic (class 4) mutations and the remaining 604 were not reported in LOVD.
All analysed observations were prospective, commencing when the patients were subjected to their first prospectively planned colonoscopy after being identified as at risk for colon cancer. For the purpose of this report, cases with any cancer prior to or at the same age as first colonoscopy (prevalent cancers) were excluded, as were all cases with <1 year of prospective observation time. This was done to avoid selection bias based on ascertainment and to ensure that no patient had any sign or symptom of cancer at inclusion.
The surveillance guidelines included follow-up aimed at diagnosis of colorectal adenomas or early CRC and in many centres endometrial cancer and ovarian cancer, as well as cancer awareness for all cancers known to be associated with LS. Surveillance and management guidelines have changed over time, and collaborating centres were subject to local/national decisions on how to practise at different times. None of these variations were used as variables in the present study. A detailed, referenced description of follow-up and compliance is provided in online supplementary table S2. The table and the references included there show that from the outset the reporting centres used different intervals between colonoscopies, but that from around 1996 onwards all except for the Finnish centre followed the emerging international guidelines advocating a 2-year interval or less. Intervals between gynaecological examinations were in general shorter. As previously published in the references given in the table, all visible adenomas at colonoscopies were removed. The references also show that precursor lesions were less frequently found in the endometrium or ovaries. In short, secondary prevention of colon cancer by identifying and removing precursor/early lesions was found to be promising, while this was not the case for endometrial and ovarian cancer. In consequence, all centres continued the colonoscopic surveillance, while some advised prophylactic hysterectomy and oophorectomy to prevent gynaecological cancers. All patients reported to the database had complete data sets, and there were no missing values.
Supplementary materials
Some centres had previously reported the observed incidence of cancer in their series but with different methods to those used in this report.3–11 One group had reported previously on survival.4 The intention of this report was to compile all information available on prospectively observed outcomes in LS patients without previous cancer and patients who were previously reported are included in the current report.
Annual and cumulative incidence rates
Each patient was observed from age at inclusion to age at last observation or age at first cancer, whichever came first. Each patient was counted only once, irrespective of how many synchronous cancers the patient might have had as first cancers. Cancers that occurred after the first cancer were not considered in this study. Sine surveillance before 25 years and after 70 years of age is not specified in current management guidelines and is inconsistent in practice, annual incidence rates (AIRs) outside 25–70 years were not calculated. For each patient, the numbers of years observed in each 5-year interval from 25 to 70 years of age were counted. All first cancers were scored according to age at diagnosis. The AIR for a given age group was derived by dividing the number of cancers observed by the sum of observation years in that age group.
Cumulative incidences for gynaecological cancers and prostate cancers were considered for appropriate genders separately, and observation time was censored at hysterectomy and oophorectomy when considering incidences for such cancers separately.
Cumulative incidence, denoted by Q, was computed starting at age 25, assuming zero incidence before age 25, using the formula Q(age)=Q(age−1)+[1−Q(age−1)]·AIR(age), where AIR (age) is the AIR as estimated from the corresponding 5-year interval.
SE for AIR was estimated as SEAIR=sqrt[AIR·(1−AIR)/Yrs], where Yrs denotes the number of observation years in the 5-year age group for which AIR is estimated. For cumulative incidence, the hazard rate H=−ln[1−AIR] was used with SE estimated SEH=SEAIR/(1−AIR). The SE, denoted by SEQ, of the cumulative incidence Q(age) up to the given age is computed in two steps. First, for each 5-year age interval, having hazard rate H with SE SEH, the contribution to the cumulative hazard from that interval is N·H with SE N·SEH, where N is the number of years from that 5-year interval: for example, the cumulative incidence up to age 32 contains all 5 years from the 25–29 age interval, but only 3 years from the 30–34 age interval. The accumulated hazard CH is computed by adding the N·H values across age intervals, while the corresponding SE, SECH, is found by setting SECH2 equal to the sum of (N·SEH) across age intervals. The accumulated hazard rate CH should now equal −ln[1−Q] with Q as computed above, while the SE of the cumulative incidence is computed as SEQ=SECH(1−Q). We estimated 95% CIs as AIR±1.96 SEAIR and Q±1.96 SEQ.
Survival
Follow-up continued after the occurrence of first cancers, and all patients were either reported to be alive or validated to be alive in population register on the day each patient was censored. Crude survival was calculated by the Kaplan–Meier algorithm as time from first cancer to last observation/death. Cancer stages at diagnosis and causes of death were not considered in this report.
