Article Text

Abstract
Objective The HCV life cycle and the lipid metabolism are inextricably intertwined. In the blood, HCV virions are associated with lipoproteins, forming lipoviroparticles (LVPs), which are the most infectious form of the virus. Apolipoprotein E (apoE), a key LVP component, plays an essential role in HCV entry, assembly and egress. ApoE is also a cell host factor involved in lipoprotein homeostasis. Although the majority of apoE is associated with lipoproteins, a lipid-free (LF) form exists in blood. However, the role of LF-apoE in both lipid metabolism and HCV life cycle is poorly understood.
Design In this study, using the cell culture-derived HCV model system in human hepatoma Huh7.5.1 cells and primary human hepatocytes (PHH), we investigated the effect of LF-apoE on the early steps of HCV life cycle and on the lipid metabolism of hepatic cells.
Results A dose-dependent decrease in HCV replication was observed when Huh7.5.1 cells and PHH were treated with increasing amounts of LF-apoE. We showed that LF-apoE acts on HCV replication independently of previously described apoE receptors. We observed that LF-apoE induced a marked hepatic cholesterol efflux via the ATP-binding cassette subfamily G member 1 (ABCG1) protein that in turn inhibits HCV replication. LF-apoE also increases both apolipoprotein AI and high-density lipoprotein production.
Conclusions Our findings highlight a new mechanism in lipid metabolism regulation and interaction of the lipid metabolism with the HCV life cycle, which may be important for viral pathogenesis and might also be explored for antiviral therapy.
- HCV
- LIPID METABOLISM
- LIPOPROTEIN-CHOLESTEROL
- LIPOPROTEIN METABOLISM
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
HCV is associated with very-low-density and low-density lipoproteins (VLDL and LDL) to form an infectious lipoviroparticle (LVP).
Apolipoprotein E (apoE) is a component of LVP that plays a key role in HCV life cycle.
ApoE is a host factor involved in lipoprotein homeostasis existing in both lipoprotein-associated and lipid-free (LF) form.
The role of LF-apoE in both lipid metabolism and HCV life cycle is poorly understood.
What are the new findings?
LF-apoE dose-dependently decreases HCV replication.
LF-apoE acts on HCV replication independently of previously described apoE receptors.
LF-apoE induces ATP-binding cassette subfamily G member 1 (ABCG1) protein-dependent cholesterol efflux that inhibits HCV replication.
LF-apoE increases apolipoprotein AI-high-density lipoprotein (HDL) production.
How might it impact on clinical practice in the foreseeable future?
Our findings highlight a new interaction between HCV and lipid metabolism, which might be explored for antiviral therapy.
Our findings represent new opportunity for the development of therapeutic strategies to treat metabolic disorders by increasing HDL production.
Introduction
HCV infection is a major cause of chronic hepatitis, liver cirrhosis and hepatocellular carcinoma worldwide.1 Novel direct-acting antivirals cure the large majority of infected patients without major side effects. Nevertheless, several challenges remain: high costs limit access to therapy, and certain difficult-to-treat patients subgroups may need adjunctive therapeutic approaches.2 Furthermore, vaccine development is hampered by viral evasion of host immune responses and to date, no vaccine is available.3
HCV almost exclusively infects human hepatocytes and many studies have shown a close link between the HCV life cycle and the hepatic lipid metabolism.4 The hallmark of HCV is its association in the bloodstream of infected patients with the very-low-density lipoproteins (VLDL) or the low-density lipoproteins (LDL) to form an infectious lipoviroparticle (LVP).5 Apolipoprotein E (apoE), a host factor present at the LVP surface,6 plays a crucial role in HCV attachment and entry7 ,8 as well as assembly and egress.9 ,10
ApoE is a glycoprotein of 299 amino acids involved in the transport, production and uptake of lipoproteins.11 In humans, the three major isoforms apoE2, apoE3 and apoE4 are mainly produced by hepatocytes, glial cells and macrophages. ApoE3, the wild-type form of apoE, differs from apoE2 and apoE4 by one amino acid substitution at positions 158 and 112, respectively, dramatically altering their function.11 ApoE2 displays reduced affinity for LDL receptor (LDLR) and is involved in type III hyperlipidaemia, whereas apoE4 exhibits a reduced stability and is associated with Alzheimer's disease.11 ,12
ApoE is a component of plasma chylomicrons, VLDL, intermediate-density lipoproteins (IDL) and high-density lipoproteins (HDL),13 which mediates their internalisation, acting as a ligand for cellular receptors such as LDLR, heparan sulfate proteoglycans (HSPGs), LDL receptor-related protein 1 (LRP1) and scavenger receptor B1 (SR-B1).14 While the major part of plasma apoE is associated with lipoproteins, apoE also exists in a lipid-free (LF) form.15–17 The structure of apoE consists of two folded domains, the receptor binding-domain (N-ter) and the lipid-binding domain (C-ter), separated by a hinge region. The physical properties of the C-ter domain allow apoE to reversibly switch between a lipoprotein-associated form and a LF form, meaning that apoE can be easily exchanged between the different classes of lipoproteins.11 Thus, LF-apoE is generated by dissociation from apoE-containing lipoproteins but can also be secreted by hepatocytes and macrophages.15–17
Despite remarkable progress towards understanding apoE function in lipid metabolism, the role of LF-apoE remains poorly understood. Since apoE plays a crucial role in the HCV life cycle, we aimed to decipher the impact of LF-apoE on HCV life cycle.
Experimental procedures
Cells lines and primary human hepatocytes
Huh7.5.1 cells were propagated as described.18 Primary human hepatocytes (PHH) were isolated from liver resections with informed consent from all subjects and approval of the Strasbourg University Ethical Committee (Approval # CPP 10-17) as described previously.18 The Huh7 Luc-ubi-neo-ET cells carrying HCV replicon genotype 1b were propagated as described.19 The Huh7.5.1/internal ribosome entry site (IRES)-Luc stable cell line was obtained by stable transfection of the pIV1122 plasmid under G418 selection.20
Plasmids and viruses
Plasmids such as pFK-Japanese fulminant hepatitis 1 (JFH1), pFK-J6-JFH1 chimaera (Jc1), pFK-Jc1 with luciferase reporter (Luc-Jc1), pFK-Luc-JFH1 and pFK-Luc-JFH1ΔE1E2 constructs have been described21–24 (see online supplementary figure S1). The plasmid pIV1122 was generated by inserting the firefly luciferase reporter gene under the control of the HCV IRES.20
Supplemental material
HCV production and infectivity
Luc-Jc1, Jc1, Luc-JFH1, JFH1 and Luc-JFH1ΔE1E2 RNA were obtained following T7 in vitro transcription. To obtain HCV-replicating cells, Huh7.5.1 cells were electroporated with viral RNA as described.9 Three days postelectroporation, HCV replication was assessed by quantification of firefly luciferase activity, or RT-qPCR quantification of viral RNA, as previously described.20 ,25 PHH were infected with the JFH1 virus as described.26 For details concerning reagents and antibodies, RNA interference experiments, proliferation assays and iodixanol density gradient ultracentrifugation see online supplementary information.
