Article Text

Abstract
Objective Low-grade chronic inflammation emerges as a potent driver of insulin resistance and glucose dysregulation in obesity and associated non-alcoholic fatty liver disease (NAFLD). The liver, subcutaneous fat and the immune system participate in disturbances of metabolism. Type I interferon (IFN) signalling initiated by innate and adaptive immunity modulates inflammatory responses consequent to infection. However, little is known about the role of type I IFN signalling in metabolic diseases and the development of NAFLD.
Design We determined the impact of type I IFN signalling by tissue-specific deletion of interferon (α and β) receptor 1 (Ifnar1) in hepatocytes (Ifnar1Δhep), adipocytes (Ifnar1Δat), intestinal epithelial cells (Ifnar1ΔIEC) or myelocytes (Ifnar1Δmyel) on glucose metabolism, obesity and hepatic disease in mice exposed to a high-fat or methionine-choline-deficient (MCD) diet. Furthermore, we investigated the expression of type I IFN-regulated genes in patients with obesity undergoing laparoscopic adjustable gastric banding (LAGB).
Results Long chain fatty acids induce type I IFN responses in murine hepatocytes and macrophages and exposure to a high-fat diet elicited type I IFN-regulated gene expression in the liver of wild-type mice. Hepatocyte-specific, but not adipose tissue-specific deletion of Ifnar1 worsened steatosis and inflammation induced by the MCD diet. In contrast, adipose-specific, but not hepatocyte-specific deletion of Ifnar1 deteriorated metabolic dysregulation induced by a high-fat diet, indicated by increased weight gain, insulin resistance and an impaired glucose tolerance. Abrogated type I IFN signalling in myeloid or intestinal epithelial cells did not modulate susceptibility to metabolic or hepatic disease. Improved metabolic control in patients with obesity after LAGB was associated with increased expression of type I IFN-regulated genes in subcutaneous adipose tissue and liver.
Conclusions Our study implicates a role for adipose and hepatocyte type I IFN signalling in diet-induced metabolic dysregulation and hepatic disease. Further studies on type I IFN signalling in metabolic diseases are warranted.
- NONALCOHOLIC STEATOHEPATITIS
- FATTY LIVER
- INTERFERON
- GLUCOSE METABOLISM
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Type I interferon signalling modulates immune responses during infection and has been implicated in autoimmunity and cancer.
Type I interferon signals via the Janus kinase/signal transducer and activator of transcription pathway in a variety of cell types and induce a plethora of interferon-regulated genes (‘interferome’).
Type I interferon signalling suppresses inflammation during autoimmune encephalitis and hepatic injury.
Type I interferons are being used in the treatment of viral hepatitis, multiple sclerosis and cancer.
What are the new findings?
Fatty acids elicit a type I interferon response in hepatocytes and macrophages.
High-fat diet-induced metabolic dysregulation is suppressed by type I interferon signalling in adipocytes.
Methionine-choline-deficient diet-induced hepatic disease is controlled by type I interferon signalling in hepatocytes.
Improved glycaemic control induced by laparoscopic gastric banding in patients with obesity is associated with increased expression of interferon-regulated genes in hepatic and subcutaneous adipose tissue.
How might it impact on clinical practice in the foreseeable future?
Our study implicates a role for type I interferon responses in metabolic dysfunction and associated hepatic disease, which may set the basis for novel therapeutic approaches.
Introduction
Metabolic diseases are dramatically increasing worldwide exemplified by the rising incidence and prevalence (∼30%) of the metabolic syndrome and type 2 diabetes.1 ,2 The metabolic syndrome is a constellation of interrelated risk factors for cardiovascular and metabolic diseases and comprises hallmarks of glucose and lipid dysregulation, elevated blood pressure and obesity (eg, increased waist circumference).1 Besides dietary and lifestyle changes, genetic risk is thought to contribute to developing a metabolic syndrome.3 The underlying mechanisms of diet-induced obesity and insulin resistance are incompletely understood. Recently, accumulating evidence supported that obesity and insulin resistance result in and are modulated by low-grade inflammation.4 Hepatic disease associated with the metabolic syndrome (eg, steatosis) can progress to an inflammatory condition termed non-alcoholic steatohepatitis, in which pathogen-associated molecular patterns drive toll-like receptor-mediated inflammatory processes.5 However, our understanding of signalling hubs that modulate inflammation and promote metabolic diseases is incomplete.
Type I interferon (IFN) signalling modulates immune responses during infection and has been implicated in disease processes such as autoimmunity and cancer.6 ,7 For more than two decades, therapeutic modulation of type I IFN signalling in diseases of the GI tract has been established.8 Toll-like receptors are key transducers of type I IFNs, which instigate immune responses on binding the IFN-α/β receptor composed of two subunits encoded by Ifnar1 and Ifnar2. IFN-α/β receptors are expressed on a variety of cell types and activate Janus kinase/signal transducer and activator of transcription (JAK/STAT) signalling, which regulates the transcription of IFN-stimulated genes that allow cell type-specific immunomodulatory responses of innate and adaptive immunity.9 For example, Plasmodium parasite infection potently induces type I IFN responses that are critical to control hepatic infection.10 IFNAR1 signalling may also suppress inflammatory responses as demonstrated in a model of autoimmune encephalitis.11 In the liver, IFNAR1 signalling protects against fibrosis, ischaemia/reperfusion and toll-like-receptor-induced injury.12–15 Data on the role of type I IFNs in metabolic diseases remain scarce.
