Article Text

Abstract
Objective The gross majority of colorectal cancer cases results from aberrant Wnt/β-catenin signalling through adenomatous polyposis coli (APC) or CTNNB1 mutations. However, a subset of human colon tumours harbour, mutually exclusive with APC and CTNNB1 mutations, gene fusions in RSPO2 or RSPO3, leading to enhanced expression of these R-spondin genes. This suggested that RSPO activation can substitute for the most common mutations as an alternative driver for intestinal cancer. Involvement of RSPO3 in tumour growth was recently shown in RSPO3-fusion-positive xenograft models. The current study determines the extent into which solely a gain in RSPO3 actually functions as a driver of intestinal cancer in a direct, causal fashion, and addresses the in vivo activities of RSPO3 in parallel.
Design We generated a conditional Rspo3 transgenic mouse model in which the Rspo3 transgene is expressed upon Cre activity. Cre is provided by cross-breeding with Lgr5-GFP-CreERT2 mice.
Results Upon in vivo Rspo3 expression, mice rapidly developed extensive hyperplastic, adenomatous and adenocarcinomatous lesions throughout the intestine. RSPO3 induced the expansion of Lgr5+ stem cells, Paneth cells, non-Paneth cell label-retaining cells and Lgr4+ cells, thus promoting both intestinal stem cell and niche compartments. Wnt/β-catenin signalling was modestly increased upon Rspo3 expression and mutant Kras synergised with Rspo3 in hyperplastic growth.
Conclusions We provide in vivo evidence that RSPO3 stimulates the crypt stem cell and niche compartments and drives rapid intestinal tumorigenesis. This establishes RSPO3 as a potent driver of intestinal cancer and proposes RSPO3 as a candidate target for therapy in patients with colorectal cancer harbouring RSPO3 fusions.
- CANCER
- INTESTINAL STEM CELL
- SIGNALING
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Overactivation of Wnt/β-catenin signalling through APC or CTNNB1 mutation underlies the majority of colorectal cancer cases.
A subset of human colon tumours harbour gene fusions of RSPO2 or RSPO3, associated with enhanced expression of these genes.
R-spondin proteins promote Wnt/β-catenin signalling.
Inhibition of RSPO3 reduces the growth of existing RSPO3-fusion-positive tumours in xenograft models.
What are the new findings?
Rspo3 expression causes rapid development of adenoma and adenocarcinoma in the intestine, establishing RSPO3 as an efficient, causal driver of intestinal cancer. RSPO3 drives extensive crypt hyperplasia and expands multiple crypt components, including Lgr5+ stem cells, Paneth cells, non-Paneth cell label-retaining cells and Lgr4+ cells.
The robust RSPO3-induced phenotype is associated with a modest increase in Wnt/β-catenin signalling.
How might it impact on clinical practice in the foreseeable future?
The potency of RSPO3 to stimulate the crypt stem cells and niche cells, and to actively drive tumorigenesis, proposes RSPO3 as a useful candidate target for therapy in patients with colorectal cancer harbouring RSPO3 fusions.
Introduction
Although in the majority of colorectal cancer cases the Wnt/β-catenin pathway is deregulated through mutations in adenomatous polyposis coli (APC) or β-catenin (CTNNB1), two studies reported gene fusions of the R-spondin family members RSPO2 and RSPO3 in a subset of human colon tumours, accompanied by enhanced expression of these genes.1 ,2 R-spondins are secreted proteins that potentiate Wnt/β-catenin signalling following binding of the leucine-rich repeat-containing G-protein coupled receptors (LGR), in the intestine represented by LGR5 and LGR4. Whereas LGR5 predominantly marks the cycling stem cells at the crypt bottom that fuel the continuous renewal of the intestinal epithelium, LGR4 is present throughout the entire crypt, on cycling stem cells, transient amplifying cells and Paneth cells.3 ,4 Paneth cells fulfil a crucial role in the intestinal crypts by providing the stem cells with a supporting niche.5 In addition to the cycling stem cells, a pool of more quiescent +4 stem cells has been identified that coexpress Lgr5 and Paneth cell markers.6–8 These label-retaining cells (LRCs) serve as Paneth cell precursors and as ‘reserve’ stem cells, able to acquire cycling capacities upon damage.7 ,8
RSPO2 and RSPO3 fusions are mutually exclusive with APC and CTNNB1 mutations in human colon tumours, suggesting that a gain in RSPO can substitute for these most common mutations as an alternative driver of intestinal tumorigenesis.1 Moreover, all the RSPO-fusion-positive colon tumours were described to harbour a mutation in either KRAS or BRAF, mutations that often co-occur in APC mutant settings as well.1 Involvement of RSPO3 in growth of pre-existing tumours was recently shown in RSPO3-fusion-positive xenograft models, where anti-RSPO3 treatment inhibited tumour growth.9 ,10 Whether indeed solely a gain in RSPO3 has the potency to act as a causal driver of intestinal cancer remained to be determined. Here, we demonstrate that RSPO3 is an oncogenic driver, rapidly causing intestinal cancer and extensive crypt hyperplasia, concomitantly stimulating stem cells and supportive niche cells.
Results
Rspo3 expression induces extensive intestinal hyperplasia and tumorigenesis
To investigate the in vivo consequence of a gain in RSPO3, we generated a conditional Rspo3 transgenic mouse model. The used Rspo3 transgene was verified to encode secreted and biologically active RSPO3 protein using β-catenin luciferase reporter assays (see online supplementary figure S1A). In generating the mouse model, the Rspo3 coding sequence was inserted in the inverse orientation, flanked by two pairs of oppositely oriented, homologous Lox sites and behind the ubiquitous CAGGS promoter (figure 1 and see online supplementary figure S1B). This design enables conditional Rspo3 expression upon Cre activity. Cre was provided by crossing our Rspo3invf/+ (Rspo3inv) mice to Lgr5-GFP-CreERT2 (Lgr5) mice11 and activation of the inducible CreERT2 recombinase was achieved by tamoxifen injection at the age of 2 months, unless mentioned otherwise. Single transgenic Lgr5 and Rspo3inv animals served as controls and were treated identically as double transgenic Lgr5;Rspo3 mice. Efficient expression of sense-oriented transgenic Rspo3 upon tamoxifen injection was confirmed in double transgenic Lgr5;Rspo3 animals exclusively and throughout the small and large intestine (see online supplementary figure S1D). Correspondingly, we confirmed RSPO3 protein expression, observed in double transgenic mice only (see online supplementary figure S1E).

Simplified schematic representation of the Rspo3inv mouse model. The Rspo3 coding sequence is inserted in the inverse orientation between two pairs of oppositely oriented Lox sites and behind the CAGGS promoter. Cre activity causes inversion of the Rspo3 transgene into the sense orientation using either one of the homologous LoxP or Lox511 pairs, leading to the middle two transgenic Rspo3 products. Subsequent excision of the remaining sequences in between the homologous Lox sites that are now in the same direction provides a product with sense oriented, irreversible Rspo3 expression. Both intermediate and locked Rspo3 recombination products encode Rspo3 sense mRNA.