LF-apoE3 treatments
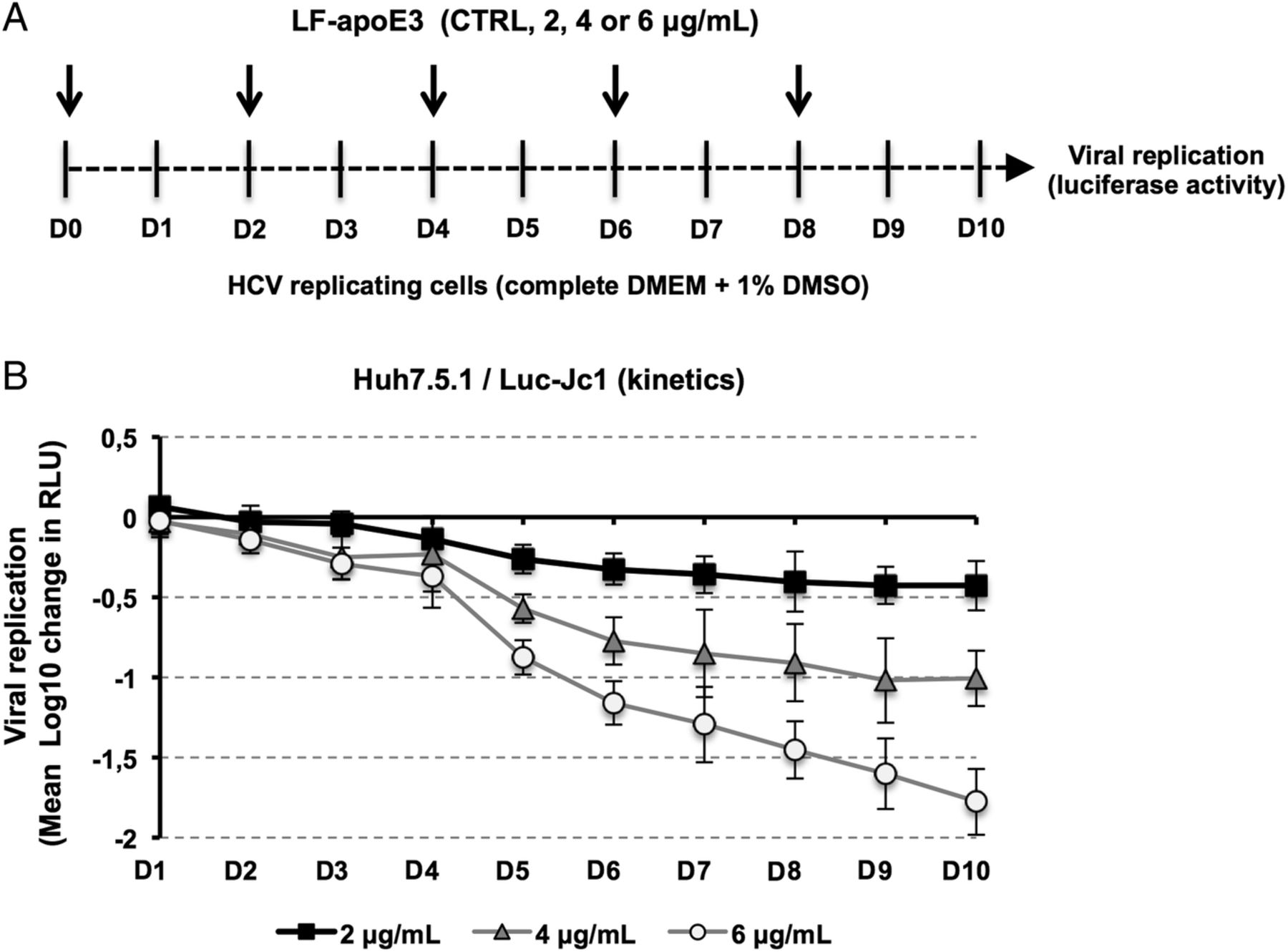
Human recombinant LF-apoE3 lyophilised powder (Sigma–Aldrich) was dissolved to 0.5 mg/mL in 5 mM phosphate-buffered saline (PBS; pH 7.8) containing 0.5 mM dithiothreitol to prevent disulfide bridges. LF-apoE3 was diluted at 2, 4 or 6 μg/mL (final concentration) in fresh medium containing 10% fetal bovine serum (FBS) and added to HCV-replicating cells. Control cells (CTRL) were treated with LF-apoE3 solvent. Viral replication was determined 48 hours post-treatment by quantification of luciferase activity or by RT-qPCR. For experiments requiring different serum conditions, HCV-replicating cells were washed with PBS and LF-apoE3 was added to fresh medium containing 10%, 2% or 0% of FBS. Viral replication was determined 24 hours post-treatment. For kinetic experiments, HCV-replicating cells were propagated in complete medium including 1% dimethylsulfoxide (DMSO) to slow down the cell proliferation. Cell culture medium was replaced every 2 days. Luciferase activity was measured every day for 10 days to assess HCV replication (figure 2A). PHH were infected with JFH1 virus as described.26 Six hours postinfection, cells were washed three times with cold PBS+heparin and two times with cold PBS. Reference cells were lysed at this step to determine the basal amount of viral RNA (BL=baseline). LF-apoE3 was added to the culture medium at 2, 4 or 6 μg/mL. Viral replication was quantified by RT-qPCR 48 hours post-treatment.
Imaging studies using fluorescent-labelled LF-apoE3
Human recombinant LF-apoE3 was labelled using the Alexa fluor 647 Labelling Kit purchased from Life Technologies, according to the manufacturer's instructions. HCV-replicating cells were treated with LF-apoE3 at 6 μg/mL. After 1 hour at 37°C, cells were gently washed three times with PBS and fixed with paraformaldehyde (PFA) 4% purchased from Electron Microscopy Sciences, for 20 min at room temperature. Cells were then washed three times with PBS, and Fluoroshield supplemented with 4′,6′-diamidino-2-phenylindole (Sigma–Aldrich) was added prior to mounting. Fluorescent imaging was performed using a Leica SP5 II confocal microscope. In parallel, a similar experiment was performed with LF-apoE3 incubated for 1 hour at 37°C with anti-apoE antibodies, before addition to the cell culture medium.
Cholesterol efflux assay
HCV-replicating cells were grown in 12-well plates (≈80% confluence) and were treated or not with the liver X receptor (LXR) agonist (T0901317) at 2 μM for 8 hours at 37°C. Cells were washed three times with PBS and incubated in PBS (600 μL) containing LF-apoE3, LF-apoE2 or LF-apoE4 at 2, 4 or 6 μg/mL (final concentration). Media were collected 4 hours post-treatment. Cholesterol amount was measured using the ‘Amplex Red Cholesterol Assay Kit’ purchased from Life Technologies, according to the manufacturer's instructions.
Iodixanol density gradient ultracentrifugation
HCV-replicating cells (grown in 10% FBS medium or grown in serum-free medium) or PHH (grown in serum-free medium) were treated with LF-apoE3 at 6 μg/mL or PBS (CTRL). Culture media were collected 48 hours post-treatment. Lipoprotein separation was performed using a flotation iodixanol gradient protocol adapted from Yee et al27 (see online supplementary information).
Statistics
Data sets were analysed using the Mann–Whitney U test. p<0.01 was considered statistically significant. Significant p values are indicated by asterisks in the individual figures.
Results
LF-apoE3 specifically impairs HCV replication in a dose-dependent manner
To address the functional impact of LF-apoE on HCV replication, we first defined LF-apoE concentrations used in our cell culture model. In plasma, apoE concentration is in the range of 30–80 μg/mL, apoE being mainly found in association with lipoproteins.28 ApoE also exists in an LF form at low levels in physiological conditions. It has been shown that LF-apoE concentrations in a range of 2–10 μg/mL are sufficient to induce biological effects such as anti-atherosclerotic effects or signalling pathways activation in vitro and in vivo.17 ,29–33 In accordance with these previous findings, we chose to perform our experiments using LF-apoE3 at 2, 4 and 6 μg/mL. Importantly, these apoE concentrations did not affect Huh7.5.1 cell proliferation, as verified by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MMT) cell proliferation assay (see online supplementary figure S2).
To study the role of LF-apoE on HCV replication, we added LF-apoE3, LF-apoE2 or LF-apoE4 to the culture medium of HCV-replicating cells and quantified HCV replication after 48 hours. As shown in figure 1A, increasing amounts of LF-apoE3 resulted in a dose-dependent decrease in viral replication. At a LF-apoE3 concentration of 6 µg/mL, an up to 60% inhibition of replication was observed compared with CTRL cells. In contrast, treating HCV-replicating cells with increasing amounts of LF-apoE2 or LF-apoE4 did not affect HCV replication (figure 1A), suggesting that the inhibitory effect is specific to the wild-type form of apoE (LF-apoE3). This observation was confirmed by a decrease in HCV RNA as quantified by RT-qPCR, as well as a decrease in the levels of the Core and NS5A viral proteins (figure 1B). Of note, a similar inhibition profile was observed when JFH1-infected PHH were treated with increasing amounts of LF-apoE3 (figure 1C).

Lipid-free apolipoprotein E3 (LF-apoE3) specifically impairs HCV replication in a dose-dependent manner. Addition of LF-apoE3 to the cell culture medium of HCV-replicating cells, but not LF-apoE2 and LF-apoE4, leads to a dose-dependent decrease in viral replication. (A) Luc-J6-JFH1 chimaera (Jc1)-replicating cells were treated with increasing amounts of LF-apoE3, LF-apoE2 or LF-apoE4. Viral replication was determined by quantification of luciferase activity after 48 hours. (B) Quantification of Jc1 viral RNA levels following LF-apoE3 treatment by RT-qPCR and detection of viral proteins Core and NS5A by western blot analysis. (C) LF-apoE3 inhibits Japanese fulminant hepatitis 1 (JFH1) replication in primary human hepatocytes (PHH). JFH1-infected PHH were treated with LF-apoE3, and viral RNA was quantified by RT-qPCR 48 hours post-treatment. The grey dotted line indicates the basal amount of viral RNA present in reference PHH at time 0 (BL=baseline) (refer to Experimental procedures section). (D) LF-apoE3 does not inhibit HCV replication by affecting cell reinfection. Luc-JFH1ΔE1E2 RNA and Luc-JFH1 replicating cells were treated with LF-apoE3, and viral replication was quantified after 2 days by measuring luciferase activity. (E) LF-apoE3 has no effect on HCV internal ribosome entry site (IRES) translation. Huh7.5.1/IRES-Luc cells were treated with LF-apoE3, and HCV IRES activity was determined 2 days post-treatment by luciferase activity measurement. (F) LF-apoE3 inhibits HCV genotype 1b replication. Huh7 Luc-ubi-neo-ET cells, which stably express a genotype 1b subgenomic replicon, were treated with LF-apoE3 for 48 hours. Replication was determined by measuring luciferase activity. Results are presented as % luciferase activity relative to HCV-replicating control cells (CTRL) (100%). Means±SD from three (A and D–F) or two (B) independent experiments performed in triplicate are shown. In (C), one representative experiment is presented. *p<0.01; **p<0.001.