To investigate the role played by type I IFN signalling in metabolic processes and inflammation, we generated mice with tissue-specific deletion of Ifnar1 in hepatocytes, adipocytes, myelocytes and intestinal epithelial cells. We exposed tissue-specific knockout mice to a high-fat or methionine-choline-deficient (MCD) diet and analysed metabolic parameters and hepatic disease. We found that adipocyte-specific deletion of Ifnar1 worsened metabolic dysregulation induced by a high-fat diet. Hepatocyte-specific deletion of Ifnar1 worsened inflammation and steatosis in mice fed the MCD diet. Myeloid cell-specific or intestinal epithelial cell-specific deletion of Ifnar1 did not modulate susceptibility to metabolic or hepatic disease. Weight loss and glycaemic control induced by laparoscopic gastric banding (LAGB) in patients with severe obesity is associated with increased type I IFN responses in hepatic and adipose tissue. Our data link adipocyte and hepatocyte type I IFN responses with metabolic and hepatic disease.
Materials and methods
Human studies
Assessment of patients considered for laparoscopic adjustable gastric banding (LAGB) were performed at the Department of Medicine, Innsbruck, Austria. Twenty-eight patients (21 females, 7 male) with a body mass index (BMI) >35 kg/m2 underwent LAGB between 2003 and 2007. Patients with an alcohol intake of >20 g/week were excluded. Other exclusion criteria were statin treatment and other cause of chronic liver diseases such as autoimmune hepatitis, primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), haemochromatosis, Wilson's disease, alpha-1 antitrypsin (A1AT) deficiency and hepatitis B or C. The study protocol was approved by the ethics committee of the Medical University Innsbruck and patients provided written informed consent before LAGB. Liver and abdominal subcutaneous tissue biopsies were taken intraoperatively at LAGB procedure. Six months after LAGB, liver and abdominal subcutaneous adipose tissue biopsies and blood samples from the fasting state were collected and analysed in a longitudinal manner. Clinical parameters were assessed with characteristics shown in table 1.
Clinical characteristics of patients undergoing LAGB
Animal studies
Ifnar1flox/flox mice11 ,16 were crossed onto Cre-expressing mice that conferred tissue specificity: (1) hepatocyte-specific deletion of Ifnar1 in Ifnar1fl/fl;Alb-Cre mice17 (Ifnar1Δhep); (2) adipocyte-specific deletion of Ifnar1 in Ifnar1fl/fl;Fabp4-Cre mice18 (Ifnar1Δat); (3) myeloid cell-specific deletion of Ifnar1 in Ifnar1fl/fl;LysM-Cre mice19 (Ifnar1Δmyel); (4) intestinal epithelial cell-specific deletion of Ifnar1 in Ifnar1fl/fl;Villin-Cre mice (Ifnar1ΔIEC),20 all on a C57/Bl6 background. Mice were bred in the specific pathogen-free (SPF) animal facility in Innsbruck and experiments were performed in compliance with the respective ethics approval (BMWF-66.011/0035-II/3b/2013). Cre-expression was maintained heterozygous, which allowed to use knockout and wild-type littermates. In experiments with a fat diet, mice aged 10 weeks were introduced to a low-fat pellet diet (containing 10% kcal derived from fat; Research Diets, D12450B) for 2 weeks. Then, mice were either kept on a low-fat diet (controls) or exposed to a high-fat pellet diet (containing 45% kcal derived from fat; Research Diets, D12451) for another 14 weeks.21 We did not note differences in body weight at baseline between knockout and wild-type littermates (data not shown). Mice were weighed every other week and a glucose tolerance test (GTT) and an insulin tolerance test (ITT) were performed in the last week of the experiments. In mice fed an MCD diet (MP Biomedicals, California, USA), mice aged 6–8 weeks were subjected to the MCD pellet diet or the methionine-choline control diet (MP Biomedicals) for 24 days as previously described.22 Mice were weighed every other day. Liver and epididymal white adipose tissue (eWAT) was weighed at the closure of the high-fat diet experiments and stored for further analyses at –80°C. We analysed eWAT as it can be reliably identified and weighed and because inflamed eWAT consequent to a high-fat diet has been implicated in the pathogenesis of non-alcoholic fatty liver disease.23 ,24
GTT and ITT
For the GTT, fasting glucose was determined with an ACCU-CHEK glucose meter (Roche Diagnostics) after a starving period overnight (10–12 hours). Then, 2 g/kg glucose (D-glucose, Gibco, Invitrogen) was orally administered by gastric gavage and blood glucose levels were measured at indicated time points with the glucose meter. For the ITT, mice were starved for 4 hours and then 0.75I U/kg insulin (Actrapid, Novo Nordisk) was injected intraperitoneally and blood glucose levels were measured at indicated time points with the glucose meter.
Histology
Tissue sections were formalin-fixed, paraffin-embedded, sectioned and stained with H&E. Samples were analysed in a blinded fashion to sample identity by an independent pathologist. The inflammatory score was composed of the number of foci which harboured cellular infiltrates per high power field (HPF) with five HPFs analysed. Images were captured on a Zeiss AxioCam.