Results
Patients included and cancers diagnosed
In total, 1942 MMR mutation carriers were included: 944 MLH1, 616 MSH2, 305 MSH6 and 77 PMS2. Of these, 1057 were females and 885 were males. They were observed for a total of 13 782 years (mean observation time 7.1 years), including 6518 male and 7264 female observation years. Countries of origin are detailed in table 1.
Numbers of patients included, observation years, mean observation years and mean age at inclusion for total series, by country of origin and by gene and gender
Mean ages at inclusion were 35.6 years for MLH1 mutation carriers, 37.7 years for MSH2 mutation carriers, 43.0 years for MSH6 mutation carriers and 47.1 years for PMS2 carriers. Observation years were 7954 for MLH1 mutation carriers, 4021 MSH2, 1555 MSH6 and 313 for PMS2. Details when stratifying on both gene and gender are given in table 1. The numbers were considered sufficient to stratify the findings on gender, mutated gene, 5-year age cohorts and cancers in specific organs. Although the numbers of PMS2 mutation carriers were limited, they are included in all results presented.
Cancers diagnosed
Among the 1942 patients, 314 had prospectively identified first cancers (table 2), of which 151 were colorectal, 72 endometrial and 19 ovarian. Of note, 21 patients had two synchronous first cancers.
First cancers prospectively diagnosed by International Classification of Disease, ninth revision (ICD-9) diagnoses, groups of diagnoses and stratified on gender in Lynch syndrome (LS) patients without prior or prevalent cancer at first colonoscopy
In total, 186 females had had their uterus removed and 153 had had removal of the ovaries prior to any cancer diagnosis, reducing observations years when calculating the AIR for endometrial and ovarian cancer (see online supplementary table S1).
Supplementary materials
Cancer incidences
Calculated cumulative incidence for any cancer at age 70 years was 75% in females and 58% in males. This sex difference was evident from 50 years of age onwards. Sex-specific cumulative cancer incidence is detailed in table 3.
Calculated sex-specific cumulative cancer incidences at 40, 50, 60 and 70 years for Lynch syndrome (LS) patients in the total series in LS patients without prior or prevalent cancer at first colonoscopy