Since previous studies have shown that apoE-derived peptides prevent HCV binding to hepatocytes,8 ,34 LF-apoE3 could affect HCV by simply inhibiting cell reinfection by newly formed virions. To address this question, we used the Luc-JFH1ΔE1E2 strain that corresponds to the JFH1 genome lacking the E1 and E2 envelop proteins and that is defective for virus production and cell reinfection.24 The full-length Luc-JFH1 RNA was used in parallel as a CTRL. As shown in figure 1D, the addition of LF-apoE3 led to a dose-dependent decrease in Luc-JFH1ΔE1E2 replication similarly to Luc-JFH1, indicating that LF-apoE3 acts on HCV replication independently of virion production and infection (figure 1D).
During HCV life cycle, viral replication and translation are closely linked. To further map the step affected by LF-apoE3, we investigated whether LF-apoE3 affects HCV translation. We used a stable cell line expressing a luciferase reporter gene under the control of the HCV IRES (Huh7.5.1/IRES-Luc).20 We observed that LF-apoE3 had no effect on luciferase expression, indicating that LF-apoE3 did not impact HCV IRES-driven translation but acts on HCV replication (figure 1E).
To confirm whether these findings are genotype-independent, we investigated the role of LF-apoE3 on a different HCV strain. We used Huh7 cells stably expressing a genotype 1b subgenomic replicon.19 As shown in figure 1F, LF-apoE3 decreases HCV genotype 1b replication in a dose-dependent manner, similarly to genotype 2a strains.
Finally, in order to evaluate the impact of extended LF-apoE3 treatment on HCV replication, we performed a kinetic experiment by treating HCV-replicating cells with LF-apoE3 for 10 days. We observed that LF-apoE3 decreases HCV replication up to 2 logs (for the 6 μg/mL condition) over a 10-day period (figure 2), confirming the robust effect for persistent infection. Taken together, these data indicate that LF-apoE3 specifically inhibits HCV replication in a genotype-independent manner.
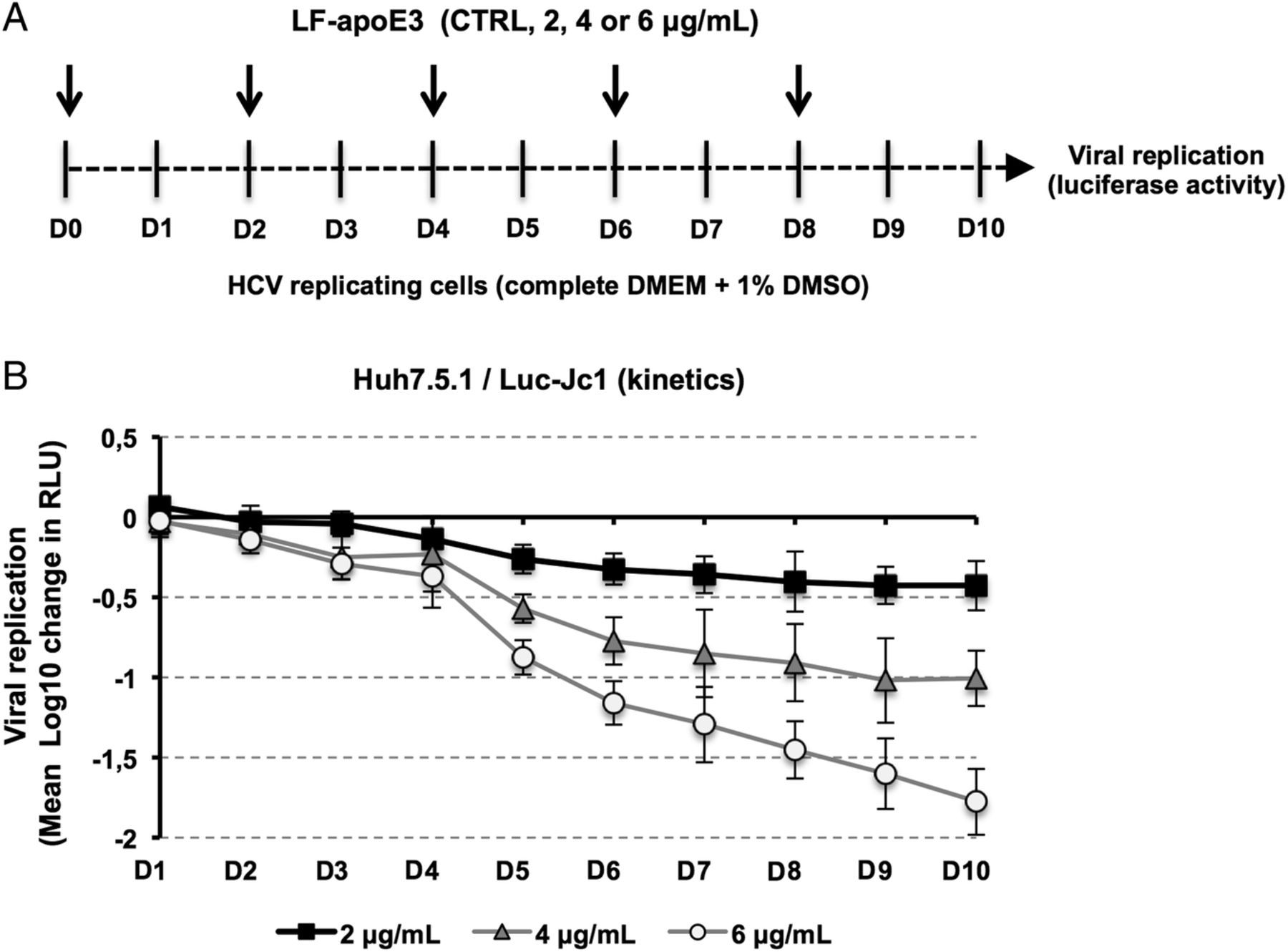
Kinetics of HCV replication after lipid-free apolipoprotein E (LF-apoE)3 treatment for 10 days. (A) Scheme of the experimental procedure. HCV-replicating cells were propagated in complete medium added with 1% of dimethylsulfoxide (DMSO) to slow down cell proliferation. Renewal of media and LF-apoE3 treatment were performed every 2 days as indicated by the arrows (D0, D2, D4, D6 and D8). Every day, HCV replication was quantified by a luciferase assay (D1–D10). (B) HCV replication is expressed as Δ Log 10 of luciferase activity (relative light unit, RLU) obtained by subtraction of the Log 10 of RLU obtained in non-treated control cells (CTRL) and Log 10 of RLU in LF-apoE3-treated cells for each day. Means±SD from three independent experiments performed in triplicate are shown.
LF-apoE3 impairs HCV replication independently of serum components
Because apoE is an exchangeable apolipoprotein able to dissociate and reassociate with lipoproteins in the bloodstream,28 we investigated whether LF-apoE3’s effect on HCV replication was due to a direct effect triggered by LF-apoE3 itself or to an indirect effect through association with a serum component present in the cell culture medium. Therefore, we treated HCV-replicating cells for 24 hours with LF-apoE3, in media containing 10, 2 or 0% FBS. As shown in online supplementary figure S3A, LF-apoE3 decreases HCV replication in a dose-dependent manner, independent of the serum concentration, suggesting that LF-apoE3 acts on HCV replication independently of serum components. Addressing this question using a different approach, we used human purified VLDL and a mix of IDL/LDL to determine whether the inhibition of HCV replication is mediated by LF-apoE3 or also by lipoprotein-associated apoE3. As shown in online supplementary figure S3B, both VLDL and IDL/LDL did not affect HCV replication, suggesting that only LF-apoE3 mediates inhibition of HCV replication.