RNA and protein expression analysis
RNA was isolated by homogenisation of cells or tissue samples using TRIzol reagent (Life Technologies). Complementary DNA was transcribed with the Reverse Transcription System (Invitrogen). Quantitative PCR (qPCR) was performed using SybrGreen (Eurogentec) and the Mx3000 qPCR cycler (Agilent Technologies) with the following mouse primer sequences: Ifnar1_forward CCGCTGACTGTGTACTGTG; Ifnar1_reverse ATTTCCAGACGCTCCTCAAA; Ifnar2_forward TCCCCACAGCAAGTGTCA; Ifnar2_reverse GCGCCTGAATCTTCTTCATGC; Ifnα_forward: CCT GTG TGA TGC AGG AAC C; Ifnα_reverse: TCA CCT CCC AGG CAC AGA; Ifnß_forward: CAG CTC CAA GAA AGG ACG AAC; Ifnß_reverse: GGC AGT GTA ACT CTT CTG CAT; Irf7_forward CTGGAGCCATGGGTATGCA; Irf7_reverse AAGCACAAGCCGAGACTGCT. To study IFN-regulated genes in patients with obesity after LAGB, we used the rts-hifnr primer set (InvivoGen). Gene expression was normalised to GAPDH in human studies and to β-actin in mouse studies. Both house-keeping genes were not regulated by experimental conditions (data not shown). Primer sequences for house-keeping genes, inflammatory cytokines and genes involved in lipogenesis are available on request.
Culture and stimulation of hepatocytes and macrophages
Fl83B hepatocytes and RAW264.7 macrophages were purchased from American Type Culture Collection. Cell lines were cultured in F-12K medium and Dulbecco's Modified Eagle's Medium, respectively, both supplemented with 10% fetal calf serum. Fatty acids (Sigma-Aldrich) were dissolved in ethanol. Cells were stimulated with oleic acid (500 µM), palmitic acid (500 µM), cholesterol (500 µM) or docosahexaenoic acid (500 µM) for indicated time points, while 96% ethanol stimulation served as a control as previously described.25 We stimulated with 500 µM long chain fatty acids as this concentration appears in the range of serum lipid concentrations in rodents exposed to a high-fat diet.26
Quantification of fatty acids
Non-essential fatty acids were quantified in serum by a coupled enzymatic reaction system acyl-CoA synthetase-acyl-CoA oxidase (ACS-ACOD) (Wako) according to routine laboratory methods on a Hitachi 902 analyser. For quantification of triglycerides, 100 mg of liver tissue was homogenised in phosphate buffered saline and bovine serum albumin-coated tubes. Liver homogenates or serum were analysed for triglyceride concentration by GPO-PAP method kit (Roche Diagnostics, 11488872) on a Hitachi 902 chemistry analyser.
Statistical analysis
Statistical significance, considered at p<0.05, was determined by an unpaired two-tailed Student's t-test, a one-way or a two-way repeated measures analysis of variance followed by post hoc Bonferroni test where appropriate. Results are shown as mean values with SEM. Data were analysed with GraphPad Prism Software.
Results
Inflammation is closely linked to the emergence of metabolic and therewith associated hepatic disease that arises from dietary changes.4 ,27 We hypothesised that type I IFN signalling impacts on metabolic inflammation and consequently metabolic derangements. In a first step, we tested if dietary components contained within soybean oil and lard (which is used in human diets and for murine metabolic experimentation) elicited a type I IFN response. Indeed, oleic acid and palmitic acid potently elicited IFN-α and IFN-β responses in hepatocytes and macrophages (figure 1A–D), whereas the expression of Ifnar1 and Ifnar2 remained mostly unaffected (see online supplementary figure S1A). Exposure to a high-fat diet did not induce IFN-α in the serum and type I IFNs neither regulated Ifna nor Ifnar1/2 expression in hepatocytes or macrophages (data not shown). In line with type I IFN responses elicited by long chain fatty acids, wild-type mice exposed to a high-fat diet exhibited increased expression of interferon-regulatory factor 7 (Irf7), a type I IFN-induced gene, in hepatic tissue (figure 1E). These studies led us to investigate the impact of type I IFN signalling on diet-induced metabolic diseases by genetic deletion of Ifnar1 in a tissue-specific manner. First, we crossed Ifnar1flox/flox mice11 ,16 with Alb-Cre mice17 to generate hepatocyte-specific Ifnar1 knockout mice. Ifnar1fl/fl;Alb-Cre (Ifnar1Δhep) mice deleted Ifnar1, but not Ifnar2, specifically in hepatocytes (see online supplementary figure S1B), were viable, born at Mendelian ratios and did neither exhibit a spontaneous metabolic nor hepatic phenotype (data not shown).

Long chain fatty acids induce type I interferon (IFN) responses in hepatocytes and macrophages. (A–D) Type I IFN expression analysis of Fl83B hepatocytes (A and B) and RAW264.7 macrophages (C and D) after stimulation with oleic acid (500 µM), palmitic acid (500 µM), cholesterol (500 µM), docosahexaenoic acid (500 µM) or ethanol (control) for indicated time points determined by quantitative PCR (qPCR) and normalised to c-β-actin. Results of three independent experiments shown. (E) IFN-regulated Ifr7 expression determined by qPCR in wild-type mice with indicated diets. Data are expressed as mean±SEM (A–E), n=3–5. One-way analysis of variance (A–E). *p<0.05, ** p<0.01, ***p<0.001. HFD, high-fat diet; LFD, low-fat diet.
supplementary figures
Hepatic IFNAR1 signalling protects against liver inflammation and steatosis induced by the MCD diet
To investigate the impact of hepatocyte type I IFN signalling on metabolic disease, we exposed Ifnar1Δhep mice to a high-fat diet. Although Ifnar1Δhep mice gained significantly less weight on a high-fat diet (see online supplementary figure S1C), we did neither observe a protection from high-fat diet-induced metabolic dysregulation (figure 2A, B, see online supplementary figure S1D–G) nor steatosis (see online supplementary figure S1H, I) when compared with wild-type mice. However, Ifnar1Δhep mice exposed to the MCD diet exhibited increased steatosis (figure 2C–E) along with increased hepatic triglyceride concentrations (figure 2F). These changes were not associated with altered expression of lipogenic genes (figure 2G and see online supplementary figure S1J). Furthermore, Ifnar1Δhep mice exposed to the MCD diet exhibited increased hepatic inflammation (figure 2H–K), although they were clinically indistinguishable from wild-type littermates (see online supplementary figure 1K–M). These data established that hepatocyte-restricted type I IFN signalling protected against hepatic disease induced by the MCD diet but not metabolic dysregulation induced by a high-fat diet.