Supplemental material
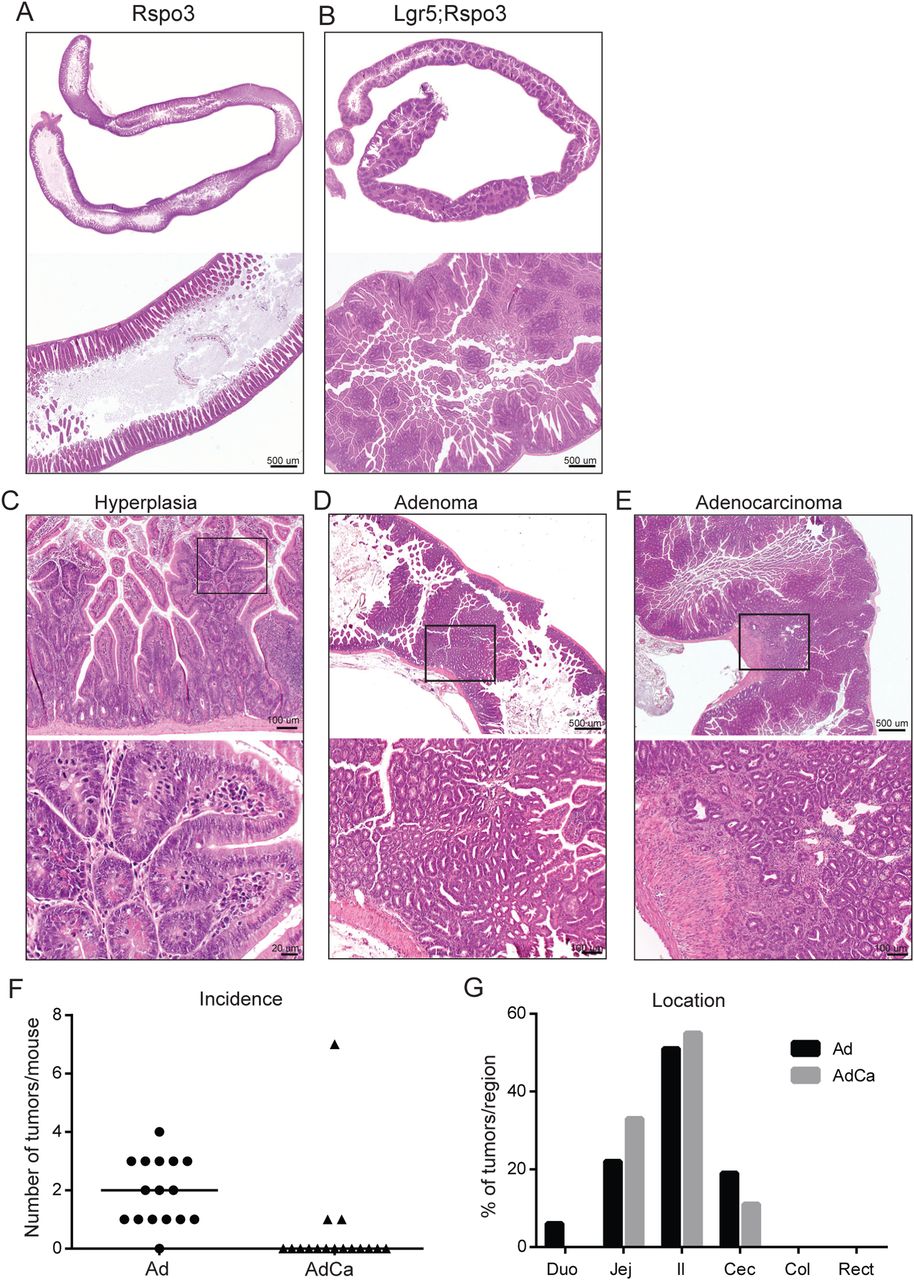
Transgenic Rspo3 expression caused abdominal enlargement and often led to rectum prolapses in double transgenic animals within several weeks, urging their analysis mostly within 2 months following induction. The intestines of Lgr5;Rspo3 mice appeared elongated and distended, and microscopic analysis revealed mucosal thickening caused by extensive epithelial hyperplasia (figure 2A, B). This robust phenotype was observed exclusively though in all double transgenic animals. Hyperplastic lesions extended upwards from crypts to the lumen, integrating with pre-existing villi and appearing as multiple microadenomatous nodules, typically showing dislocated Paneth cells and high rates of mitosis and local necrotic changes (figure 2B, C). These lesions were observed throughout the entire small intestine, in the caecum and proximal colon (see online supplementary figure S2A). In addition, the majority of animals developed adenomas (figure 2D), some of which even progressed to adenocarcinomas as indicated by local invasion (figure 2E). On average 2.5 (±2.3) tumours, mostly adenomas, were detected per Lgr5;Rspo3 mouse, most of which developed in the jejunum, ileum and caecum (figure 2F, G). When tamoxifen was injected at 25 days of age, the phenotype and tumour incidence (average 2.6±2.4, see online supplementary figure S2B) were comparable to that of mice injected at 2 months of age. These data provide evidence that Rspo3 drives rapid intestinal tumorigenesis and extensive hyperplastic growth.