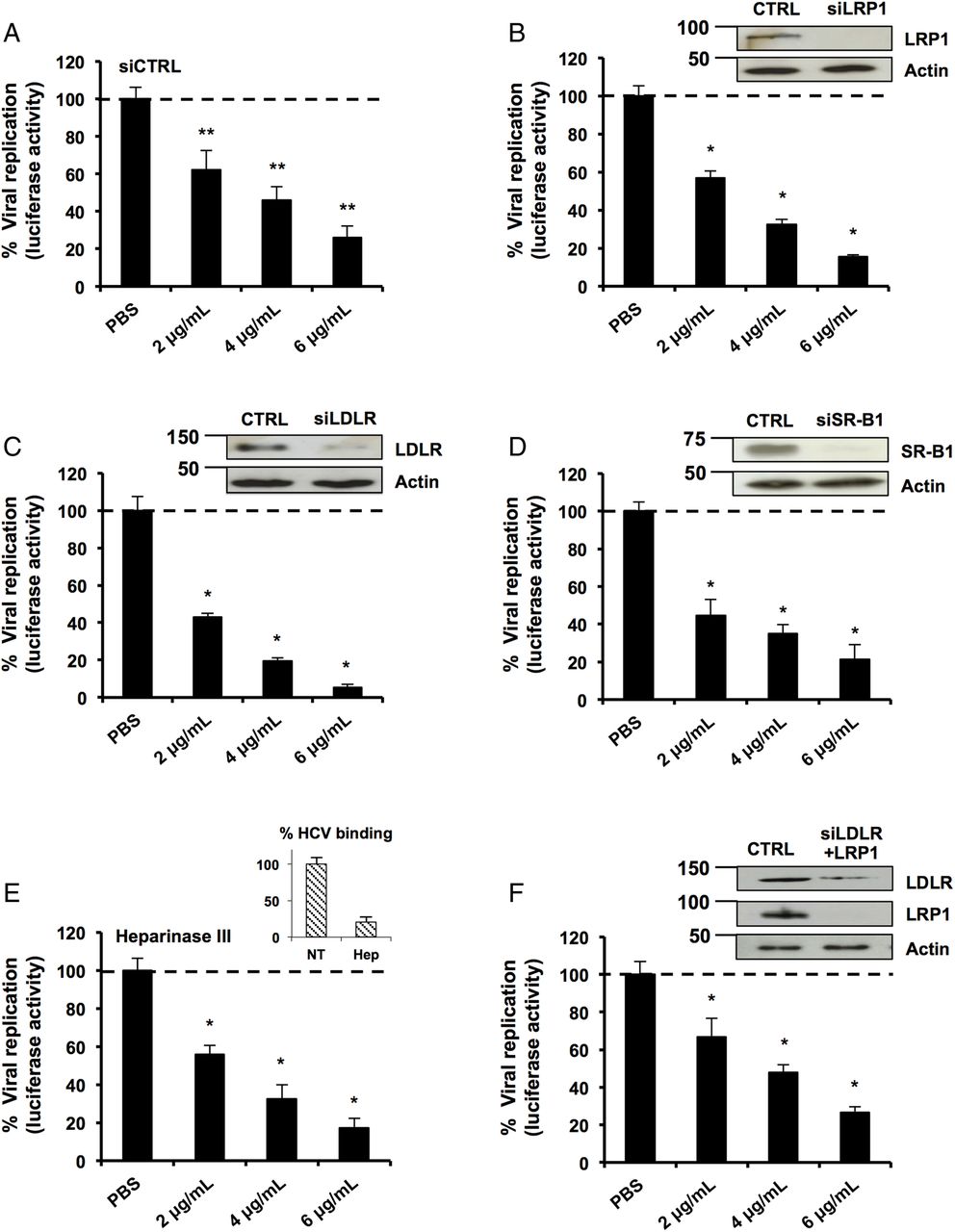
Classical apoE receptors and apoE recycling process are not involved in the LF-apoE3-induced inhibition of HCV replication
We then investigated the mechanism of action leading to inhibition of HCV replication. To interact with the hepatocyte, apoE binds to several receptors such as LDLR, LRP1, SR-B1 and HSPGs.8 ,30 ,35 To address the possible role for these receptors, we used RNA interference to modulate the expression levels of LDLR, SR-B1 and LRP1 in hepatic cells. As shown in figure 3A–D, the silencing of these receptors did not restore HCV replication in LF-apoE3-treated cells. A similar effect was observed after treatment with heparinase III that degrades HSPGs (figure 3E). To eliminate the effect of receptor compensation,35 we used combinations of small interfering RNAs to simultaneously target LDLR and LRP1 (figure 3F). Despite efficient silencing, LF-apoE3's inhibition of replication was not affected. Taken together, our data clearly demonstrate that LF-apoE3 acts on HCV replication independently of conventional apoE receptors.

Classical apolipoprotein E (apoE) receptors are not involved in the lipid-free (LF)-apoE3-induced inhibition of HCV replication. (A–D) Knockdown of classical apoE receptors does not impair LF-apoE3’s inhibitory effect on HCV replication. HCV-replicating cells were reverse-transfected with a non-targeting small interfering RNA (siRNA) (siCTRL) (A) or siRNAs targeting the different receptors: siLRP1 (B), siLDLR (C) and siSR-B1 (D). Following 48 hours of silencing, cells were treated with LF-apoE3. Viral replication was determined by luciferase activity 48 hours post-treatment. Knockdowns were confirmed by western blot (B–D). (E) HCV-replicating cells were treated with Heparinase III to remove heparan sulfate proteoglycans (2 UI/mL) for 2 hours and treated with LF-apoE3. Luciferase activity was measured after 24 hours. (F) Double knockdown of low-density lipoprotein receptor (LDLR) and LDL receptor-related protein 1 (LRP1) does not impair LF-apoE3's inhibitory effect on HCV replication. LDLR and LRP1 were simultaneously knocked down using the same experimental strategy employed in (A–D). Results are presented as % luciferase activity relative to silenced HCV-replicating cells treated with phosphate-buffered saline (PBS, set at 100%). Means±SD from three (A) or two (B–F) independent experiments performed in triplicate are shown. *p<0.01; **p<0.001.
Following apoE-containing lipoprotein endocytosis in hepatocytes, a major part of apoE is recycled while the lipids are metabolised.36 Rab GTPases 5 (Rab5) and 11 (Rab11) are involved in apoE-recycling process by regulating early endosome and recycling endosomes trafficking, respectively.36–38 To study whether the apoE-recycling process is involved in the observed inhibition of HCV replication, we silenced Rab5 and Rab11 and measured HCV replication following LF-apoE3 treatment. As shown in online supplementary figure S4A, B, silencing of Rab5 and Rab11 did not impair the inhibitory effect of LF-apoE3 on HCV replication. Thus, LF-apoE3 inhibits HCV replication by a yet undefined intracellular pathway that is not affected by hampering apoE recycling.