Hepatic type I interferon (IFN) signalling suppresses steatosis and inflammation induced by the methionine-choline-deficient (MCD) diet. (A and B) Mice were exposed to a high-fat (HFD) diet and were analysed after 16 weeks, (A) glucose tolerance test, (B) insulin tolerance test. (C–K) Mice were exposed to the MCD diet for 24 days, (C) histological steatosis assessment based on H&E sections expressed in percent, (D) representative macroscopic images from liver specimen. (E) Representative H&E images. (F) Triglyceride concentration in mg/g liver. (G) Expression of Fas in the liver of indicated genotypes determined by quantitative PCR (qPCR) and normalised to c-β-actin. (H) Histological assessment of hepatic inflammation on H&E sections. (I–K) Expression of inflammatory cytokines (IL-1β, Cxcl1, TNF-α) in the liver of indicated genotypes determined by qPCR and normalised to c-β-actin. Data are expressed as mean±SEM, n=10 (A, B), n=5 (C–K). Two-way repeated measures analysis of variance (A, B), unpaired two-tailed Student's t-test (C, F–K). * p<0.05, ** p<0.01.
Adipose tissue IFNAR1 signalling suppresses metabolic dysregulation induced by a high-fat diet
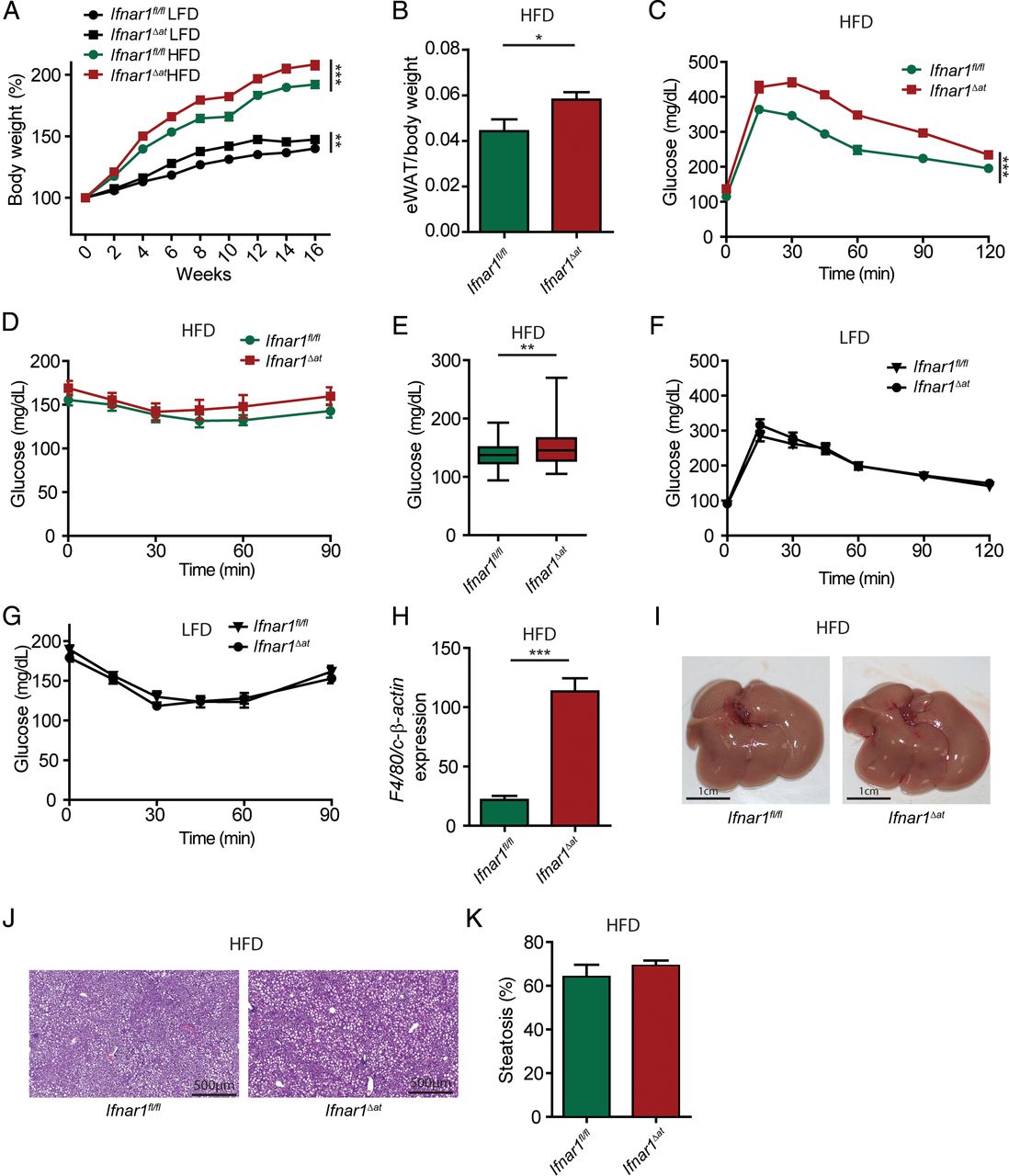
As a high-fat diet strongly induced the expression of Ifnar1 in adipose tissue (see online supplementary figure S2A), we aimed at investigating the role of adipocyte IFNAR1 signalling on diet-induced disease. To do so, we crossed Ifnar1fl/fl mice with Fabp4-Cre mice18 to generate mice with adipose-specific deletion of Ifnar1 (Ifnar1Δat) (see online supplementary figure S2B). Ifnar1Δat mice were viable, born at Mendelian ratios and did not exhibit a spontaneous metabolic or hepatic phenotype (data not shown). Ifnar1Δat mice exposed to a high-fat diet gained significantly more weight compared with controls (figure 3A) and exhibited increased eWAT (figure 3B). Importantly, Ifnar1Δat mice on the high-fat diet demonstrated decreased glucose tolerance and a slightly increased insulin resistance compared with controls (figure 3C–E). Although Ifnar1Δat mice gained more weight on a low-fat diet (figure 3A), but not on a normal chow diet (see online supplementary figure S2C), we did neither observe impaired glucose tolerance nor increased insulin resistance compared with wild-type mice on a low-fat diet (figure 3F, G). These data established that IFNAR1 signalling in adipose tissue suppressed metabolic dysregulation induced by a high-fat diet. This observation was independent from systemic triglyceride or non-essential fatty acid concentrations (see online supplementary figure S2D,E). However, we noted increased expression of F4/80, a marker of macrophages, in adipose tissue from Ifnar1Δat mice (figure 3H). Notably, we did not observe increased steatosis in Ifnar1Δat mice on a high-fat diet (figure 3I–K). In line with these data, we did not find increased susceptibility to MCD-induced hepatic disease in Ifnar1Δat mice (see online supplementary figure S2F–I).

