Article Text

Abstract
Objective Interferons (IFNs) mediate direct antiviral activity. They play a crucial role in the early host immune response against viral infections. However, IFN therapy for HBV infection is less effective than for other viral infections.
Design We explored the cellular targets of HBV in response to IFNs using proteome-wide screening.
Results Using LC-MS/MS, we identified proteins downregulated and upregulated by IFN treatment in HBV X protein (HBx)-stable and control cells. We found several IFN-stimulated genes downregulated by HBx, including TRIM22, which is known as an antiretroviral protein. We demonstrated that HBx suppresses the transcription of TRIM22 through a single CpG methylation in its 5′-UTR, which further reduces the IFN regulatory factor-1 binding affinity, thereby suppressing the IFN-stimulated induction of TRIM22.
Conclusions We verified our findings using a mouse model, primary human hepatocytes and human liver tissues. Our data elucidate a mechanism by which HBV evades the host innate immune system.
- HEPATITIS B
- INFECTIOUS DISEASE
- LIVER
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Interferons (IFNs) mediate direct antiviral activity and play a crucial role in the host immune response against viral infections.
IFN therapy for HBV infection is less effective than for other viral infections, implying that HBV has mechanisms to evade or counteract the IFN-mediated antiviral effects. However, the precise mechanism by which HBV escapes from IFNs remains largely unclear.
TRIM22 is an IFN-stimulated gene and known as an antiviral protein against retroviruses, such as influenza virus, HIV and HBV.
What are the new findings?
Proteome-wide analysis identifies HBV X protein (HBx)-mediated desensitised proteins by IFN induction.
HBV (via its protein HBx) suppresses the transcription of TRIM22 through a single CpG methylation in its 5′-UTR, which reduces the IFN regulatory factor-1 binding, thereby suppressing the IFN-stimulated induction of TRIM22.
Suppression of TRIM22 by HBx is involved in viral escape from IFN-mediated antiviral response.
HBV suppresses TRIM22 expression in a mouse model, primary human hepatocytes and human liver tissues.
How might it impact on clinical practice in the foreseeable future?
Our findings provide new insights that might be useful for counteracting the anti-IFN strategies of the virus and a potential way to improve the therapeutic effect of IFNs for HBV clearance.
Our data elucidate the mechanism by which HBV evades the host innate immune system, and may provide a new and attractive therapeutic strategy for the cure of HBV infection.
Introduction
The battle between viral infection and the host defence system determines pathogenesis or cure of disease. During viral infections, hosts recognise viruses and effectively eradicate them through innate and adaptive immunity. However, viruses have various strategies to counteract and evade host immune systems.1
At early stages of infection, interferons (IFNs) are the major effector cytokines against various pathogens during innate immune response. All types of IFNs (type I: IFNα, IFNβ and IFNω; type II: IFNγ; type III: IFNλ1, IFNλ2 and IFNλ3) are induced mainly through pathogen-associated molecules activated by pattern recognition receptors.2 IFNs secreted in response to virus infection ultimately induce multiple interferon-stimulated genes (ISGs), which have multiple antiviral and immune-modulating activities via IFNs’ signalling pathways.3 Therefore, IFNs and their signalling pathways are targeted by viruses, which allow them to escape host immunity.1
HBV infection causes chronic hepatitis, liver cirrhosis and even hepatocellular carcinoma. It is a major health problem, with over 350 million carriers worldwide.4 HBV, a non-cytopathic hepatotropic DNA virus, has a distinct replication strategy through reverse transcription using pregenomic RNA (pgRNA) as a template in capsids.5 The synthesis of pgRNA and other viral RNAs is regulated by viral enhancers and core promoters.6 The HBV X protein (HBx) is multifunctional, and its role is related to dysregulating cellular functions and to pathogenesis.7 Notably, recent studies have revealed that HBx epigenetically regulates host proteins associated with HBV-mediated liver pathogenesis.8 ,9
Extensive studies have shown that IFNs suppress HBV replication. IFNα inhibits HBV replication through epigenetic regulation of cccDNA10 and MyD88-mediated pgRNA degradation.11 IFNγ mainly mediates the antiviral effect of HBV-specific cytotoxic T lymphocytes.12 Both type I and type II IFNs inhibit HBV replication by disturbing the formation of replication-competent nucleocapsids containing pgRNA13 and accelerating their decay.14 HBV replication is also restricted by the antiviral function of several ISGs such as MxA and APOBEC3G.15 ,16 Therefore, pegylated-IFNα is included in the current antiviral therapy regimen against HBV infection.17
However, only 30%–44% of patients with chronic hepatitis B show satisfactory virological and serological responses to treatment with pegylated-IFNα.18 ,19 Furthermore, the susceptibility of HBV to IFNs is lower than that of HCV.20 These studies strongly imply that HBV has mechanisms to evade or counteract the IFN-mediated antiviral effects, which increase virus survival and cause resistance to IFN therapy. Some lines of evidence have shown that viral polymerase and HBx proteins are involved.21–23 However, the precise mechanism by which HBV escapes from IFNs remains largely unclear.