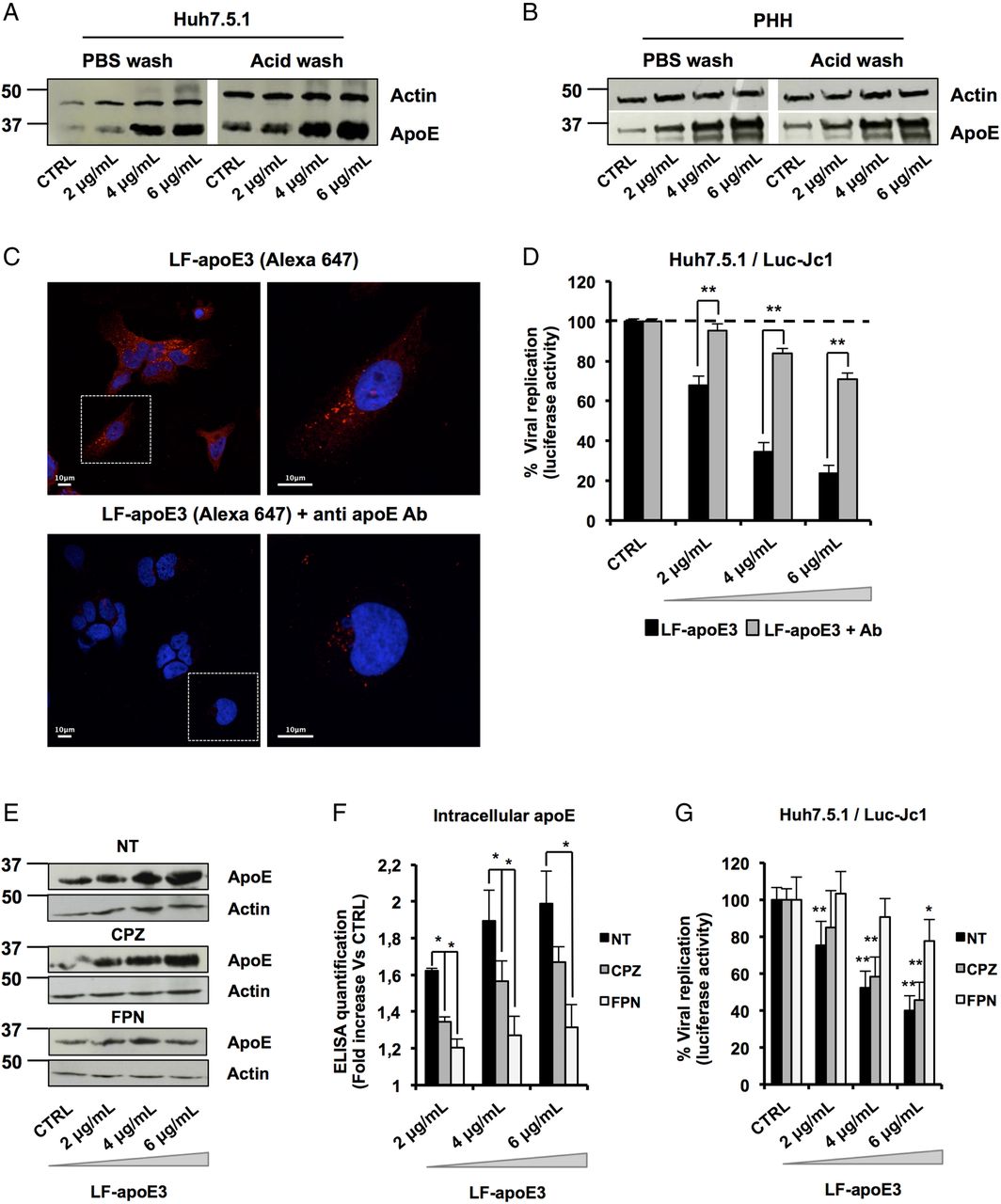
LF-apoE3 requires lipid raft integrity to inhibit HCV replication
To further decipher the mode of action of LF-apoE3 on HCV replication, we investigated whether LF-apoE3 is taken up by hepatic cells. First, we performed western blot (WB) employing cell lysates of Huh7.5.1 cells and PHH treated with LF-apoE3. As shown in figure 4A, B, we observed a dose-dependent accumulation of apoE in treated Huh7.5.1 cells and PHH (figure 4A, B). To demonstrate that LF-apoE3 is indeed taken up by the cells, we removed cell surface-bound proteins by washing the LF-apoE3-incubated cells with a cold acid buffer (0.2 M glycine buffer+0.15 M NaCl, pH 3.0) prior to perform WB analysis.39 ApoE dose-dependently accumulated in the cells even after acid wash at 37°C, indicating that LF-apoE3 enters hepatic cells and does not remain on the cell surface (figure 4A, B). As a CTRL, the efficacy of acid cell washing was assessed at 10°C, a temperature at which LF-apoE3 binds the membrane but cannot enter the cells and is thus removed by acid buffer washing (data not shown).
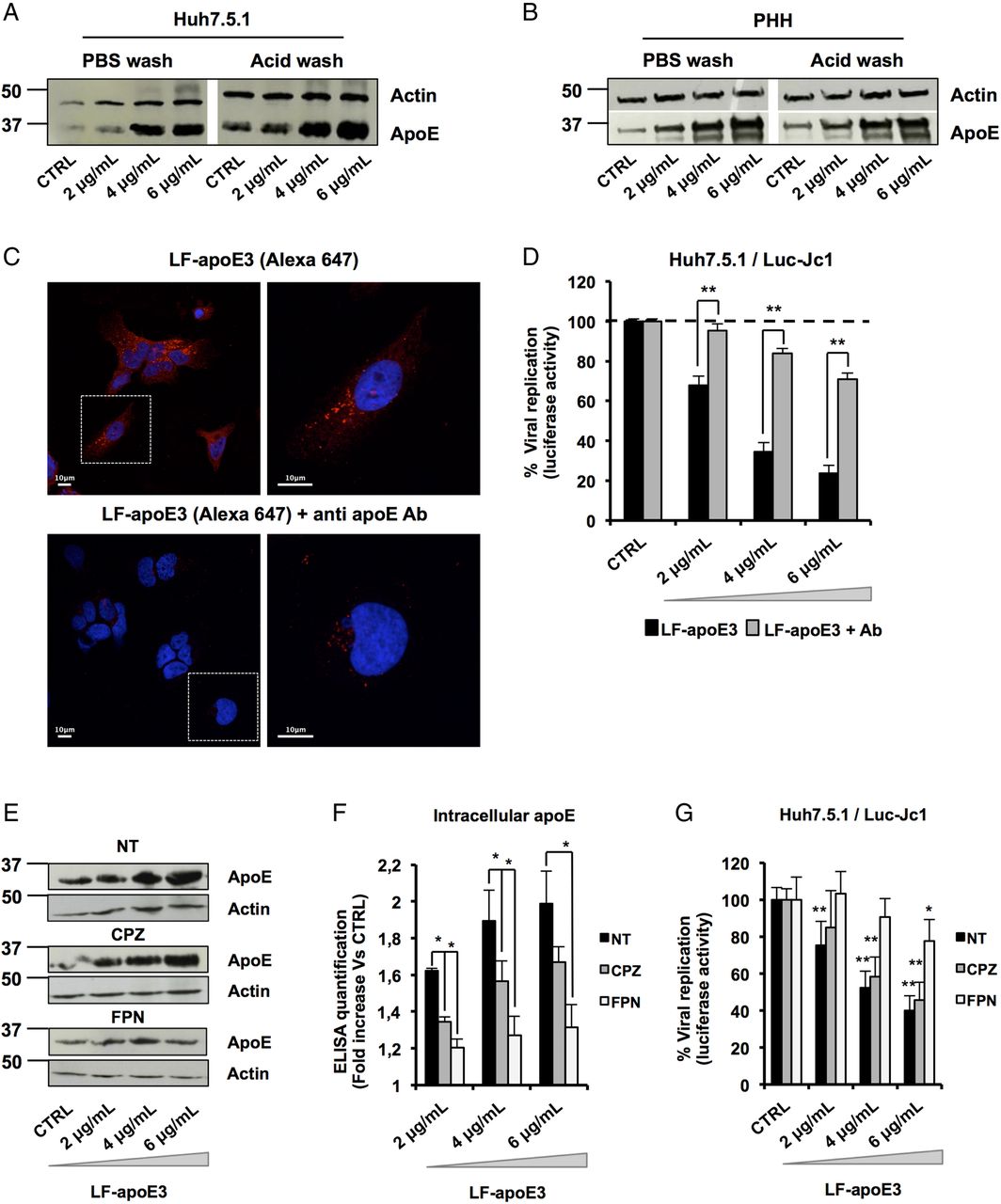
Lipid-free apolipoprotein E3 (LF-apoE3) requires lipid raft integrity to inhibit HCV replication. (A and B) Exogenous LF-apoE3 accumulates in hepatic cells. Huh7.5.1 cells (A) or primary human hepatocytes (PHH) (B) treated with increasing amounts of LF-apoE3 for 1 hour at 37°C. Cells were then washed three times with cold phosphate-buffered saline (PBS) or cold acid buffer (0.2 M glycine buffer+0.15 M NaCl, pH 3.0). Western blot (WB) analysis was performed on cell lysates to detect LF-apoE3 uptake. (C) Exogenous LF-apoE3 labelled with Alexa Fluor 647 is taken-up by Huh7.5.1 cells. Labelled LF-apoE3 alone or premixed with anti-apoE antibody (Ab) during 1 hour was added to HCV-replicating cells for 1 hour at 37°C. Cells were then fixed in paraformaldehyde (PFA) 4% during 20 min at RT and analysed by fluorescence using Leica SP5 II confocal microscope. 4′,6′-Diamidino-2-phenylindole (DAPI)-labelled nuclei are shown in blue. (D) HCV replication was quantified 48 hours post-treatment with LF-apoE3 or LF-apoE3 plus Ab. Results are expressed as % luciferase activity relative to CTRL HCV-replicating cells (100%). Means±SEM from three independent experiments done in triplicate are shown. (E–G) Disruption of lipid raft integrity abolishes LF-apoE3 uptake by hepatocytes. (E) HCV-replicating cells were treated with chlorpromazine (CPZ) (5 μg/mL) or filipin (FPN) (5 μg/mL) for 1 hour before LF-apoE3 treatment. After 1 hour, cells were lysed and WB analysis (E) or ELISA (F) was performed to visualise apoE accumulation in cells. Results are expressed as fold increase of apoE in treated cells, compared with non-treated cells (NT). Means±SD from three independent experiments done in duplicate are shown. (G) In parallel, HCV replication was quantified by measuring luciferase activity 24 hours post-treatment. To facilitate the comparison between the different conditions, the control of each condition (CTRL cells) was set at 100% and the results are presented as a % luciferase activity relative to this CTRL. Means±SD from three independent experiments done in triplicate are shown. *p<0.01; **p<0.001.