Adipose type I interferon (IFN) signalling suppresses metabolic dysregulation induced by a high-fat diet (HFD). (A–K) Mice were exposed to a HFD or low-fat (LFD) (control) diet and were analysed after 16 weeks. (A) Body weight in per cent at indicated time points. (B) Epididymal white adipose tissue (eWAT) weight. (C) Glucose tolerance test, (D) insulin tolerance test. (E) Glucose concentration over the course of the ITT. (F, G) Mice were exposed to a LFD and were analysed after 16 weeks, glucose tolerance test (F), insulin tolerance test (G). (H) Expression of the macrophage marker F4/80 in adipose tissue of indicated genotypes determined by quantitative PCR and normalised to c-β-actin. (I) Representative macroscopic images of the liver after 16 weeks on a HFD, with representative H&E images (J) and steatosis quantification in (K). Data are expressed as mean±SEM (A–K), n=10 (A–K). Two-way repeated measures analysis of variance (A, C, D, F and G), unpaired Student's t-test (B, E, H, K). *p<0.05, **p<0.01, ***p<0.001.
Neither myeloid nor intestinal epithelial IFNAR1 signalling modulates susceptibility to metabolic or hepatic disease induced by a high-fat or MCD diet
Dysbiosis has been implicated in hepatic and metabolic diseases.28 We have recently described intestinal dysbiosis in mice that specifically lack Ifnar1 in intestinal epithelial cells (Ifnar1fl/fl;Villin-Cre or Ifnar1ΔIEC mice).20 Thus, we tested whether intestinal epithelial Ifnar1 deletion impacted on the susceptibility to developing hepatic or metabolic disease consequent to exposure with a high-fat or MCD diet. However, Ifnar1ΔIEC mice exhibited comparable weight gain (figure 4A), clinical parameters (see online supplementary figure S3A, B) and a similar impaired glucose tolerance compared with wild-type controls on a high-fat diet (figure 4B, C and see online supplementary figure S3C,D). Similarly, epithelial deletion of Ifnar1 did not lead to increased susceptibility to hepatic disease in mice on the MCD diet (figure 4D, E and see online supplementary figure S3E–G).
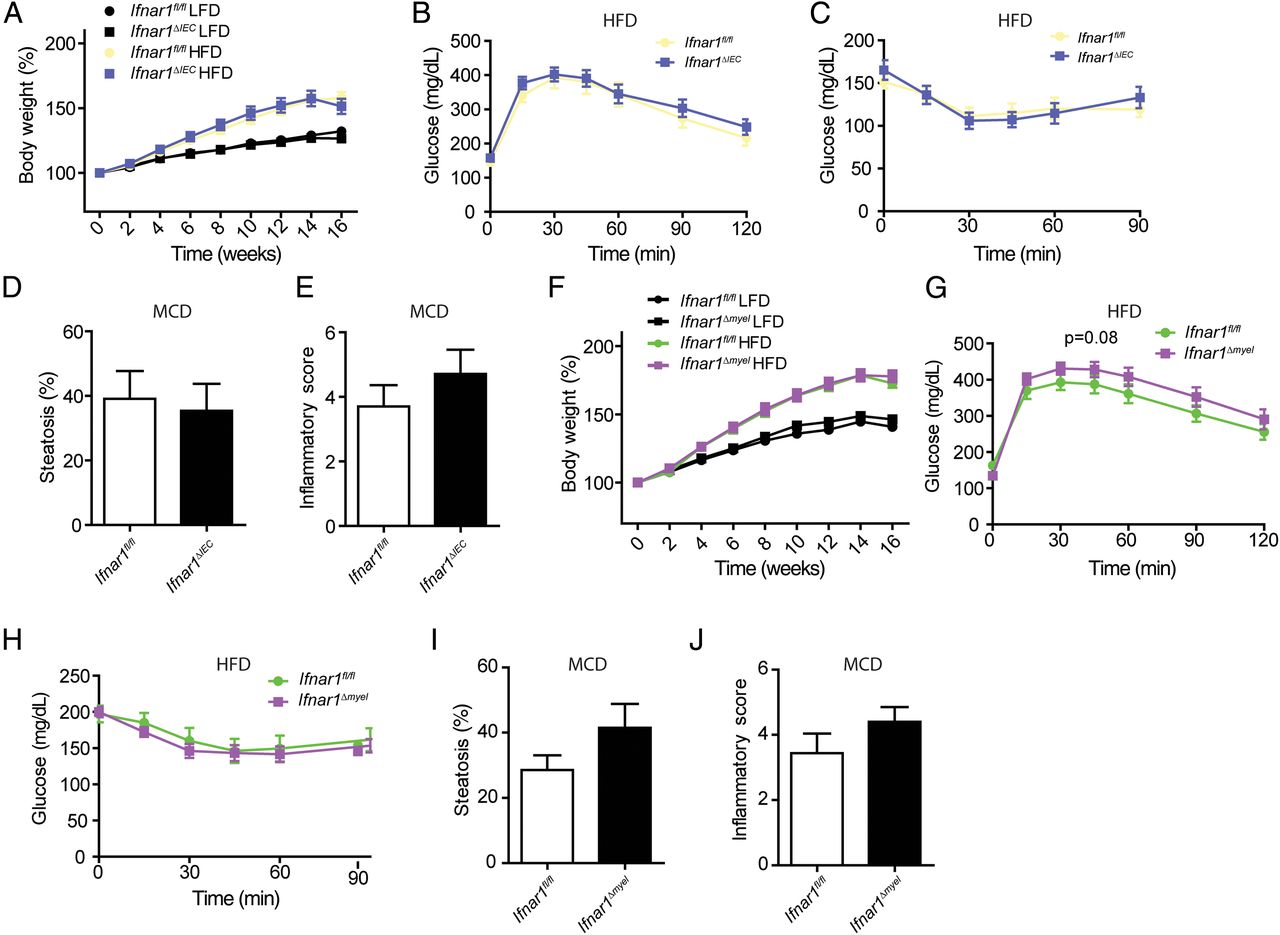
Myeloid and intestinal epithelial type I interferon (IFN) signalling does not impact on metabolic dysregulation or hepatic disease. (A–C) Indicated genotypes were exposed to a high-fat diet (HFD) or low-fat diet (LFD) (control) diet and were analysed after 16 weeks. (A) Body weight in per cent at indicated time points. (B) Glucose tolerance test, (C) insulin tolerance test. (D and E) Mice were exposed to a methionine-choline-deficient (MCD) diet for 24 days, quantification of steatosis (D) and histological assessment of hepatic inflammation (E) based on H&E sections. (F–H) Indicated genotypes were exposed to a HFD or LFD (control) diet and were analysed after 16 weeks. (F) Body weight in per cent at indicated time points. (G) Glucose tolerance test, (H) insulin tolerance test. (I and J) Mice were exposed to the MCD diet for 24 days, quantification of steatosis (I) and histological assessment of hepatic inflammation (J) based on H&E sections. Data are expressed as mean±SEM (A–J), n=7–10 (A–J). Two-way repeated measures analysis of variance (A–C, F–H), unpaired Student's t-test (D, E, I and J).