Proteins of the tripartite motif (TRIM) family are involved in various biological processes including oncogenesis, apoptosis and antiviral immune response. In particular, many members of the TRIM family induced in response to IFNs are associated with innate immunity and antiviral defence.24 Among them, TRIM22, one of the closest paralogs of TRIM5α, displays antiretroviral activity similar to that of TRIM5α.25 Several studies have found that TRIM22 inhibits the transcription and replication of HIV through suppression of its long terminal repeat and disruption of the intracellular trafficking of the viral Gag protein.26 ,27 TRIM22 also restricts influenza virus and encephalomyocarditis virus by degrading a nucleoprotein and viral 3C protease, respectively, through its E3 ligase activity,28 ,29 suggesting the antiviral role of TRIM22 in a wide range of viral infections. Most importantly, TRIM22 has anti-HBV activity because it suppresses the HBV core promoter.30 Genome-wide microarray analysis revealed that viral clearance in HBV-infected chimpanzees is associated with TRIM22.31 Although the antiviral functions of TRIM22 have been extensively studied, the mechanism of viral evasion from TRIM22 is not yet known.
In this study, we demonstrate that HBV downregulates TRIM22 via epigenetic control, which makes it possible for the virus to evade the antiviral effect of IFNs during innate immune response. Our study provides new insights that might be useful for counteracting the anti-IFN strategies of the virus and also provides a foundation for improving the outcome of IFN therapy in patients with HBV infection. Our data also explain why coinfection with HBV and HIV has a worse prognosis than infection with HBV alone.
Results