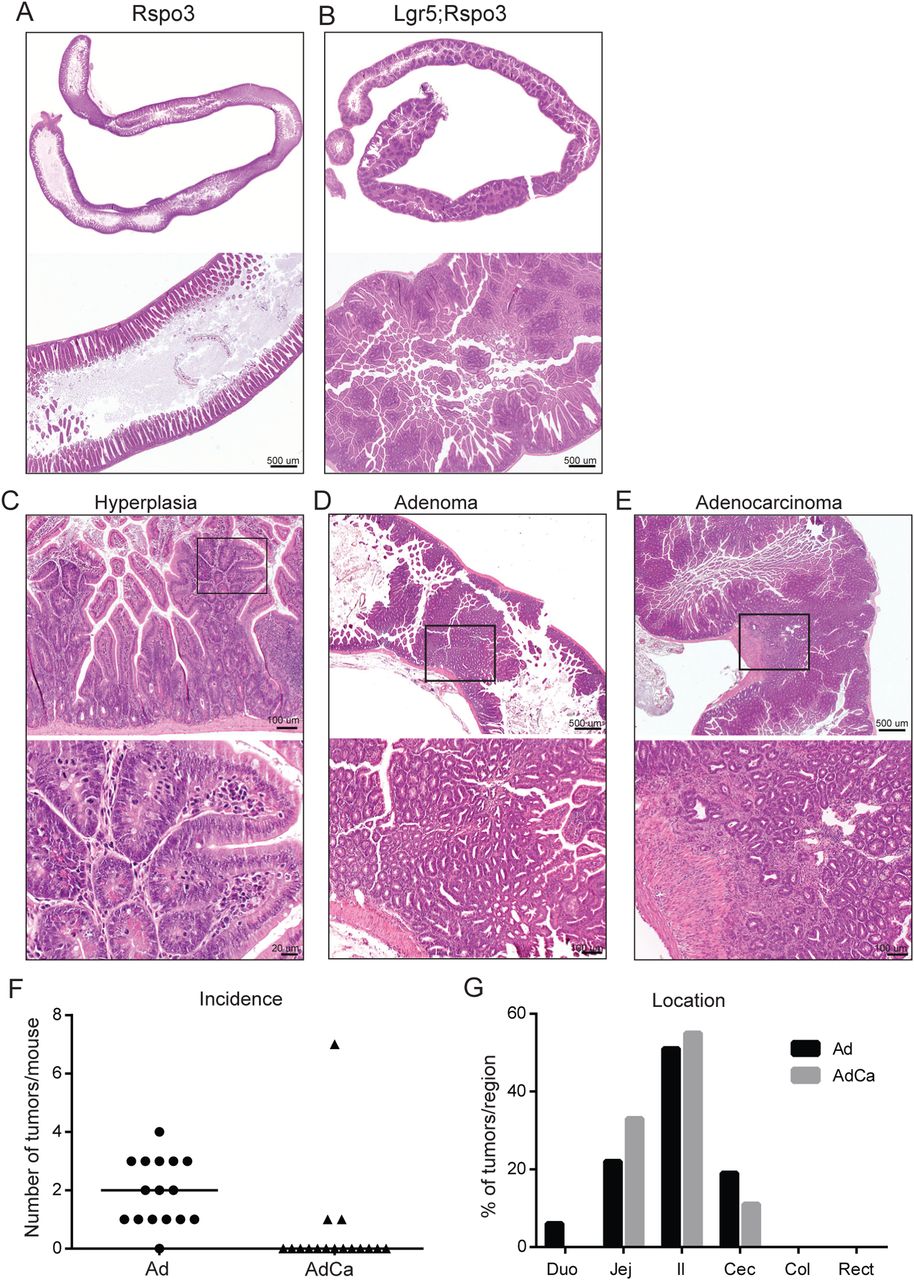
Rspo3 induces extensive intestinal hyperplasia and tumorigenesis. H&E stainings of small intestine of (A) Rspo3inv control and (B-E) Lgr5;Rspo3 mice, including representative examples of (C) hyperplastic, (D) adenomatous and (E) adenocarcinomatous growth, (C–E) lower panels showing an enlargement of the boxed regions. (F) Tumour occurrence in Lgr5;Rspo3 animals (n=16) as illustrated by (F) the amount of tumours per mouse, lines indicating the median, and (G) tumour distribution (percentage of tumours detected per region).
RSPO3 expands Lgr5+ stem cells and Paneth cells
We examined the occurrence of crypt cells by staining for green fluorescent protein (GFP) representing Lgr5-GFP-CreERT2 expressing cells and lysozyme indicating Paneth cells. In Lgr5 control mice, we observed GFP expression specifically in the crypt bottom stem cells and in a stochastic fashion, as has been reported (figure 3A upper panel).11 In the intestines of Lgr5;Rspo3 animals, we noticed an expansion of the Lgr5-GFP+ stem cell zone in hyperplastic and neoplastic lesions (figure 3A). In parallel, we observed a clear increase in the abundance of Paneth cells in the Rspo3-induced lesions in Lgr5;Rspo3 mice (figure 3B). Paneth cells were dislocated, presenting higher upwards towards the lumen, reaching higher up than the Lgr5+ cells. More specifically in adenoma, Paneth cells were observed dispersed throughout the entire tumour, whereas Lgr5-positive cells were present mainly in the lower parts of these lesions. Notably, Paneth cells were prominently present at the invading front of adenocarcinomatous lesions, as were Lgr5+ cells though less consistent. Even in the proximal large intestine, RSPO3 induced the appearance of lysozyme-positive Paneth cells, which are normally absent in the mouse large intestine (figure 3C). In accordance with the abnormal growth and crypt expansion observed in Rspo3 expressing mice, proliferation was clearly enhanced, as well as the amount of Sox9-positive cells (figure 4A, B). Nuclear β-catenin was detected in some Paneth cells though appeared hardly affected by transgenic Rspo3 expression (figure 4C), suggesting a relatively moderate gain in Wnt/β-catenin signalling in our Lgr5;Rspo3 model compared with Apc mutant mouse models. Indeed, we did detect clear nuclear β-catenin in tumours of age-matched and background-matched ApcMin mice (see online supplementary figure S3A). Taken together, RSPO3 imposes hyperplastic, adenomatous and adenocarcinomatous growth in the mouse intestine, accompanied by expansion of the crypt compartment, including Lgr5+ stem cells, Paneth cells and proliferative progenitor cells.

Rspo3 induces expansion of Lgr5 stem cells and Paneth cells. Immunohistochemical staining of (A) GFP and (B) lysozyme in representative hyperplastic and neoplastic lesions in the small intestine. (C) Lysozyme staining showing Paneth cell metaplasia in hyperplastic proximal colon (middle panel) and in adenoma at the caecum-colon transition (lower panel).