To test the impact of IFNAR1 signalling in myeloid cells (such as monocytes/macrophages) on metabolic dysregulation and hepatic disease, we generated Ifnar1fl/fl;LysM-Cre19 (Ifnar1Δmyel) mice and exposed these mice to a high-fat or MCD diet. Ifnar1Δmyel mice on a high-fat diet were not susceptible to metabolic disease when compared with wild-type controls (figure 4F–H and see online supplementary figure S4A–D). Similarly, Ifnar1Δmyel mice on the MCD diet were not susceptible to hepatic disease when compared with wild-type controls (figure 4I, J and see online supplementary figure S4E–G).
Weight loss induced by LAGB restores type I IFN responses and metabolic dysregulation in patients with severe obesity
Bariatric surgery and LAGB has been used to induce weight loss and to improve metabolic dysregulation in patients with severe obesity29 and we previously reported that LAGB improved glycaemic control along with a reduction in BMI.30 ,31 To explore a role of type I IFN signalling in metabolic dysregulation, we investigated the impact of weight loss on type I IFN responses in patients with severe obesity. To do so, we used a cohort of 28 patients (21 females and 7 males) with a median age of 38 years and determined type I IFN responses in the liver and subcutaneous fat before and after LAGB (table 1). Notably, our cohort exhibited signs of improved glucose metabolism and hepatic injury as indicated by improved fasting glucose and homeostasis model assessment index and reduced liver enzyme elevation after LAGB-induced weight loss (table 1 and ref. 32). Subcutaneous fat assessed before surgery exhibited increased expression of most type I IFN-regulated genes when compared with liver tissue (figure 5A). Importantly, improved metabolic control after LAGB (as investigated 6 months after surgery) was associated with increased expression of some classical IFN-regulated genes such as interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) and ISG15 ubiquitin-like modifier (G1P2) in the liver and fat, respectively (figure 5B, C). These data suggest that type I IFN responses are modulated during diet-induced metabolic inflammation in humans. To further investigate this, we tested for an association between the expression of type I IFN response genes and tumour necrosis factor α (TNF-α) or interleukin (IL)-6, respectively.33 Indeed, in hepatic tissue, we found a positive correlation between type I IFN-regulated gene expression and TNF-α and a negative correlation with IL-6 expression (see online supplementary figure S5A, B).