Proteome-wide screening for HBx-mediated desensitised proteins by IFN treatment: identification of TRIM22 as a possible target for immune evasion
Response to IFNs and their subsequent antiviral activity that induces ISGs can be impaired by a variety of viruses, protecting them from IFNs.1 In HBV, the HBx protein is a key player regulating the expression of host proteins by epigenetic modification.8 ,9 In this regard, we investigated the possible targets involved in HBV protection from IFNs. First, we performed proteome-wide analysis using LC-MS/MS after treatment of HBx-expressing and control (transfected with pcDNA) cells with or without IFNγ (figure 1A).
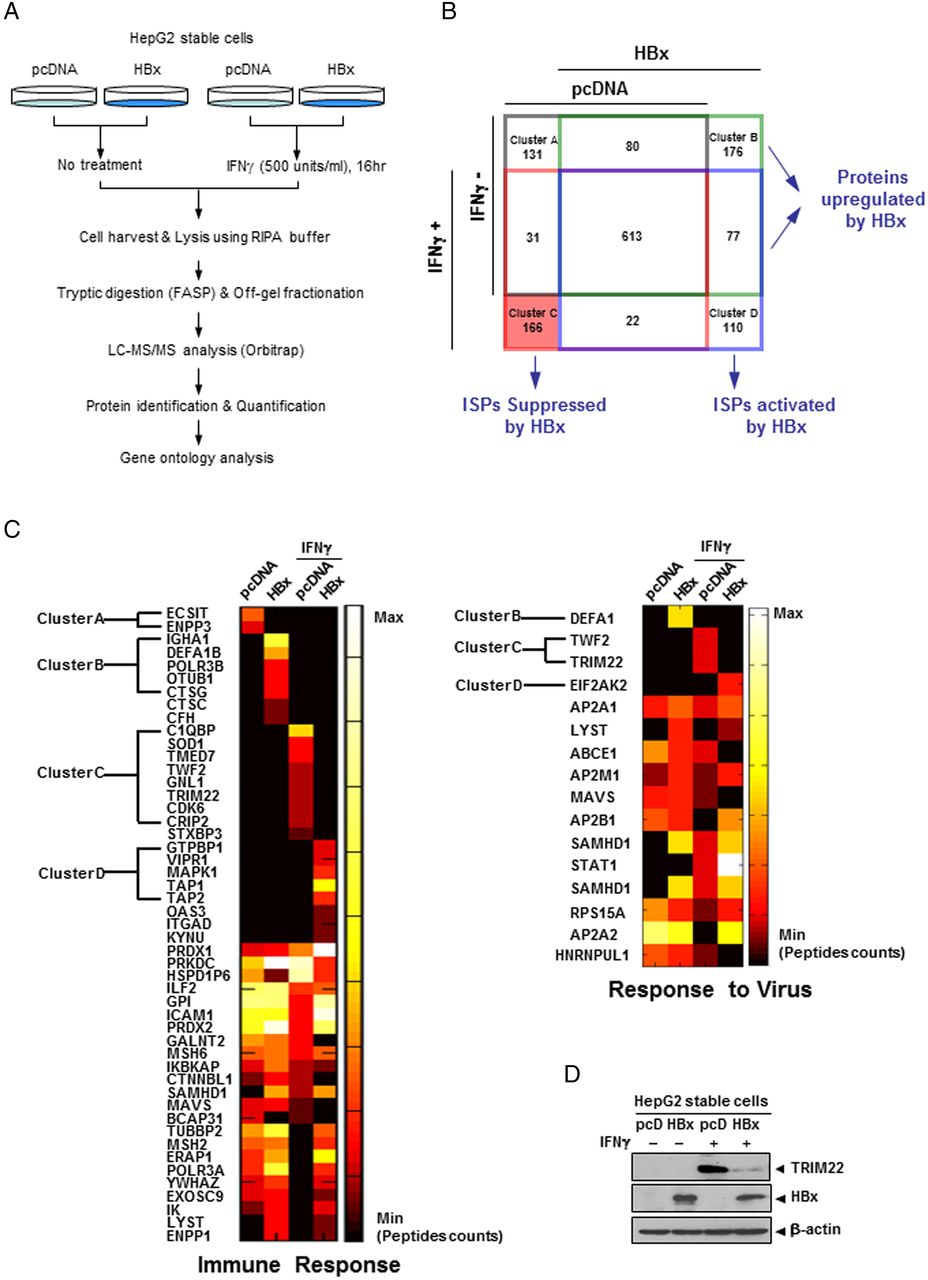
Proteome-wide screening to identify proteins desensitised by interferon (IFN) treatment in cells stably expressing HBV X protein (HBx). (A) The schematic outline of the screening procedure. (B) Venn diagram of identified proteins with four groups (grey outline, IFNγ−/pcDNA; green outline, IFNγ−/HBx; red outline, IFNγ+/pcDNA; blue outline, IFNγ+/HBx). Arabic numbers show the number of identified proteins in each cluster. Cluster C (red box) denotes the IFN-stimulated proteins (ISPs) that are suppressed by HBx. (C) Comparative analysis (heat map) of identified proteins categorised in ‘Immune Response’ and ‘Response to Virus’ groups by reference-based gene ontology analysis. Spectrum count was normalised by dividing the number of spectra of a specific protein by the total number of spectra found in each group. Note that TRIM22 in Cluster C is included in both categories. (D) Validation of TRIM22 expression in cells of each group used for LC-MS/MS analysis. Cells were treated with IFNγ (500 units/mL) for 16 hours, harvested and lysed with RIPA buffer. Proteins were detected by western blotting.