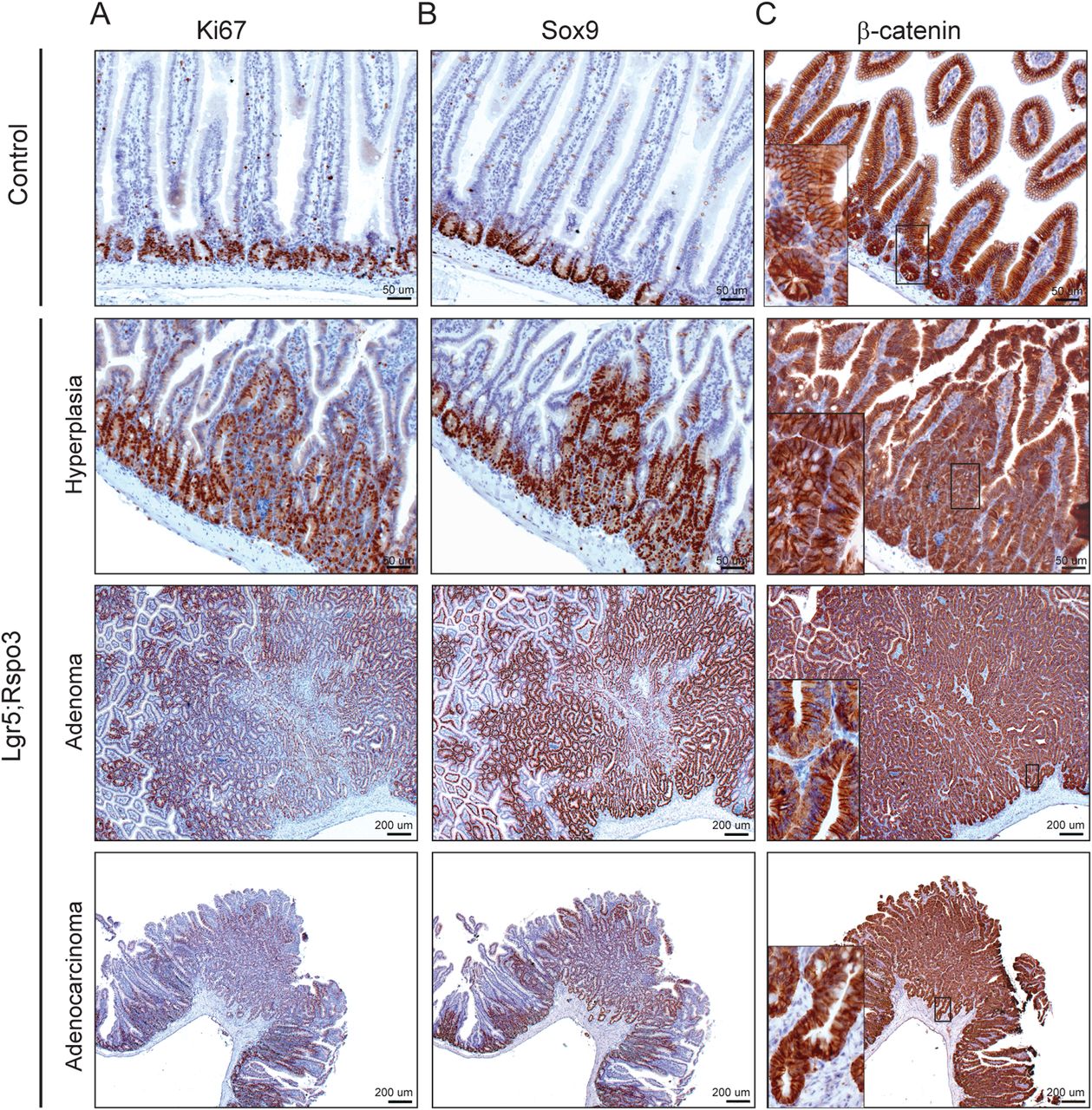
Rspo3-induced lesions are highly proliferative. Immunohistochemical staining of (A) Ki67, (B) Sox9 and (C) β-catenin in representative Rspo3-induced lesions. Inserts show predominant membranous β-catenin expression besides mild nuclear detection in Paneth cells.
RSPO3 causes expansion of additional crypt cell types
In the setting of Apc deletion in Lgr5 cells, Lgr5+ stem cells have been reported as the cells of origin in the developing adenoma.12 ,13 We performed lineage tracing experiments to examine a possible Lgr5+ stem cell derivation of the Rspo3-induced lesions. Lgr5;Rspo3 mice were cross-bred to mTmG Cre reporter mice, enabling detection of an intense GFP signal in the progeny upon Cre-mediated switching, well distinguishable from the weaker GFP staining derived from the Lgr5-GFP-CreERT2 cassette. In Lgr5;mTmG mice without transgenic Rspo3, ribbons of GFPhi staining presented from the crypts upwards into the villi (figure 5A). However, upon Rspo3 expression the overall occurrence of GFPhi cells seemed reduced, appearing less frequent and only in small foci, leaving the vast majority of the Rspo3-induced hyperplasias and tumours untraced (figure 5A). A similar reduction in lineage tracing was observed upon Rspo3 expression using Rosa26-LacZ mice as a Cre reporter (figure 5B). Although stochastic expression and suboptimal efficiency of Cre could cause under-representation of Lgr5-derived lineage tracing, this holds true both for mice expressing as well as lacking transgenic Rspo3, and thus cannot explain the relative reduction in Lgr5-Cre-driven lineage tracing in Rspo3 expressing mice. This indicates that Lgr5+ cells are not the sole cells of origin and alternative cell types must be involved in the development of the Rspo3-driven lesions.

Rspo3 expands additional crypt cell types. (A) GFP immunostaining in the small intestine of Lgr5;mTmG mice with or without transgenic Rspo3. GFPhigh cells indicate mTmG reporter activity, distinguishable from GFPmid cells derived from the Lgr5-GFP-CreERT2 cassette. (B) X-gal staining of Lgr5;R26-LacZ mice with or without transgenic Rspo3. In situ hybridisation showing Lgr4 and Lgr5 mRNA expression (C) in an Rspo3-induced adenoma of an Lgr5;Rspo3 mouse and (D) in a xenograft of a human RSPO3-fusion-positive colon tumour. (E) Representative example of bromodeoxyuridine (BrDU)-lysozyme double immunostaining in Lgr5 single and Lgr5;Rspo3 double transgenic jejunum, 2 weeks after repetitive BrDU injections. Arrows indicate lysozyme-positive label-retaining cells (LRCs) (black) and lysozyme-negative LRCs (grey). (F) Quantification of BrDU-retaining cells in the jejunum, being either lysozyme-negative or lysozyme-positive (2 weeks after BrDU), showing averages and SDs over three independent experiments. *p<0.05, **p<0.01, Student's t-test.
Given that RSPO3 is a secreted protein, most likely it will act not only in an autocrine but also in a paracrine fashion on adjacent cells expressing the LGR5 and LGR4 receptor. Lgr4 has been reported to be expressed throughout the entire crypt compartment, present on crypt base columnar cells, Paneth cells and transient amplifying cells.4 Lgr4 in situ hybridisation revealed that Lgr4+ cells presented abundantly throughout the entire Rspo3-induced lesions, demonstrating the efficient expansion of these cells upon Rspo3 expression (figure 5C). This abundance of Lgr4+ cells is in contrast to the more sparsely present Lgr5+ cells, which were restricted to the lesion bases (figures 3B and 5C). Importantly, a comparable distribution pattern of LGR4+ and LGR5+ cells was observed in xenografts of RSPO3-fusion-positive human colon tumours. In these patient-derived tumours, LGR5 expression was limited and focal, whereas LGR4 was expressed abundantly throughout the entire tumours, confirming the expansion of LGR4+ cells in the human situation (figure 5D).
In addition to the cycling stem cells, more quiescent stem cells were recently identified that coexpress Lgr5 and Paneth cell markers. In contrast to the crypt bottom stem cells, these cells are label-retaining, a feature that also holds true for Paneth cells. Moreover, these quiescent cells which are located at the +4 position serve as Paneth cell precursors and as ‘reserve’ stem cells, acquiring cycling stem cell capacities under specific circumstances.7 ,8 To investigate the influence of RSPO3 on these quiescent cells, we performed repetitive bromodeoxyuridine (BrDU) injections, and allowed label retention for 1 week or 2 weeks, which was the maximum we could achieve given the Rspo3-induced morbidity. Subsequently, double staining was performed for BrDU and lysozyme, allowing discrimination between non-Paneth and Paneth cell LRCs. Transgenic Rspo3 expression clearly enhanced the presence of total LRCs, 1 week and 2 weeks following BrDU injections (see online supplementary figure S3B and figure 5E, F respectively). Moreover, among these LRCs, lysozyme-negative LRCs were especially enhanced upon Rspo3 expression, indicating a relative increase in non-Paneth cell LRCs, or, the presumptive Paneth cell precursors that may acquire stem cell activities.