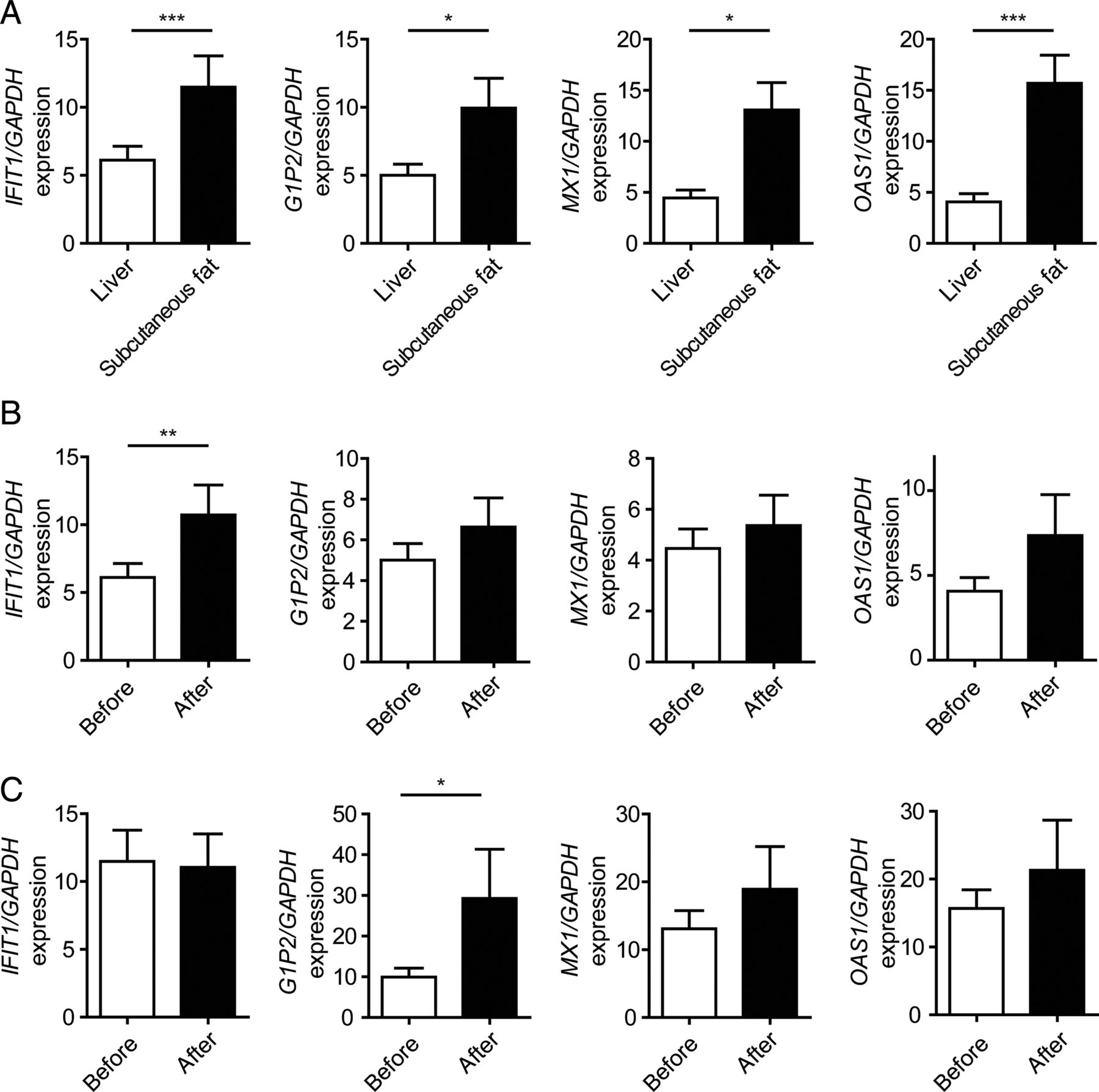
Expression of interferon (IFN) target genes in patients before and after laparoscopic gastric banding. (A) Expression of type I IFN-regulated genes in liver and subcutaneous adipose tissue before laparoscopic adjustable gastric banding (LAGB) relative to GAPDH determined by quantitative PCR (qPCR). (B and C) Expression of indicated genes relative to GAPDH in hepatic (B) and subcutaneous adipose (C) tissue before and after LAGB determined by qPCR. Data are expressed as mean±SEM, n=28. Two-tailed unpaired Student's t-test (A), Wilcoxon signed-rank test (B and C). * p<0.05, **p<0.01, ***p<0.001. IFIT1, interferon-induced protein with tetratricopeptide repeats 1; G1P2, ISG15 ubiquitin-like modifier; MX1, MX dynamin-like GTPase 1; OAS1, 2’-5’-oligoadenylate synthetase 1.
Discussion
Inflammation emerges as a potent driver of metabolic diseases associated with obesity.27 ,34 As we found that fatty acids elicited type I IFN responses in hepatocytes and macrophages, we assessed type I IFN signalling in diet-induced metabolic disease in vivo. Wild-type mice on a high-fat diet exhibited an increased hepatic type I IFN response exemplified by increased Irf7 expression which has similarly been observed in an experimental model of steatohepatitis.35 These data led us to investigate the role played by type I IFN signalling in hepatic and metabolic disease processes, which we addressed by analysing high-fat- or MCD-fed tissue-specific Ifnar1 knockout mice.