The calculated cumulative cancer incidence by age 70 years was high in MLH1 and MSH2 (72%) carriers and lower (at borderline significance) in MSH6 (54%) and PMS2 (18%) carriers. The calculated cumulative incidence of cancer at age 70 years for LS patients from different current ages onwards and different mutated MMR genes are given in table 4.
Calculated cumulative incidences for any cancer from 25, 40, 50 and 60 years of age (current age) to 70 years of age for Lynch syndrome (LS) patients by mutated gene, in LS patients without cancer at current age in LS patients without prior or prevalent cancer at first colonoscopy
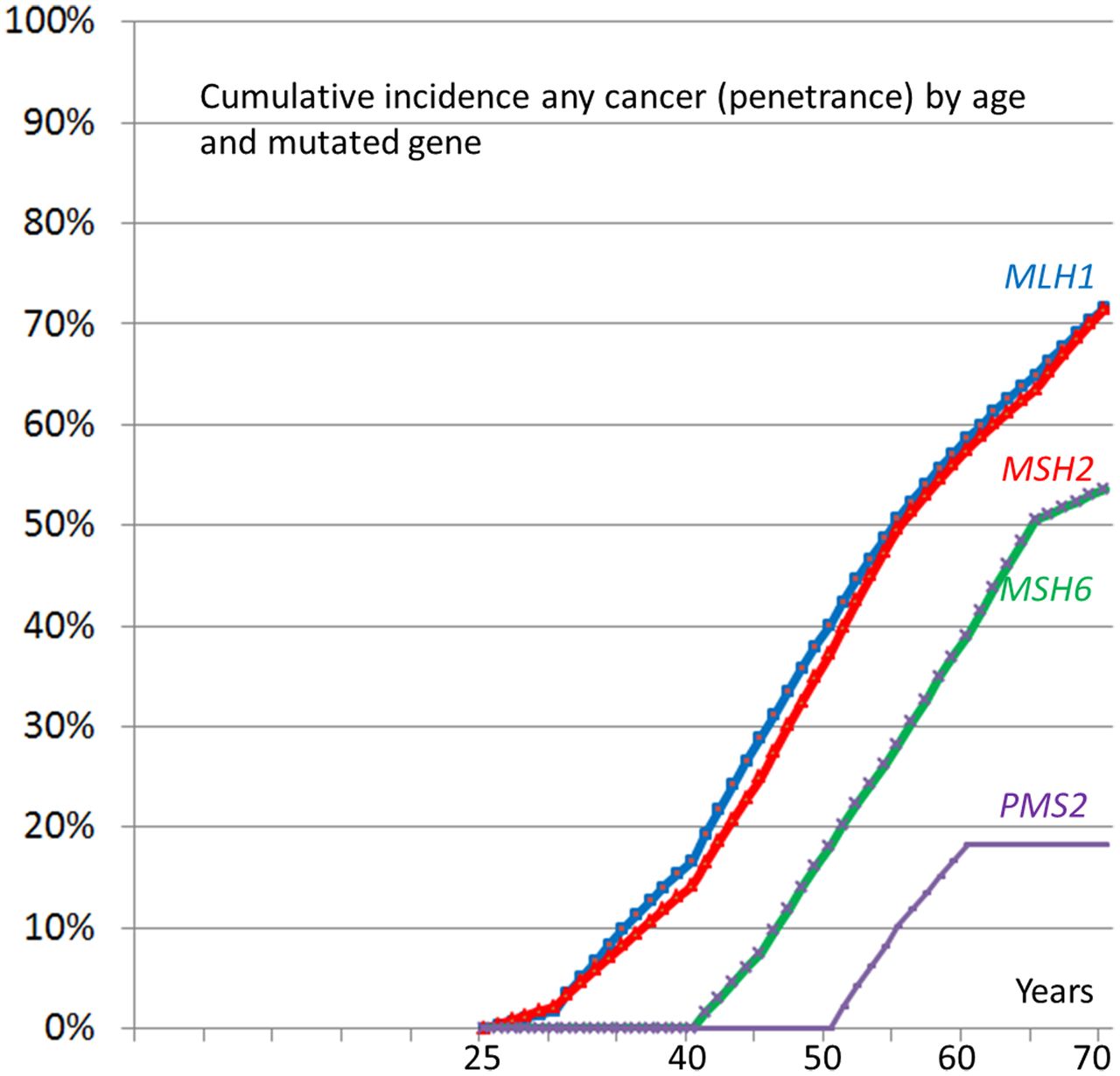
Figure 1 shows the calculated complete distributions of time to any first cancer from 25 to 70 years of age by gene.

Calculated cumulative incidences by age and mutated gene for any cancer.
Table 5 shows cumulative cancer incidences from 25 years to different greater ages by mutated MMR gene, cancer type and gender. CRC cumulative incidence was high in MLH1 (46%) and MSH2 (35%) mutation carriers, lower in MSH6 (20%) and lower PMS2 (10%) mutation carriers. Endometrial cancer cumulative incidence was high for MLH1 (34%), MSH2 (51%) and MSH6 (49%) mutation carriers, and this cancer was also frequent in older female PMS2 mutation carriers (24%). Ovarian cancer was only identified in MLH1 (11%) and MSH2 (14%) mutation carriers and most cases were diagnosed before age 50. Upper GI and urinary tract cancers were infrequent and mostly diagnosed after age 60. In all, cancers were frequently seen in MLH1 and MSH2 mutation carriers from 25 years onwards but not before 40 years in MSH6 and PMS2 mutation carriers.
Calculated cumulative cancer incidences from 25 to 40, 50, 60 or 70 years of age for any cancer and for selected groups of cancers occurring as first cancer, by gender and by mutated gene
Figures 2 and 3 show the calculated complete cumulative distributions from 25 to 70 years of age for CRC and endometrial cancer, respectively, as the first cancer by gene, corresponding with the rows for CRC and endometrial cancer in table 5. To facilitate genetic counselling and clinical practice, an interactive website providing the complete distributions of all cancer groups in table 5 from any age selected by the user and counting to any greater age is available at http://www.lscarisk.org.

Calculated cumulative incidences by age and mutated gene for colorectal cancer (CRC) as the first cancer.

{kind=link}
{kind=link}
{kind=link}
Calculated cumulative incidences by age and mutated gene for endometrial cancer as the first cancer by gene.