Together these data indicate that Rspo3 induces lesions that are only partially derived from Lgr5+ cells, though in parallel causes an abundant expansion of Lgr4+ cells, label-retaining Paneth cells and their presumptive quiescent precursors.
RSPO3 imposes a crypt-associated gene expression profile
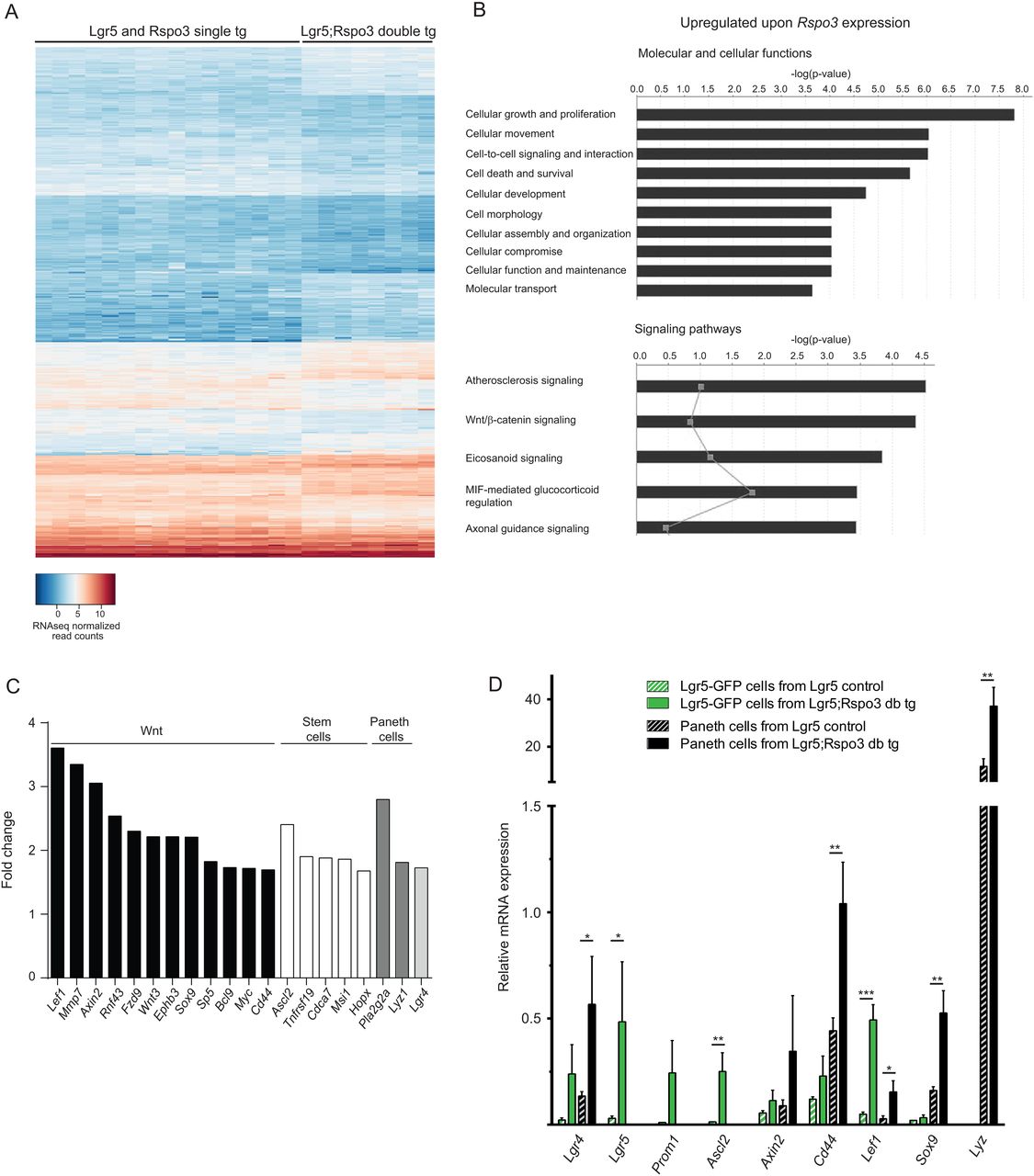
RNAseq analysis was performed on jejunum tissue of Lgr5 and Rspo3inv single transgenic control mice and Lgr5;Rspo3 double transgenic animals (n=8 per genotype). Unsupervised clustering on gene expression profiles showed tight clustering of Lgr5;Rspo3 tissues (see online supplementary figure S4). Lgr5 and Rspo3inv single transgenic controls clustered together, indicating their similarity and justifying to pool them as controls. We found 587 differentially expressed genes of which 255 were increased and 332 decreased (figure 6A, filtered p<0.05 and fold change >1.5). Gene ontology analysis revealed that most significantly enriched molecular and cellular functions upon Rspo3 expression were related to cellular growth, proliferation, movement and signalling (figure 6B). Wnt/β-catenin signalling appeared as the second most significantly upregulated signalling pathway, while an increased expression of phosholipase genes Pla2g5, Pla2g2a, Pla2g2f, Pla2g4c and Pla2g12a underlied high ranking of the majority of the other pathways, except for the axonal guidance pathway (figure 6B and see online supplementary figure S5A). Taking a closer look at individual genes involved in cellular proliferation, growth and Wnt/β-catenin signalling in the intestine, we found a broad panel of genes among the upregulated genes. In addition to 12 upregulated Wnt signalling related genes, we observed increased transcripts of crypt stem cell markers Ascl2, Hopx, Cdca7, Tnfrsf19 and Msi1 and of Paneth cell associated genes Lyz1 and Pla2g2a (figure 6C). Also, enhanced Lgr4 expression was observed, corresponding to the above described expansion of Lgr4+ cells. Conversely, differentiation markers of non-crypt cell lineages were downregulated, among which Sis and Muc2, and most significantly downregulated functions involve metabolism and associated signalling pathways (see online supplementary figure S5B–D). These expression data match our findings of abnormal cellular growth and crypt hyperplasia, including expansion of Lgr4+, Paneth and stem cells and also indicate significantly increased Wnt/β-catenin signalling.

Rspo3 induces a crypt-associated gene expression profile. (A) Heat map of normalised per-sample expression values (2log and n=8 per genotype), including differentially expressed genes upon transgenic Rspo3 expression (filtered p<0.05 and fold change >1.5). (B) Gene ontology analysis showing most significantly enhanced molecular and cellular functions (upper panel) and signalling pathways (lower panel) upon Rspo3 expression. Grey points indicate the gene ratio per signalling pathway. (C) Wnt pathway and crypt-associated genes enriched upon Rspo3 expression. (D) Relative RNA expression levels in sorted Lgr5-GFP and Paneth cells of either Lgr5 control or Lgr5;Rspo3 transgenic mice as determined by qPCR. Bars indicate the averages with SDs from three mice per genotype sorted during independent experiments. *p<0.05, **p<0.01, ***p<0.001, Student's t-test.