A total of 1406 proteins were identified from four different groups (IFNγ−/pcDNA, IFNγ−/HBx, IFNγ+/pcDNA and IFNγ+/HBx cells), each of which included 855, 946, 832 and 822 proteins, respectively. They were identified using LC-MS/MS analysis following FASP digestion and off-gel fractionation (see online supplementary information for details). Within each group, proteins identified only in one other group were assigned to Cluster A, B, C or D (figure 1B). According to this classification, 110 proteins in Cluster D were upregulated by both IFNγ and HBx, and 166 proteins in Cluster C were upregulated by IFNγ (IFN-stimulated proteins, ISPs), but were suppressed by HBx.
supplementary data
All identified proteins were classified by Gene Ontology using DAVID bioinformatics resources (http://david.abcc.ncifcrf.gov). Among the putative biological functions, we focused on two categories, ‘Immune Response’ and ‘Response to Virus’. Differential expression of proteins in each category is shown in figure 1C. The information about eight ISPs from Cluster C suppressed by HBx is summarised in online supplementary table S1. Notably, only TRIM22 and TWF2 were detected in both categories (Immune Response and Response to Virus). Since TRIM22 was reported to have antiviral activity against several viruses including HBV,25 we focused on TRIM22 as a potential target of HBV. Western blot analysis confirmed that TRIM22 is induced by IFNγ; however, this effect was considerably desensitised by HBx (figure 1D).
HBV desensitises the expression of IFN-induced TRIM22 through the viral protein HBx
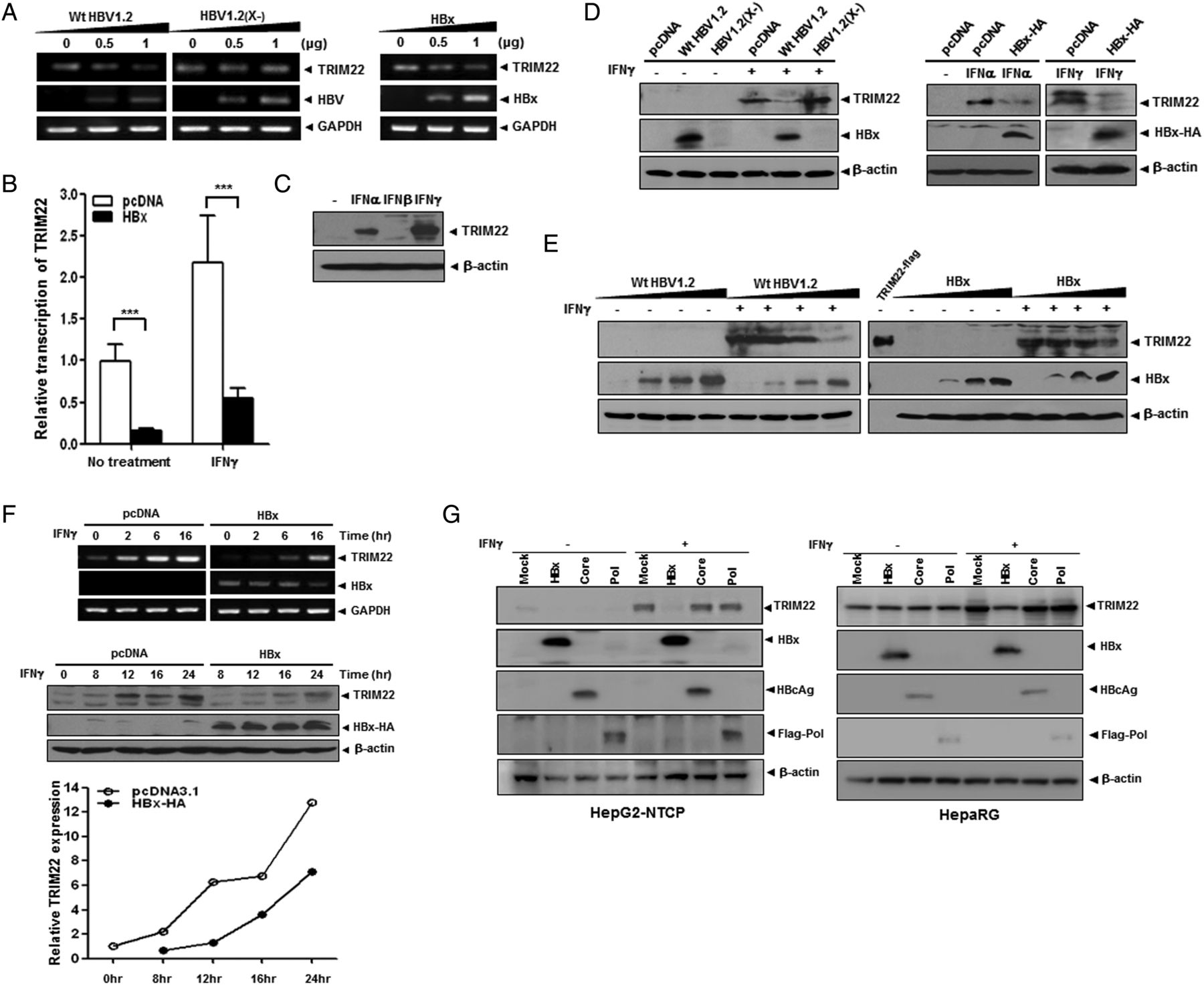
To investigate whether HBV and HBx regulate the expression of TRIM22, we first examined the basal levels of TRIM22 mRNA after transfection with replication-competent WT HBV1.2mer (WT HBV1.2), HBx-deficient HBV genome (HBV1.2(X-)) or pcDNA-HBx-HA. Both WT HBV1.2 and HBx-HA decreased the expression of TRIM22 in a dose-dependent manner, whereas HBV1.2(X-) did not significantly change the level of TRIM22 mRNA (figure 2A and see online supplementary figure S1A). Furthermore, HBx suppressed the induction of TRIM22 upon IFNγ treatment (figure 2B and see online supplementary figure S1B). Among IFNs, IFNγ strongly induced TRIM22 expression, whereas IFNβ showed little effect (figure 2C and see online supplementary figure S2). The activities of IFNs were verified by the induction of ISGs including TRIM22 (see online supplementary figure S2).