Time since last colonoscopy to CRC
Table 6 demonstrates time since last colonoscopy in the 145 cases with CRC. Mean time since last colonoscopy was 31.8 months, median 27 months and range 7–123 months. Considering 2-year and 3-year interval between colonoscopies as compliant with protocol if interval <2.5 and 3.5 years, respectively, 84 cases (58%) were diagnosed within 2.5 since last colonoscopy and 115 (79%) within 3.5 years.
Time since last colonoscopy in months; and number, cumulative number and cumulative % of colorectal cancer (CRC) cases diagnosed in each time interval
Survival
Overall 5-year and 10-year survival were excellent, reflecting survival for the most frequent cancers, CRC and endometrial cancer (table 7). Ovarian cancer survival also appeared to be excellent in LS patients, although the small number of cases adds uncertainty to the observation. A majority of patients also achieved 10-year survival of upper GI and urinary tract cancers, although survival rates were not as good as for the other cancers.
5-year and 10-year crude survival after first cancer diagnosed by cancer type in Lynch syndrome (LS) patients without prior or prevalent cancer at first colonoscopy
Discussion
Our study was designed to answer three critical clinical questions in a large cohort of LS mutation carriers participating in surveillance programmes: what is the cumulative risk by age to first cancer, in which organs are first cancers most likely to occur, and what are the outcomes for these cancers? It is essential that once identified LS patients are offered reliable estimates of their future cancer risk and appropriate, individualised surveillance. This is the first study to present prospective empirical observations from multiple centres and including sufficient numbers to meet these needs. This study reports time to first cancer (penetrance of the mutated genes) and survival after first cancer diagnosed. As the database is expanded, we will later examine time to next cancer in those surviving first cancer.
We show that cumulative incidence of any cancer at age 70 years is high for all MMR gene mutation carriers, and slightly higher for females (75%) than males (58%). The cumulative incidences were 72% for MLH1 and MSH2 mutation carriers but lower in MSH6 (52%) and PMS2 (18%) mutation carriers. MSH6 and PMS2 carriers developed no cancers before 40 years of age. However, the lower number of MSH6 and PMS2 mutation carriers studied made the risk estimates for these genes less certain, particularly at younger ages.
Despite the screening colonoscopy carried out, CRC was the most frequent first cancer observed and had a cumulative incidence at age 70 of 46% in MLH1 mutation carriers, and 35% in MSH2 mutation carriers with lower incidence for carriers of mutations in MSH6 and PMS2. The results for CRC confirmed that colonoscopy fails to prevent a substantial number of CRCs as previously reported.3–10 Retrospective studies have provided with different estimates of cancer incidence mostly without colonoscopic surveillance ranging from 22% to 74%. Among the few previous reports from prospective studies,3–10 none used methods adjusting for age and many did not discriminate between the patients carrying mutations in the different MMR genes. It is commonly agreed that CRCs may be prevented by colonoscopy, but lacking a control group we could not calculate what proportion this might have been. Also, the CIs of penetrance estimates for the mutated genes both in our and previous studies were too wide to consider a later onset of CRC in our study than in previous reports.
In this first report, we did not include stage at diagnosis of CRC and we could not determine effects of surveillance colonoscopy on the pathological stage at which CRCs were diagnosed, although early diagnosis may be a contributing factor to the excellent long-term survival of CRC that we observed in LS patients. When carrying out the study, we considered <2.5 years and <3.5 years between colonoscopies as compliant with a guideline of 2-year and 3-year intervals, respectively. As seen in table 6, non-compliance could not explain the majority of the CRC cases. We would like to have more information before considering time since last colonoscopy in more detail, and the database is currently being expanded to include stage at diagnosis, prognosis, synchronous and para-synchronous adenomas and other parameters as a follow-up study based on the current results. Meanwhile, we advocate continuation of the existing surveillance programmes.
Some CRC in LS may have good prognosis13 and without a control group it remains theoretically possible that the CRCs occurring in our cohort would have been cured even without surveillance for asymptomatic disease, but a randomised trial denying LS patients access to colonoscopy to test such an hypothesis is unlikely to be justifiable. On the other hand, the purpose of and evidence base for ever more frequent surveillance needs to be revisited, and we will expand our database to address this issue via further studies.
Does the incidence of CRC in LS patients under surveillance indicate that our current approach has failed or is the good survival evidence of success? For LS patients, it is of course survival and quality of life that are the key concerns. This study shows that the former is being achieved while assessment of the latter will require further studies.