To assess whether the increased mRNA levels of crypt-associated genes solely reflects the expansion of crypt cells or also concerns differential gene expression at the cellular level, Lgr5-GFP stem cells and Paneth cells were sorted by flow cytometry6 from Lgr5 single transgenic control and Lgr5;Rspo3 double transgenic mice. Expression of Lgr5 and lysozyme being restricted to the sorted Lgr5-GFP and Paneth cell populations, respectively, confirmed correct identity of the sorted cells. Gene expression analysis using quantitative PCR revealed enhanced expression of stem cell markers Lgr5, Ascl2 and Prom1 in sorted Lgr5+ stem cells upon transgenic Rspo3 expression, and transgenic Paneth cells expressed increased levels of lysozyme. Interestingly, Wnt target genes Axin2, Cd44, Lef1 and Sox9 were upregulated not only in Lgr5+ stem cells, but also in Paneth cells, as was the case for Lgr4 as well. Together these data indicate that RSPO3 promotes the expression of Wnt target genes, stem cell and Paneth cell genes by expanding the population of concerning cells, and additionally by stimulating the expression of these stemness-associated genes at the cellular level.
RSPO3 alters organoid morphology and enables RSPO1-independent growth
Investigating the growth of intestinal organoids from our Lgr5;Rspo3 transgenic mice, we noticed that shortly after seeding, the vast majority of Lgr5;Rspo3 transgenic organoids typically presented as large cysts, or spheres, as has been described for other Wnt activating culture conditions5 and clearly distinct from the appearance of control organoids (figure 7A, B). Lgr5;Rspo3 spheres did not maintain this cystic phenotype over time but rather showed crypt budding and differentiation after several days. The crypt regions developing in Lgr5;Rspo3 organoids showed aberrant morphology, with the lumen appearing open and being relatively large (figure 7A, B and see online supplementary figure S6). Furthermore, whereas control organoids rapidly died following depletion of exogenous RSPO1 from the medium, Lgr5;Rspo3 organoids survived and maintained their typical morphological phenotype as observed in the presence of RSPO1 (figure 7C), indicating the functional replacement of exogenous RSPO1 by the transgenic RSPO3. Robust proliferation capacity of the Lgr5;Rspo3 organoids in the absence of exogenous RSPO1 was confirmed by Ki67 staining (figure 7D).
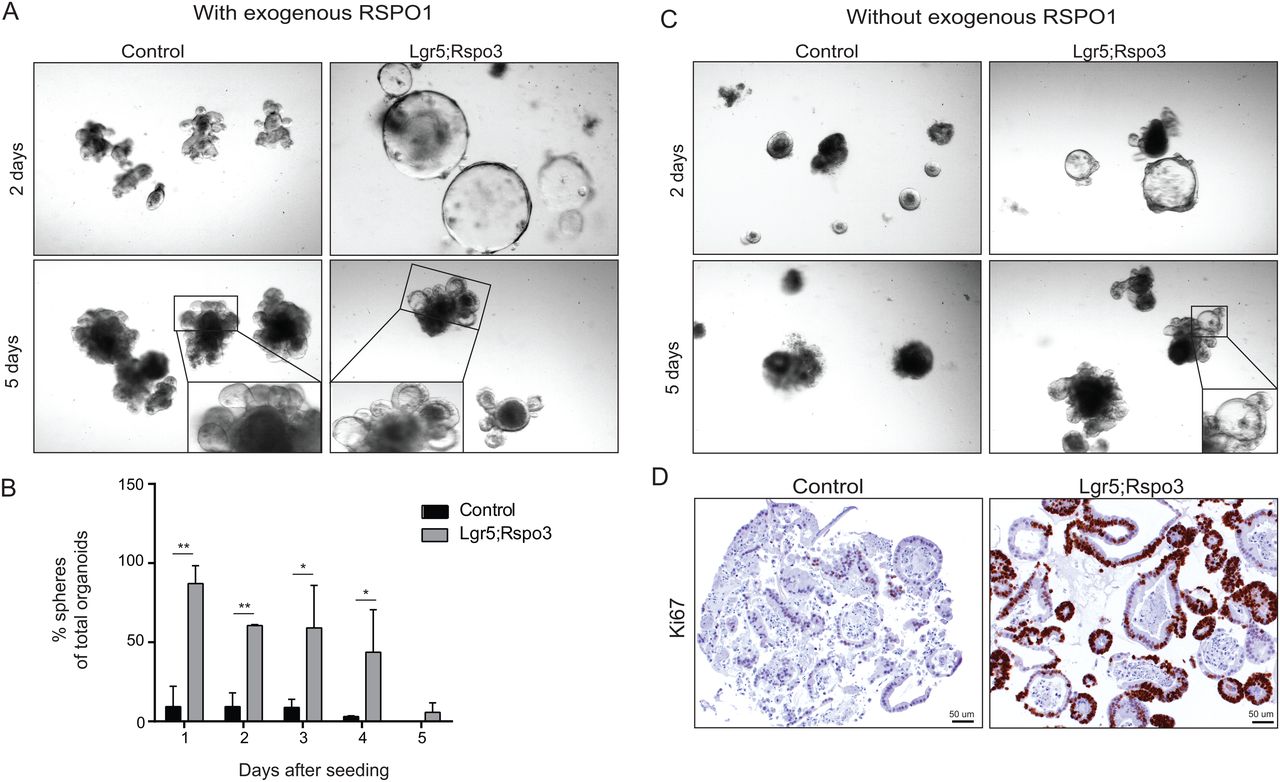
RSPO3 alters organoid morphology and allows RSPO1-independent growth. (A) Organoids grown from control and Lgr5;Rspo3 transgenic mice, 2 (upper) and 5 (lower) days after seeding, showing the increase in spheroid growth, aberrant crypt formation and open lumen upon Rspo3 expression. (B) Percentage of spheres of total organoids in culture, bars indicate the averages with SDs from minimally three experiments. *p<0.05, **p<0.01, Student's t-test. (C) Organoids from control mice disintegrate following the depletion of exogenous RSPO1, whereas Lgr5;Rspo3 organoids survive and show similar phenotype as with exogenous RSPO1. (D) Ki67 staining visualising proliferation absence and presence in control versus Lgr5;Rspo3 organoids, respectively, in the absence of exogenous RSPO1.