To further confirm LF-apoE uptake, we traced fluorescent-labelled LF-apoE3 by confocal microscopy and observed that LF-apoE3 indeed enters into Huh7.5.1 cells (figure 4C). Moreover, preincubation of LF-apoE3 with anti-apoE antibodies blocked LF-apoE3 entry (figure 4C). Furthermore, we observed that the inhibition of HCV replication was lost by blocking LF-apoE3 entry with an apoE antibody (figure 4D). These data indicate that, on one hand, LF-apoE3 uptake is an essential requirement for efficient inhibition of HCV replication and on the other hand that the effect on HCV replication is LF-apoE3-specific.
To investigate the potential pathways of LF-apoE3 entry, we treated cells with chlorpromazine (CPZ), an inhibitor of clathrin-mediated endocytosis, or filipin (FPN), an inhibitor of lipid raft mobility, and followed both LF-apoE3 entry and its impact on HCV replication (figure 4E–G). As shown in figure 4E, LF-apoE3 entry was strongly impaired in FPN-treated cells, compared with non-treated cells suggesting that apoE uptake is mainly lipid-raft dependent. This result was confirmed by intracellular apoE quantification using an ELISA assay (figure 4F). Moreover lipid-raft-dependent entry of LF-apoE3 is mainly required for LF-apoE3-mediated inhibition of HCV replication (figure 4G). In our experimental conditions, CPZ treatment also inhibits LF-apoE3 entry but to a lesser extent compared with FPN treatment. This effect could be due to association of a small part of LF-apoE3 with lipoproteins produced by the cells (figure 4F–G). Collectively, these data suggest that both lipid-raft-dependent and clathrin-dependent pathways may contribute to cell entry of LF-apoE3. However, given the magnitude of pharmacological inhibition, it is likely that lipid-raft-dependent entry is more relevant for LF-apoE3-mediated inhibition of HCV replication.
LF-apoE3 induces an ATP-binding cassette subfamily G member 1 protein-dependent cholesterol efflux that inhibits HCV replication
In 2003, Shi et al40 demonstrated that HCV replication occurs in close proximity to lipid rafts, consisting of cholesterol-enriched microdomains. Thus, depletion of cellular cholesterol might dramatically impair HCV replication.41 Since apoE plays a key role in the hepatic lipid metabolism,11–17 we hypothesised that the inhibition of HCV replication might occur through cholesterol depletion. To address this point, we quantified extracellular cholesterol released from hepatic cells treated with LF-apoE3. Furthermore, we compared the effect of LF-apoE3 with LF-apoE2 and LF-apoE4 on cholesterol release. As shown in figure 5A, we observed a dose-dependent increase in extracellular cholesterol after LF-apoE3 treatment, suggesting a LF-apoE3-dependent cholesterol efflux from hepatic cells. In contrast, LF-apoE2 and apoE4 only induced a modest increase in cholesterol efflux.
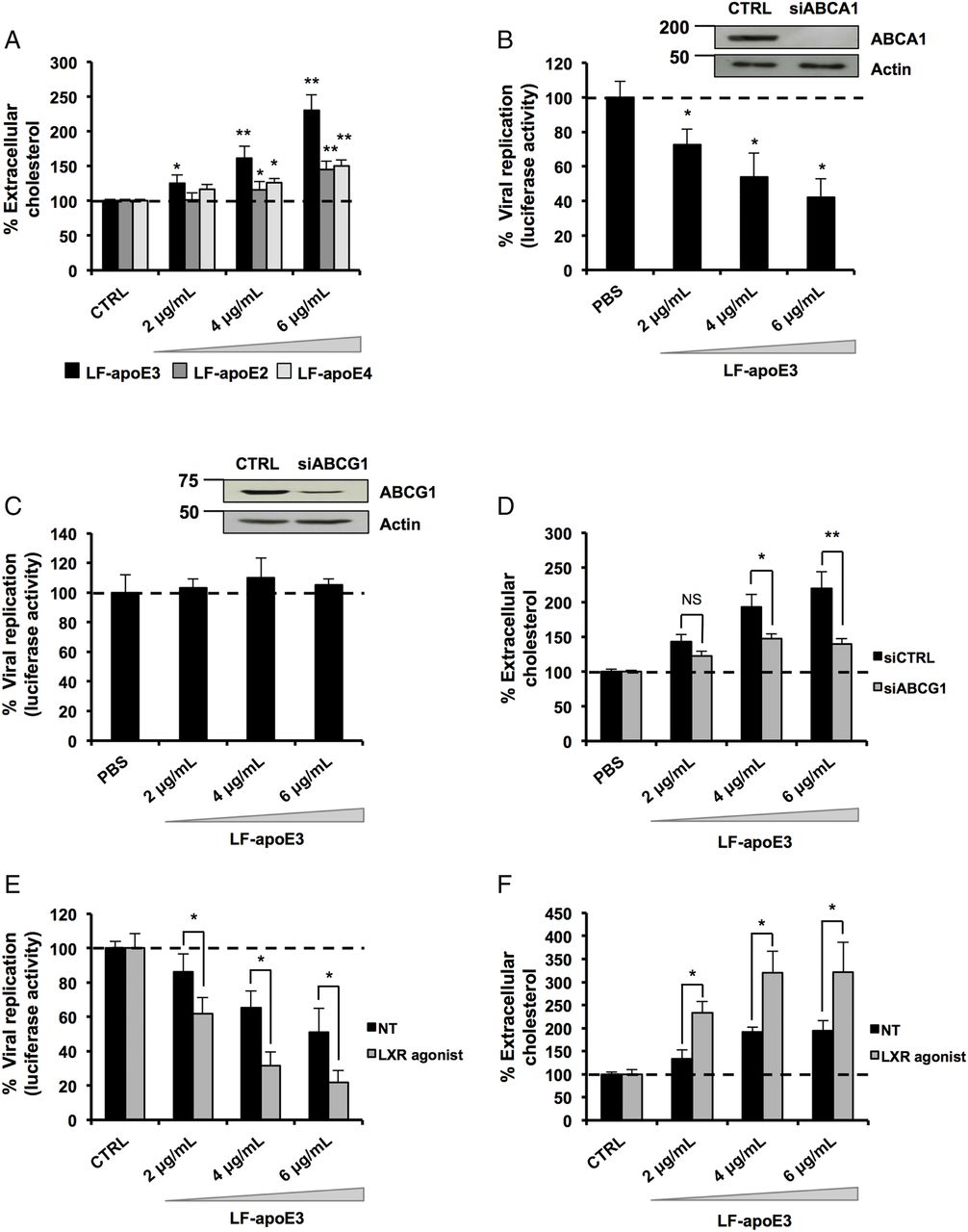
Lipid-free apolipoprotein E3 (LF-apoE3) induces an ATP-binding cassette subfamily G member 1 protein (ABCG1)-dependent cholesterol efflux that inhibits HCV replication. (A) LF-apoE3, but not LF-apoE2 and LF-apoE4, induces a marked cholesterol efflux from Huh7.5.1 cells. Cholesterol efflux assay was performed as described in Materials and methods section using LF-apoE3, LF-apoE2 and LF-apoE4. Means±SEM from two independent experiments done in triplicate are shown. (B) Knockdown of ABCG1 but not ATP-binding cassette subfamily A member 1 protein (ABCA1) alleviates the inhibitory effect of LF-apoE3 on HCV-replicating cells. Two days after ABCA1 (B) or ABCG1 (C) silencing, cells were treated with LF-apoE3 and viral replication was assessed 48 hours post-treatment by luciferase activity quantification. Results are expressed as a % luciferase activity relative to silenced non-treated HCV-replicating cells (set at 100%). Means±SD from three independent experiments done in triplicate are shown. (D) LF-apoE3 induces an ABCG1-dependent cholesterol efflux. Cholesterol efflux assay was performed using LF-apoE3 in cells transfected with small interfering RNA targeting ABCG1 or siCTRL. Means±SEM from five independent experiments done in triplicate are shown. (E and F) An increase in ABCG1 expression improves both LF-apoE3 inhibitory effects on HCV replication and LF-apoE3-dependent cholesterol efflux. HCV-replicating cells (E) or Huh7.5.1 cells (F) were treated with the liver X receptor (LXR) agonist T0901317, at 2 μM for 8 hours, to increase ABCG1 expression. (E) HCV-replicating cells were treated with LF-apoE3 and HCV replication was quantified by luciferase activity measurement. Means±SD from three independent experiments done in triplicate are shown. (F) Cholesterol efflux assay was performed on Huh7.5.1 or Huh7.5.1 cells treated with LXR agonist. Means±SEM from two independent experiments done in triplicate are shown. *p<0.01; **p<0.001.