By genetic deletion of Ifnar1 specifically in adipocytes, we unravel an unappreciated role for type I IFN signalling in the development of metabolic dysregulation induced by a high-fat diet. Ifnar1Δat mice gained more weight on a high-fat diet along with an expansion of eWAT, and Ifnar1Δat mice exhibited an impaired glucose tolerance along with increased insulin resistance compared with controls. Importantly, considering the crosstalk between adipose and hepatic metabolism,34 ,36 we did not observe increased susceptibility for hepatic disease in adipose-specific Ifnar1 knockout mice neither on the high-fat diet nor on the MCD diet.
In contrast, IFNAR1 signalling in hepatocytes suppressed hepatic disease consequent to the MCD diet, which robustly elicits type I IFN responses.35 The protective effects of IFNAR1 is in line with recent reports that demonstrate anti-inflammatory functions of IFNAR1 signalling in murine models of fibrosis, ischaemia/reperfusion and toll-like receptor-induced hepatic injury.12–14 Ifnar1Δhep mice were more susceptible to steatosis on the MCD diet but not to steatosis induced by a high-fat diet. This observation may be explained by a higher level of inflammation in the MCD model of steatohepatitis when compared with a high-fat diet.37 Importantly, although the liver is involved in glucose metabolism,38 lack of type I IFN signalling in hepatocytes did not modulate susceptibility to metabolic dysregulation or obesity.
Intestinal dysbiosis has been linked to metabolic dysregulation and hepatic disease.22 ,28 ,39 The intestinal epithelium resides at the interface of the microbiota and may similarly regulate host metabolism.40 Despite a colonic dysbiosis of intestinal epithelial-specific Ifnar1 knockout mice raised in the same location as previously reported,20 Ifnar1ΔIEC mice did neither exhibit increased susceptibility to the high-fat nor the MCD diet. Along the same line, myelocyte type I IFN signalling did not modulate susceptibility to hepatic disease. This finding was rather unexpected as myeloid cells (eg, macrophages) promoted non-alcoholic steatosis and inflammation41 and IFNAR1 signalling on myeloid cells suppressed inflammation in autoimmune encephalitis.11 Our observation may be explained by a tissue-specific immunomodulatory function of type I IFN signalling in myelocytes.42
As such, our data indicate that metabolic dysregulation induced by a high-fat diet is suppressed by type I IFN signalling in adipocytes but not hepatocytes. Vice versa, hepatic disease induced by the MCD diet is suppressed by type I IFN signalling in hepatocytes, but not adipocytes (figure 6). We noted increased F4/80 expression in Ifnar1Δat mice on a high-fat diet, suggesting that IFNAR1 in adipose tissue controls macrophage inflammation,43 which warrants further investigation. The downstream mechanisms by which type I IFN signalling modulates hepatic and adipose inflammation could also involve cytokine (eg, TNF-α) production, modulated receptor responses (eg, IL-1 receptor antagonist induction)14 or a broader STAT3-mediated transcriptional regulation of IFN target genes that repress the production of inflammatory mediators.9 Importantly, IFNAR1 signalling and the JAK/STAT pathway could also have direct (ie, cell-intrinsic) effects on functions of adipocytes (eg, differentiation),44 hepatocytes45 or lymphoid cells (such as natural killer cells),46 which may modulate susceptibility to metabolic disease. For example, type I IFN responses could modulate differentiation of brown/white adipose tissue and lipolysis.47

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Type I interferon (IFN) signalling in hepatic and metabolic disease. (A) Hepatocyte type I IFN signalling suppresses steatosis and inflammation induced by a methionine-choline-deficient (MCD) diet but not high-fat diet. Importantly, hepatocyte type I IFN signalling does not modulate metabolic dysregulation induced by a high-fat diet. (B) Adipose type I IFN signalling suppresses metabolic dysregulation induced by a high-fat diet. However, adipose type I IFN signalling does not regulate susceptibility to hepatic disease induced by a high-fat or MCD diet. Note that myeloid and intestinal epithelial type I IFN signalling did neither modulate hepatic nor metabolic disease induced by a high-fat or MCD diet. WT, wild type.
To further explore a role of type I IFN signalling in humans, we analysed the expression of type I IFN-induced targets before and after LAGB. We noted that weight loss and improved insulin resistance induced by LAGB was associated with an increased expression of IFIT1 and G1P2 in hepatic and subcutaneous adipose tissue, respectively. This suggested that LAGB-induced type I IFN responses may impact on glucose metabolism in patients with obesity. Indirect correlation of IL-6 expression with type I IFN-regulated genes in the liver further suggested that dietary factors and possibly hepatic inflammation impacts on type I IFN responses after LAGB.
In conclusion, our study implicates a role for type I IFN signalling in diet-induced metabolic and hepatic disease. Further studies are warranted to investigate the mechanism of IFNAR1-mediated modulation of metabolic inflammation.
Acknowledgments
We thank Dr Arthur Kaser and Dr Ulrich Kalinke for providing their expertise.
References
Footnotes
VW and TEA contributed equally.
Contributors VW and TEA designed, performed and analysed most experiments and prepared the manuscript, together with CG, FG and BE helped with experimentation. PM performed histology analysis and SK and ARM provided expertise. HT coordinated the project.
Funding This project was supported by the excellence initiative (Competence Centers for Excellent Technologies—COMET) of the Austrian Research Promotion Agency FFG: Research Center of Excellence in Vascular Ageing Tyrol, VASCage (K-Project Nr. 843536) funded by the BMVIT, BMWFW, the Wirtschaftsagentur Wien and the Standortagentur Tirol (to HT) and by the Austrian Science Fund (FWF) P 29379-B28 and the Tyrolian Science Fund (TWF) 0404/1812 (both to TEA).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics Committee Medical University Innsbruck, Innsbruck, Austria.
Provenance and peer review Not commissioned; externally peer reviewed.