Mutant Kras enhances RSPO3-induced hyperplastic, though not neoplastic growth
All human colon tumours with RSPO2 or RSPO3 fusion transcripts harboured a mutation in either KRAS or BRAF.1 To investigate the relevance of mutant Kras in the setting of enhanced RSPO3, we cross-bred our Lgr5;Rspo3 mice with KrasG12V mice. F1 hybrid progeny was injected with tamoxifen at 25 days of age, activating Cre and enabling expression of Rspo3 and of the mutant Kras allele. Triple transgenic mice expressing both Rspo3 and mutant Kras (Lgr5;Rspo3;Kras genotype) appeared more morbid than corresponding Lgr5;Rspo3 controls, although survival did not differ significantly between Lgr5;Rspo3;Kras and Lgr5;Rspo3 cohorts (see online supplementary figure S7). Lgr5;Kras mice displayed no signs of morbidity and accordingly, microscopic analysis of the intestines revealed no phenotypical alterations (figure 8A). However, mutant Kras activation in combination with transgenic Rspo3 expression revealed an increase in the severity of the hyperplastic phenotype (figure 8A), indicating that mutant Kras and RSPO3 synergistically stimulate hyperplastic growth. In contrast, tumour numbers were not significantly different between the Lgr5;Rspo3 and Lgr5;Rspo3;Kras cohorts (figure 8B). Restricted to the Lgr5;Rspo3;Kras cohort, three cases of adenomatosis were observed, in which animals displayed multiple adenomatous lesions throughout the small intestine, merging into one another and hampering exact quantification. Given the reported synergy between mutant Kras and mutant Apc on Wnt/β-catenin signalling,14 we performed immunohistochemical β-catenin stainings on the intestines of mice expressing transgenic Rspo3 with and without mutant Kras (figure 8C). Nuclear translocation of β-catenin was hardly observed both in Lgr5;Rspo3 and Lgr5;Rspo3;Kras intestines. Accordingly, qPCR analyses showed that RSPO3 increased the expression of Axin2, Cd44, Sox9 and Rnf43, but coexpression of the mutant Kras allele from the Lgr5-GFP-CreERT2 cells did not raise the expression levels any further (figure 8D). Together these data indicate that in this mouse model, when expressed in Lgr5+ cells, mutant Kras synergises with RSPO3 in hyperplastic growth, but not in promoting Wnt signalling or tumorigenesis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mutant Kras and Rspo3 synergise in hyperplastic though not neoplastic growth. (A) H&E stainings of small intestine of Lgr5;Kras, Lgr5;Rspo3 and Lgr5;Rspo3;Kras mice, showing augmentation of the hyperplastic phenotype in triple transgenic mice. The right-most panel shows a representative example of adenomatosis as observed in Lgr5;Rspo3;Kras mice exclusively. (B) Number of adenoma (left panel) and adenocarcinoma (right panel) observed in Lgr5;Rspo3 and Lgr5;Rspo3;Kras mice, lines indicating the median. Open triangles represent cases of adenomatosis, where tumour numbers are an estimation of ≥20. (C) β-catenin immunostaining on representative neoplastic lesions in small intestine of Lgr5;Rspo3 and Lgr5;Rspo3;Kras mice. Inserts show that nuclear β-catenin detection is not affected by mutant Kras. (D) Relative expression of Wnt target genes in the jejunum (n=8 per genotype). **p<0.01, ***p<0.001, Student's t-test.
Discussion
Recognising the prominent role of APC and CTNNB1 mutations in human colorectal cancer, many mouse studies have investigated the effects of activating the associated Wnt/β-catenin signalling pathway, showing proliferative and tumorigenic phenotypes.12 ,15–21 Gene fusions leading to enhanced RSPO3 expression have been proposed as a possible alternative to these most common APC or CTNNB1 mutation routes in driving intestinal cancer.1 We show that transgenic Rspo3 expression induces a proliferative phenotype resembling that of other Wnt activating mouse models. Moreover, we provide in vivo evidence that RSPO3 stimulates stem cell and niche compartments and drives rapid intestinal tumorigenesis. In line with reported Wnt/β-catenin promoting activities of R-spondin, transgenic Rspo3 expression potentiated the Wnt signalling pathway, as was apparent from multiple gene expression analyses and cystic organoid morphology. We demonstrated that RSPO3 enhanced Wnt/β-catenin signalling both in Lgr5+ stem cells and Paneth cells. Despite this, we hardly observed nuclear translocation of β-catenin in RSPO3-induced lesions, indicating that the gain in Wnt signalling is relatively low, especially compared with Apc mutant mouse models. In this regard, it might be interesting to investigate the involvement of alternative signalling factors, and the collective increase of five phospholipase genes propose these as interesting candidates. Among those, Pla2g2a has recently been described as an important regulatory stem cell niche factor.22
Thus, while RSPO3 raises Wnt/β-catenin signalling to a relatively modest level, it drives tumorigenesis efficiently. Importantly, our data also demonstrate that RSPO3 causes an expansion of Lgr5+ stem cells, Paneth cells, Lgr4+ cells and LRCs. The capacity to stimulate stem cells and niche cells by promoting their Wnt signalling and causing their expansion likely provides RSPO3 with tumorigenic potential. After all, persistent overstimulation of stem cells and their niche cells provides a solid basis for abnormal, uncontrolled tissue growth. This is supported by the recent finding that anti-RSPO3 treatment of existing RSPO3-fusion-positive tumour xenografts inhibits tumour growth and reduces stem cell marker expression.9 ,10 We demonstrate that stimulation of stemness by RSPO3 thus goes beyond stimulating the Lgr5+ stem cells themselves and includes stimulation of Lgr4+ cells, Paneth cells and their presumptive precursors. Besides providing Lgr5+ stem cells with a stemness-supporting niche, it is of interest to determine whether these alternative, expanded cell types contribute to RSPO3-driven tumorigenesis as a cell of origin. In this regard, very limited lineage tracing was observed from Lgr5+ cells in RSPO3-induced lesions, indicating that the lesions derive from alternative cell types. Supportively, quiescent Paneth cell precursors have been described to acquire cycling stem cell properties upon tissue damage, serving as a reserve stem cell pool, and more differentiated intestinal cell types have been shown to act as tumour initiating cells under specific circumstances.8 ,19 ,23
Taken together, we demonstrate that RSPO3 stimulates Lgr5+ stem cells, Lgr4+ cells, Paneth cells and their presumptive quiescent precursors by causing their expansion as well as promoting the expression of related genes, including Wnt/β-catenin signalling genes. Considering that R-spondins are secreted proteins, this implies that RSPO3 may potentially influence a broad spectrum of stem cell and niche cell types in patients with colorectal cancer harbouring RSPO3 fusions. Unfortunately, not much knowledge exists to date about the occurrence and functioning of alternative stem cells, Paneth cells and their precursors in the human colon, besides that part of human colon tumours harbour Paneth cells, and their presence in adenoma is associated with an increased risk for developing additional colonic adenomas.16 ,24 ,25 We provide in vivo evidence that RSPO3 stimulates the intestinal stem cell and niche compartments and drives rapid tumorigenesis, proposing RSPO3 as a promising candidate therapy target for patients with colorectal cancer carrying RSPO3 fusions.