In hepatocytes, ATP-binding cassettes subfamily A member 1 (ABCA1) and subfamily G member 1 (ABCG1) mainly drive cholesterol efflux. Their function is to transfer intracellular cholesterol to an extracellular acceptor. While ABCA1 is specialised in loading cholesterol on LF-apolipoprotein AI (apoAI) to form pre-HDL, ABCG1 transfers cholesterol directly to HDL.42 We addressed a possible role for ABCA1 and ABCG1 in LF-apoE3-mediated inhibition of HCV replication. HCV replication was measured in ABCA1-silenced or ABCG1-silenced cells after LF-apoE3 treatment. We observed that, despite efficient silencing of ABCA1, LF-apoE3 inhibited HCV replication (figure 5B). In contrast, the silencing of ABCG1 abolished the inhibitory effect of LF-apoE3 on HCV replication (figure 5C). These results suggest that LF-apoE3 induces an ABCG1-dependent cholesterol efflux resulting in inhibition of HCV replication. To confirm this hypothesis, we performed a cholesterol efflux assay on ABCG1-silenced cells treated with LF-apoE3 (figure 5D). Silencing efficacy was assessed by RT-qPCR (see online supplementary figure S5A). As observed in figure 5D, silencing of ABCG1 markedly reduces the LF-apoE3-dependent cholesterol efflux. To further confirm the role of ABCG1, we increased ABCG1 expression in HCV-replicating cells (figure 5E) and Huh7.5.1 cells (figure 5F) using a LXR agonist (T0901317) and treated the cells with LF-apoE3. Drug efficacy was confirmed by measurement of ABCG1 mRNA using RT-qPCR (see online supplementary figure S5B). We observed that the increase in ABCG1 expression markedly improved the inhibitory effect of LF-apoE3 on HCV replication (figure 5E). Moreover, this result correlates with a strong increase in LF-apoE3-dependent cholesterol efflux (figure 5F). Taken together, these results demonstrate that LF-apoE3 inhibits HCV replication through an ABCG1-dependent cholesterol efflux.
LF-apoE3 induces an increase in apoAI-HDL production
ABCG1 is known to load cholesterol on HDL.42 Since HDL production mainly depends on apoAI synthesis, we wondered whether LF-apoE3 influences the expression level of apoAI in order to favour HDL synthesis. Therefore, we monitored the levels of intracellular apoAI following LF-apoE3 treatment by WB (figure 6A) and ELISA (figure 6B) in Huh7.5.1 cells. We observed that the intracellular apoAI level markedly increased in a dose-dependent manner under LF-apoE3 treatment (figure 6A, B). In contrast, LF-apoE2 and LF-apoE4 treatment did not affect the apoAI protein level, suggesting that the increase in apoAI expression is LF-apoE3-specific (figure 6C, D). Of note, we confirmed that LF-apoE3 increases apoAI expression in PHH (figure 6E, F).

Lipid-free apolipoprotein E3 (LF-apoE3) induces an increase in apolipoprotein AI (apoAI) production in hepatic cells. Huh7.5.1 cells were treated with increasing amounts of LF-apoE3 (A and B), LF-apoE2 (C) and LF-apo4 (D) and primary human hepatocytes (PHH) were treated with LF-apoE3 (E and F). After 48 hours, western blot was performed to detect apoAI protein level (A and C–E). In parallel, intracellular apoAI was quantified after LF-apoE3 treatment in Huh 7.5.1 (B) and PHH (F) using an ELISA. Means±SD from four independent experiments done in triplicate are shown. *p<0.01; **p<0.001.
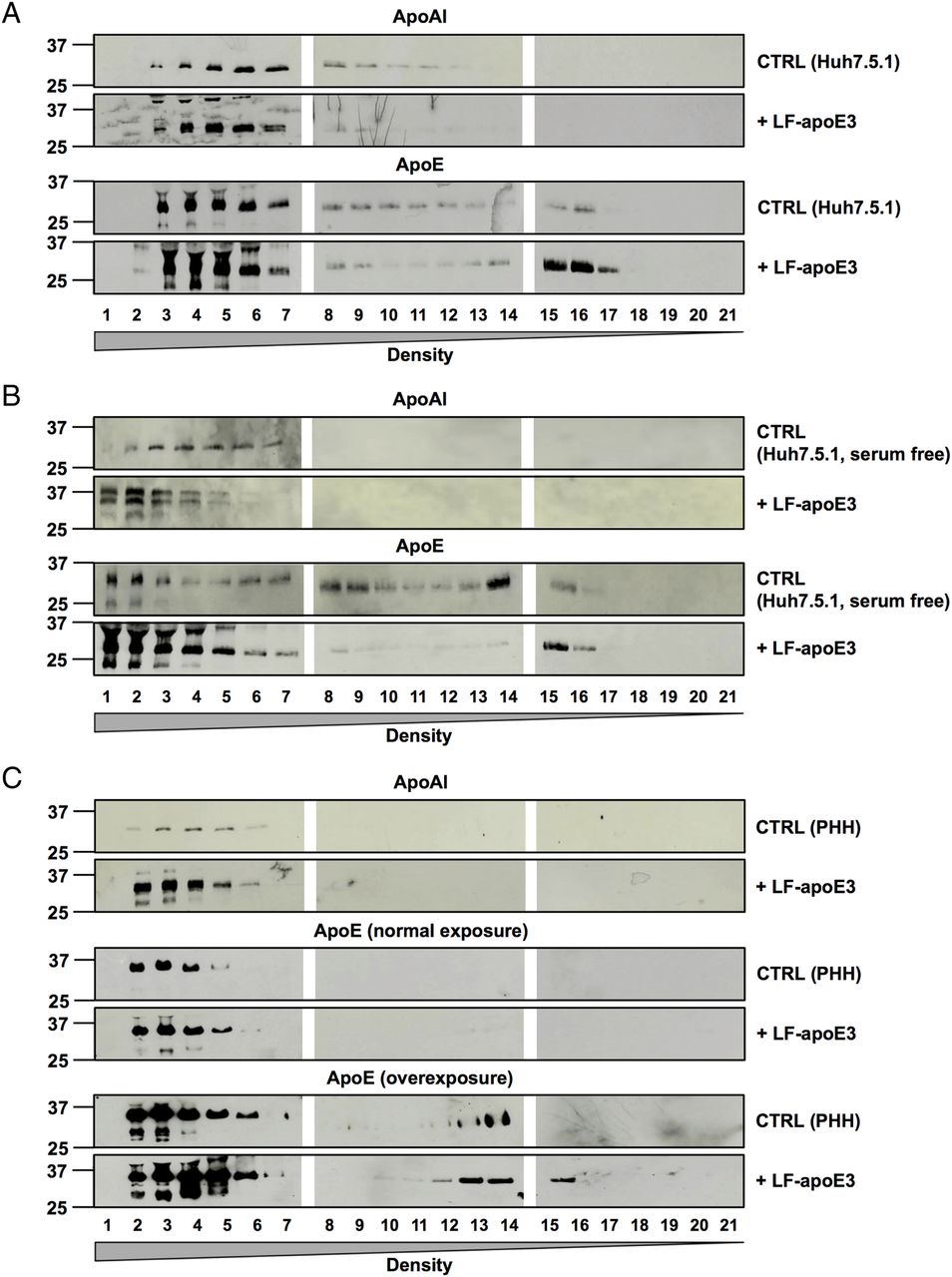
To demonstrate that LF-apoE3 indeed increases apoAI-HDL production, we performed a flotation iodixanol gradient to separate and analyse the lipoproteins produced in cell culture supernatant of CTRL cells or Huh7.5.1 cells treated with LF-apoE327 (figure 7A). Using WB analysis of the fractions harvested, we confirmed that the apoAI protein level was strongly increased in cell supernatants of LF-apoE3-treated cells, compared with CTRL cells (figure 7A, upper panels). Moreover, the apoAI increase was observed in the heavy density fractions (1–7) corresponding to the HDL (figure 7A–C, upper panels). These results indicate that LF-apoE3 increases apoAI-HDL production. A similar profile was obtained for Huh7.5.1 cells grown in serum-free conditions and for PHH (figure 7B, C). These data confirm that LF-apoE3 increases apoAI-HDL production in hepatic cells regardless of the presence of serum.