Adenomas in the general population are known to cause CRC, and adenomectomy is documented to prevent CRC.14 Previous reports suggest that incidence of adenomas in LS and late-onset colon cancer families without MMR mutations are similar7 while the carcinogenetic process from adenoma to carcinoma is accelerated in LS patients.7 ,15 Lack of functioning MMR genes cause microsatellite instability (MSI) and invasive cancers in LS show MSI.16 Hyperplastic polyps in the colon of LS patients reportedly do not show MSI,17 while adenomas show increasing loss of MMR gene product and MSI with increasing dysplasia.16 LS patients may also present MSI in the crypts of a macroscopically normal gut surface.18 Thus, one may speculate that MMR mutation carriers are capable of producing CRC not only inside an adenoma but also independently of a macroscopically visible adenoma, which in turn may rise several questions: how small the invasive early cancers may be in LS? Which is the sensitivity of colonoscopy to identify such lesions?19 How rapidly a small preinvasive lesion may progress to invasive cancer? Which is the time window, if any, in a preinvasive macroscopic detectable stage? We are currently expanding the database to address these questions.
Our prospective results do not support major differences in CRC incidence according to gender. Based on assumed carrier status in former generations, differences in CRC incidence by gender have been reported for MLH1 and MSH2,20 but these differences were less notable in a later retrospective study including proven carriers only.21
Regarding specific genes, MSH6 carriers had later onset of any cancer and of CRC than MLH1 and MSH2 carriers, in line with previous reports. The low number of MSH6 and PMS2 observation years most probably represents an ascertainment error linked to their lower penetrance22 and to the less widespread availability of diagnostic testing for these genes. Also, the age distributions may have been influenced by MSH6 mutation carriers presenting to genetic clinics at older ages after their relatives had already developed cancers. In spite of these uncertainties, our results question current guidance that advises that MSH6 carriers should commence colorectal surveillance at young ages.1 ,2 A study of a larger cohort of young mutation carriers is needed to draw firm conclusions.
The higher cumulative incidence of any cancer at age 70 years in females (75%) versus males (58%) may be a consequence of the incidences of endometrial and ovarian cancers in females. Endometrial cancer cumulative incidence at 70 years was consistently high for MLH1 (34%), MSH2 (51%) and MSH6 (49%) and notable in PMS2 carriers (24%). Ovarian cancer cumulative incidence at 70 years was lower and restricted to MLH1 (11%) and MSH2 (15%) carriers. In our series, 21/314 (7%) of the cases identified with first cancers had two synchronous cancers in different organs, suggesting that the risk of getting a new primary cancer may be increased in those having already had one or more cancers or precursor lesions1 ,2 There is limited prospectively observed evidence, however, on the magnitude of such risks. The methods used in this study are not suitable for calculation of these risks. Neither does our study provide information on cancer risks in LS patients under 25 years of age. As the clinical guidelines used in our collaborating centres suggest starting surveillance at this age, we could not consider AIR for younger patients. Because colonoscopic surveillance did not prevent all CRC in MLH1 and MSH2 carriers, it is unlikely that it did so in MSH6 and PMS2 carriers. Our findings make it very unlikely that MSH6 or PMS2 mutation carriers develop a significant number of LS-associated cancers before 25 years, but a small proportion of MLH1 and MHS2 mutation carriers may do so. Our results have no bearing on the current guidelines that suggest surveillance only after age 25 years for MLH1 and MSH2 mutation carriers.
The three major LS-associated cancer types, CRC, endometrial and ovarian, all had excellent observed survival, which is very encouraging for carriers of MMR gene mutations. Further investigation will be needed to determine the degree to which this good prognosis is the result of early detection due to surveillance, improved prognosis due to detection at early stages, lead time bias due to early detection and/or represents a better prognosis for cancers in LS patients than for cancers arising in the general population. Relevant factors might include the high degree of immunogenicity and paucity of progressive metastatic disease in LS CRCs13 ,23 and the efficacy of current treatments in the patient group.
Most ovarian cancers in LS were cured, consistent with our previous study,22 and the same was true for endometrial cancers. Also, our findings support the notion that LS ovarian cancers have a good prognosis when given current treatment.24 ,25 This good prognosis may be partly the result of early detection due to surveillance, but pathological and molecular genetic analysis have also indicated more favourable tumour characteristics in LS-associated ovarian cancers compared with sporadic disease.26 ,27 This is in sharp contrast to BRCA1/2 ovarian cancer cases that have a poor prognosis despite early detection and treatment.28 The cumulative incidence for ovarian cancer in MLH1 and MSH2 carriers up to 50 years of age was similar to reported cumulative incidence in BRCA1 carriers28 but with lower incidence later in life. The finding that ovarian cancer was restricted to MLH1 and MSH2 mutation carriers was unexpected and needs validation.