Materials and methods
Generation of Rspo3inv transgenic mouse model
A detailed description of the generation of the Rspo3 cassette that allows conditional expression of Rspo3 after Cre activation, and the generation of the FVB.129P2-Gt(Rosa)26Sor6(CAG-Rspo3)Nki/A (MGI:5697338, abbreviated to Rspo3inv) transgenic founder strain is provided in the online supplementary information. Shortly, the Rspo3 coding sequence was inserted in antisense orientation between two sets of non-homologous Lox sites in a head to head orientation, 3′ to the CAGGS promoter in a Rosa26 gene targeting cassette.26 The obtained Rspo3inv construct (figure 1 and see online supplementary figure S1A) was introduced into strain 129 derived IB10 E14ES cells by electroporation and selected embryonic stem (ES) cells were injected into 129/Ola blastocysts. Founder strains were back-crossed for five times to 129/Ola, followed by more than nine generations back-crossing to FVB.
Mouse strains and treatments
All mouse strains were maintained heterozygous. The Rspo3inv mice were provided with inducible Cre by cross-breeding with Lgr5-GFP-CreERT2 transgenic mice11 on FVB background. Cre was activated by intraperitoneal tamoxifen injection (200 µL 10 mg/mL in corn oil) of single and double transgenic progeny at the age of 2 months, followed by analysis 1–2 months later, unless mentioned otherwise. BrDU was administered by intraperitoneally injecting 100 µL (10 mg/mL in phosphate-buffered saline (PBS)), six times on two consecutive days, and 2 weeks after tamoxifen. Double transgenic Lgr5-GFP-CreERT2;Rspo3inv mice were cross-bred to mTmG mice (back-crossed to FVB),27 Rosa26-LacZ mice,28 and KrasLSL G12Vgeo mice.29 Progeny was injected with tamoxifen at day 25, followed by analysis within 1–2 months.
Histology and immunohistochemistry
Intestines were formalin-fixed and paraffin-embedded (FFPE), followed by H&E staining according to routine protocols. Cultured organoids were fixed in 4% PFA and embedded in low-melting agarose prior to paraffin embedding. Immunohistochemistry was performed on consecutive sections using antigen retrievals, antibodies and visualisation as indicated in online supplementary table SI. For quantification of BrDU/lysozyme-positive cells, Aperio ImageScope software was used to precisely discriminate single or double staining for each cell. Twenty-five GFP-positive jejunum crypts were counted per mouse, followed by averaging the three experiments. X-gal staining was performed as described in the online supplementary information.
RNA in situ hybridisation
RNA in situ hybridisation was performed using RNAscope 2.0 high definition (HD) Brown for FFPE (Advanced Cell Diagnostics (ACD)) and following manufacturer's instructions with minor adjustments. Pretreatment (1) 10 min RT, pretreatment (2) boil 15 min and pretreatment (3) 10 min in 40°C hybridisation oven. Probes Mm Lgr4 318321, Mm Lgr5 312171, Hs LGR4 460551 and Hs LGR5 311021 (ACD) were hybridised 2 hours at 40°C. Sections of xenograft tumours derived from RSPO3-fusion-positive human colon tumours were obtained from Crown BioScience.
RNA isolation, cDNA synthesis and expression analysis of whole tissue
For the purpose of RNA sequencing analysis, RNA was isolated from jejunum tissues using TRIzol reagent (Ambion Life Technologies) followed by purification using the MinElute Cleanup Kit (Qiagen). Illumina TruSeq mRNA libraries were generated using the TruSeq RNA Library Preparation Kit v2 (Illumina) and samples were sequenced 51 bp single-end using the Illumina Hiseq2000 platforms (Illumina). Everything was performed according to routine manufacturer’s protocols. Limma's Voom was used for normalisation and expression data were analysed using Ingenuity Pathway Analysis.
For RT-PCR and qPCR purposes, RNA was isolated from jejunum tissues using TissueLyser LT (Qiagen) and RNeasy Plus Mini Kit (Qiagen), followed by cDNA generation using Maxima First Strand cDNA Synthesis Kit (Fisher Scientific). RT-PCR was performed using MyTaq Red DNA Polymerase and qPCR using SensiFAST SYBR Hi-ROX Kit (both GCBiotech). Primers are shown in online supplementary table SII. Expression levels were normalised using Actb.
Crypt isolation, culture and cell preparation for flow cytometric sorting
Small intestinal crypts were isolated and cultured as described,6 ,30 using mice that received Tamoxifen at the age of 25 days. Cultured organoids were dissociated with narrowed Pasteurs' pipettes, seeded into Matrigel (Corning) and maintained for indicated durations, without or with exogenous RSPO1. RSPO1 was supplemented as 10% conditioned medium, derived from HEK293T cells transfected with an Rspo1 expression vector.31
For flow cytometric sorting, isolated crypts were incubated with TrypLE (Life Technologies) and 2000 U/mL Dnase (Sigma) 10 min, 37°C. Dissociated cells were passed through a 40 μm strainer and stained 30 min at 4°C with BV421-conjugated-TER119, BV421-conjugated-CD31, BV421-conjugated-CD45 and APC-conjugated CD24 (all BioLegend). Sorting was performed using FACS AriaIII (BD Bioscience) as described previously.6 In short, single viable epithelial cells (DAPI− and lineage−) were selected based on CD24, GFP and side scatter (SSC). Paneth cells (CD24hiSSChi) and Lgr5-GFPhi stem cells were sorted in three independent runs.
Expression analysis of sorted cells
RNA was isolated following the Trizol reagent protocol (Invitrogen), adding 1 µL Glyco-blue (15 mg/mL Ambion). RNA was converted into cDNA using the high capacity RNA-to-cDNA kit (Applied Biosystems) and employed in a preamplification step using the TaqmanPreAmp Master Mix (Applied Biosystems). Quantitative PCR was performed using TaqMan probes and assays (Applied Biosystems). Everything was performed according to manufacturer's instructions. Expression levels were corrected for Actb expression.
Acknowledgments
The authors thank the Netherlands Cancer Institute’s animal facility, animal pathology department (E Riem, J van Ooij, L de Vrije and J van der Meer) and microarray facility (I de Rink) for their service and technical assistance, and Professor Dr H te Riele for providing FFPE materials of ApcMin mice. The authors also thank A Vendel-Zwaagstra for performing the TOP/FOP β-catenin reporter assay and Professor Dr R Fodde for thoughtful discussion and supporting collaborative experiments.
References
Footnotes
Contributors ERMB and JH: conceived, designed and oversaw the project and wrote the manuscript. Mouse studies: ERMB, NCT, JH and MB. Histology: J-YS, ERMB, NCT and JH. Gene expression analysis: ERMB, MB, GJI, MS. Flow cytometric sorting: AS. Organoid studies: ERMB and MAJK.
Competing interests None declared.
Ethics approval All animal experiments were performed with approval of the Animals Ethics Committee and according to Dutch legislation.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Commentary