Lipid-free apolipoprotein E3 (LF-apoE3) induces an increase in apolipoprotein AI (apoAI) high-density lipoprotein (HDL) production. Huh7.5.1 cells grown in complete medium (A), or in serum-free medium (B), and primary human hepatocytes (PHH) (C) were treated with LF-apoE3 (6 μg/mL). After 2 days, cell culture supernatants were harvested. The different lipoproteins were separated by iodixanol gradient and ultracentrifugation. Twenty-one fractions were harvested, and apoAI and apoE protein levels were detected by western blot analysis in each fraction. For PHH, apoE in the lowest density fractions was detected after a long exposure (C).
Finally, we studied the presence of apoE in the different fractions by WB analysis. First, we observed an increase in the apoE protein level in the lowest density fractions (15–21) after LF-apoE3 treatment, suggesting that a part of LF-apoE3 associates with VLDL (figure 7A, bottom panels). The same profile was obtained using serum-free conditions and PHH (figure 7B, C, bottom panels), suggesting that LF-apoE3 associates with VLDL produced de novo by the hepatic cells. Secondly, we observed an increase in the apoE protein level in the highest density (1–7) after LF-apoE3 treatment (figure 7A–C). This corresponds to the exogenous LF-apoE3 protein since the LF-apoE protein, non-associated with lipoproteins, cannot migrate in the gradients. Nevertheless, we cannot exclude an association of LF-apoE3 with the apoAI-HDL produced by the cells. Taken together, our data show that LF-apoE3 promotes apoAI-HDL formation, which in turn becomes available for cholesterol loading by ABCG1 (see figure 8). This mechanism is responsible for the observed inhibition of HCV replication as a consequence of cellular cholesterol depletion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lipid-free apolipoprotein E3 (LF-apoE3) inhibits HCV replication by inducing an ATP-binding cassette subfamily G member 1 protein (ABCG1)-dependent cholesterol efflux and increasing apolipoprotein AI (apoAI)-high-density lipoprotein (HDL) production. In the proposed model, LF-apoE3 enters hepatocytes by interacting with a yet undefined receptor, via a lipid-raft dependent pathway. Following entry, LF-apoE3 induces an increase in apoAI-HDL production and secretion, which in turn becomes available for cholesterol loading by ABCG1. This cholesterol efflux is responsible for the observed inhibition of HCV replication.
Discussion
Using the cell culture-derived HCV model, we report here for the first time that LF-apoE3 specifically inhibits HCV replication through its regulatory role in hepatic lipid metabolism (figure 8). This study highlights a new function for apoE and points out the importance of LF-apoE–hepatocyte interactions to induce a host cell-mediated response affecting viral replication, as a consequence of a modification of the lipid metabolism homeostasis. We demonstrate that LF-apoE3 acts on HCV replication independently of the classical apoE receptors, hinting at the existence of a yet unidentified apoE receptor at the hepatocyte surface able to induce a new signalling pathway. Several studies have already proposed the presence of such a receptor, by demonstrating that LF-apoE3 induces antimitogenic effects in smooth muscle cells and inhibits LTCD4+ and LTCD8+ proliferation via a receptor distinct from the conventional lipoprotein receptors.36 ,43
In line with other studies,31 ,44–48 we showed that LF-apoE3 induces cholesterol efflux from hepatocytes. Moreover, we highlighted for the first time a functional role of ABCG1 in apoE-mediated cholesterol efflux. Since ABCG1 promotes cholesterol efflux on HDL42 and LF-apoE does not directly interact with ABCG1,49 our data suggest that LF-apoE3 acts on HDL metabolism. We demonstrated that LF-apoE3 increases apoAI-HDL production and secretion in hepatic cells. ABCA1 and ABCG1 often work together to form HDL.42 Several studies have shown that apoE can induce ABCA1-dependent cholesterol efflux.47 ,48 However, in our hands, ABCA1 was not involved in LF-apoE3 inhibition of HCV replication. Consistent with this observation, Kiss et al50 demonstrated that lipidation of apoAI leading to HDL formation occurs by both ABCA1-dependent and ABCA1-independent pathways. In 2006, Wang et al,51 demonstrated that ABCG1 can induce movement of cholesterol from endoplasmic reticulum (ER) to the plasma membrane to promote cholesterol efflux to an extracellular acceptor. We thus propose that LF-apoE3 inhibits HCV replication by mobilising cholesterol from ER via ABCG1.
Taken together, our data suggest that LF-apoE3 increases premature apoAI-HDL production by a yet undefined mechanism, which in turn leads to an ABCG1-dependent cholesterol efflux in order to form more mature HDL. This model correlates with an in vivo study showing that apoE reduces atherosclerosis by increasing plasma apoAI-HDL in mice.52 In addition, Zhang and collaborators showed that apoE induces cholesterol efflux without supplementary addition of extracellular cholesterol acceptors,53 supporting the fact that LF-apoE3 induces extracellular acceptors production such as HDL.
Such mechanisms are likely to serve as feedback regulatory loops in a physiological context. We can conceive that after intense formation of VLDL and LDL, extracellular LF-apoE3 would induce cholesterol efflux to prevent hepatotoxic accumulation of cholesterol.
With regard to HCV infection, the LF-apoE3-induced inhibition of replication might appear paradoxical. Indeed, endogenous apoE has been shown to act at multiple steps of the HCV life cycle.7–10 However, in this study, LF-apoE3 is added to the cell culture medium and is thus not included in the pool of endogenous apoE. One explanation could be that HCV might take advantage of the increase in LF-apoE3 concentration in the extracellular environment, as a consequence of high levels of VLDL-associated virion production, to fine-tune its level of replication and thus establish chronicity. Disturbing this balance may present possible means to contain HCV infection.
Collectively, our findings highlight a new mechanism in lipid metabolism regulation but also new interrelationships between mediators of the lipid metabolism and the HCV life cycle. These findings are important for a better understanding of viral pathogenesis and might be explored for antiviral therapy. Moreover, such a mechanism may also represent an opportunity for the development of therapeutic strategies to treat metabolic disorders, such as hyperlipidaemia or atherosclerosis.
Acknowledgments
We acknowledge Dr F Chisari (The Scripps Research Institute, La Jolla, CA) for the Huh7.5.1 cells, R Bartenschlager (University of Heidelberg, Germany), T Watika (University of Tokyo, Tokyo, Japan) and CM Rice (Rockefeller University, New York City, NY, for HCV strains, Dr M Harris (University of Leeds, UK) for the sheep polyclonal anti-NS5A antibody and Dr V Lohmann (Institut for Virologie, Heidelberg, Germany) for the subgenomic replicon system, genotype 1b. We acknowledge M Parnot (INSERM UMR_S1110, Strasbourg, France) for excellent technical assistance, Professor P Pessaux (INSERM UMR_S1110, Institut Hospitalo-Universitaire, Pôle Hépato-digestif, Hôpitaux Universitaires de Strasbourg, France) for providing liver resections for isolation of PHH as well as S Durand and L Heydmann (INSERM UMR_S1110, Strasbourg, France) for PHH isolation. We acknowledge Drs MB Zeisel, O Koutsopoulos and L Mailly (INSERM UMR_S1110, Strasbourg, France) for critical reading of the manuscript. We also thank Dr S Chasserot for the confocal microscopy analysis (INCI UPR3212, CNRS, Strasbourg, France), Dr S Pfeffer and Dr Franck Martin (Institut de Biologie Moleculaire et Cellulaire, UPR 9002, CNRS Strasbourg France) and Dr Jean-Christophe Ame (Institut de Recherche de l’École de Biotechnologie de Strasbourg, UMR7242, CNRS, Strasbourg, France) for setting up experimental procedures.
References
Footnotes
Contributors EC, CS, ERV and TFB wrote the manuscript. EC, CS and ML designed experiments. EC and MAO performed experiments. EC and CS analysed the data. CS initiated and directed the project.
Funding This work has been published under the framework of the LABEX HepSYS [ANR-10-LABX-0028] and benefits from funding from the state managed by the French National Research Agency as part of the Investments for the future programme. EC and ML were supported by a fellowship of the French Ministry for Research and Education (MESR). TFB acknowledges funding by the European Union (ERC-AdG-2014 HEPCIR, EU2020 HEPCAR, EU-Interreg IV-Rhin Supérieur-FEDER-Hepato-Regio-Net) and ARC-IHU (TheraHCC programme) (ARC IHU201301187). CS acknowledges funding by the Agence Nationale de Recherches sur le SIDA (ANRS) (2010-307/2011-415) and the Initiative d'excellence (IdEx) programme (IDEX W13RATCS) from the University of Strasbourg.
Competing interests None declared.
Ethics approval Strasbourg University Ethical Committee (Approval # CPP 10-17).
Provenance and peer review Not commissioned; externally peer reviewed.
Author names in bold designate shared co-first authorship