Certain characteristics of the methods we used need to be highlighted. The calculation of cumulative incidence by age is based upon the observed point estimates for annual incidence for each age group. The method adjusts for uneven distribution of numbers in the different age groups, and for the different distributions of ages of patients by mutated gene. If we had estimated penetrance by gene from inclusion to last observation using the Kaplan–Meier algorithm, equal AIR in all age groups had to be assumed for each gene. If doing so, we would have compared younger MLH1/MSH2 carriers to older MSH6/PMS2 carriers. Assimilating our data into one larger database was mandatory to arrive at sufficient numbers to use our method to calculate AIR by age and thereby estimate cumulative incidences by age, mutated gene and gender. If doing so, we avoided many ascertainment and time-trend biases present in retrospective family studies that were previously used to determine penetrance, and patient numbers were sufficient to adjust for skewed distributions by age and mutated gene.
In spite of the strengths, this study has some limitations. The cumulative incidences presented are not cumulative incidences for organ-specific cancers, but for any first cancers and for the organ in which the first cancers occurred. Calculation of cumulative incidence by age for specific cancers irrespective of the occurrence of other cancers will need a different data set and specific methods, and the database is currently being expanded to do so. The long period (>20 years) of observation is likely to span improvements in techniques for early diagnosis of cancer, as well as treatment, both of which may improve survival. Assuming that shorter time between colonoscopies and/or that recent treatment is possibly better than the average during the period reported, our results may be considered worst-case estimates.
The information provided here should help in the genetic counselling of individual patients. We have made it available via an interactive open access website for personalised healthcare at http://www.LScarisk.org. Based on the algorithm given above and the information given in online supplementary table S1, the website calculates the cumulative risk of developing cancer from any age selected by the user to any age up to 70 years for any currently unaffected patient with LS by entering age, gender and mutated gene. The website may be used for any single patient to provide personalised risk estimates for all groups of cancers detailed in table 5.
The findings presented here raise a number of questions that will be addressed by further studies: the effects of colonoscopy intervals on CRC incidence; whether or not CRC in LS may emerge outside a macroscopic visible adenoma, time to subsequent cancer in LS patients surviving their first cancers; the lifetime risk for any specific cancer in those surviving the other cancer types; the disease-specific survival for all distinct cancer types; and investigation of the penetrance and expression of different classes of mutations at each MMR gene locus. We are currently expanding the database to address these key questions. As the numbers of young MSH6 or PMS2 mutation carriers we have been able to include have been limited so far, we welcome other parties with such series to join us and contribute to future refinement of incidence rates for MSH6 and PMS2 mutation carriers.
For further information on the collaborating activities, please visit http://insight-group.org/ and http://mallorca-group.eu/. To tailor cancer risk prediction according to a given patient's age, gender and mutated gene, visit http://LsCaRisk.org.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
Twitter Follow Toni Seppälä at @Adductor and Ignacio Blanco at @consejogenetico
Contributors PM: managed database and calculated results. PM, JB, GC, IMF, FM and JS drafted the manuscript. EAR, KT, SN and EH calculated the CIs. SN and EH established the website. All participated in study design, interpretation of results, writing of manuscript and approved final manuscript.
Funding The Finnish contribution was supported by The Finnish Cancer Foundation, The Sigrid Juselius Foundation, Mary and Georg Ehrnrooth foundation and State Research Funding. The Spanish contribution has been funded by the Spanish Ministry of Economy and Competitiveness SAF2012-33636 (GC); the Carlos III Health Institute; RTICC (RD12/0036/0008); the Scientific Foundation Asociación Española Contra el Cáncer; and the Government of Catalonia (2014 SGR 338) and the Welsh Contribution by the Wales Gene Park.
Competing interests JB has a patent for high-speed low-cost tumour profiling pending to JB and QuantuMDx. DGE is a NIHR senior investigator.
Patient consent Obtained.
Ethics approval Ethical approval for health service and/or health service as research project as described.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement There are no unpublished data to the current ends. Additional results will be published from additional data. Each contributor has the authority to distribute own data to anyone without permission from the group. No patient identifiable data were exported from any participating centre.