HBV suppresses the expression of TRIM22 induced by interferons (IFNs) through the viral protein HBV X protein (HBx). (A) Inhibition of TRIM22 transcription by HBx. Huh7 cells were transfected with the indicated plasmids. After 48 hours, cells were harvested and analysed by semiquantitative RT-PCR. (B) Effect of HBx on the level of TRIM22 mRNA in cells treated with or without IFNγ (2 hours, 500 units/mL). Data are from three independent real-time PCR analyses (***p<0.001) and normalised to the expression level of GAPDH. (C) Effect of IFNs on TRIM22 expression. HepG2 cells were treated with the indicated IFNs (500 units/mL) for 24 hours and analysed by western blotting. (D) HBx-mediated suppression TRIM22 expression induced by IFNγ. Cells transfected with 2 μg of indicated plasmids were treated with IFNs (500 units/mL) for 24 hours, and the expression level of TRIM22 was analysed by western blotting. (E) Dose-dependent suppression of IFN-inducible TRIM22 by HBx. HepG2 cells were transfected with increasing amounts (0, 1, 2 and 4 μg) of the indicated plasmids and treated with IFNγ for 24 hours. Total DNA amounts were adjusted to 4 μg using pcDNA3.1. The lysate of cells overexpressing TRIM22-flag was used as a positive control. (F) Effect of HBx on the expression kinetics of TRIM22 in the presence of IFNγ. HepG2 cells were transfected with HBx-HA (2 μg) and treated with IFNγ for indicated time. The levels of TRIM22 mRNA and protein were analysed by semiquantitative RT-PCR and western blotting, respectively. The relative expression of TRIM22 protein was measured by GelQuant.NET analysis software (Biochem Lab Solutions). (G) Effect of viral proteins on TRIM22 expression in HepG2-NTCP and HepaRG cells. Cells transfected with 2 μg of indicated plasmids were treated with IFNs (500 units/mL) for 24 hours, and the expression levels of TRIM22 and viral proteins were analysed by western blotting.
The suppression of TRIM22 by HBx was further confirmed by western blot analysis using the virus and HBx-encoding plasmid (figure 2D). TRIM22 expression was significantly suppressed by HBx (figure 2E). The time course of TRIM22 induction by IFNγ was considerably disrupted in HBx-expressing cells (figure 2F and see online supplementary figure S1B). In addition, only HBx suppressed the induction of TRIM22 by IFNγ, whereas other viral proteins including polymerase and core proteins had no effect in either HepG2-NTCP or HepaRG infection systems (figure 2G).
Since the anti-HBV effect of TRIM22 occurs in the nucleus through inhibition of the core promoter,30 we checked the nuclear level of TRIM22. As shown in online supplementary figure S3, the proportion of TRIM22 in the nucleus after IFNγ treatment was not altered by the presence of HBx.
Identification of the element responsible for HBx-mediated suppression of TRIM22
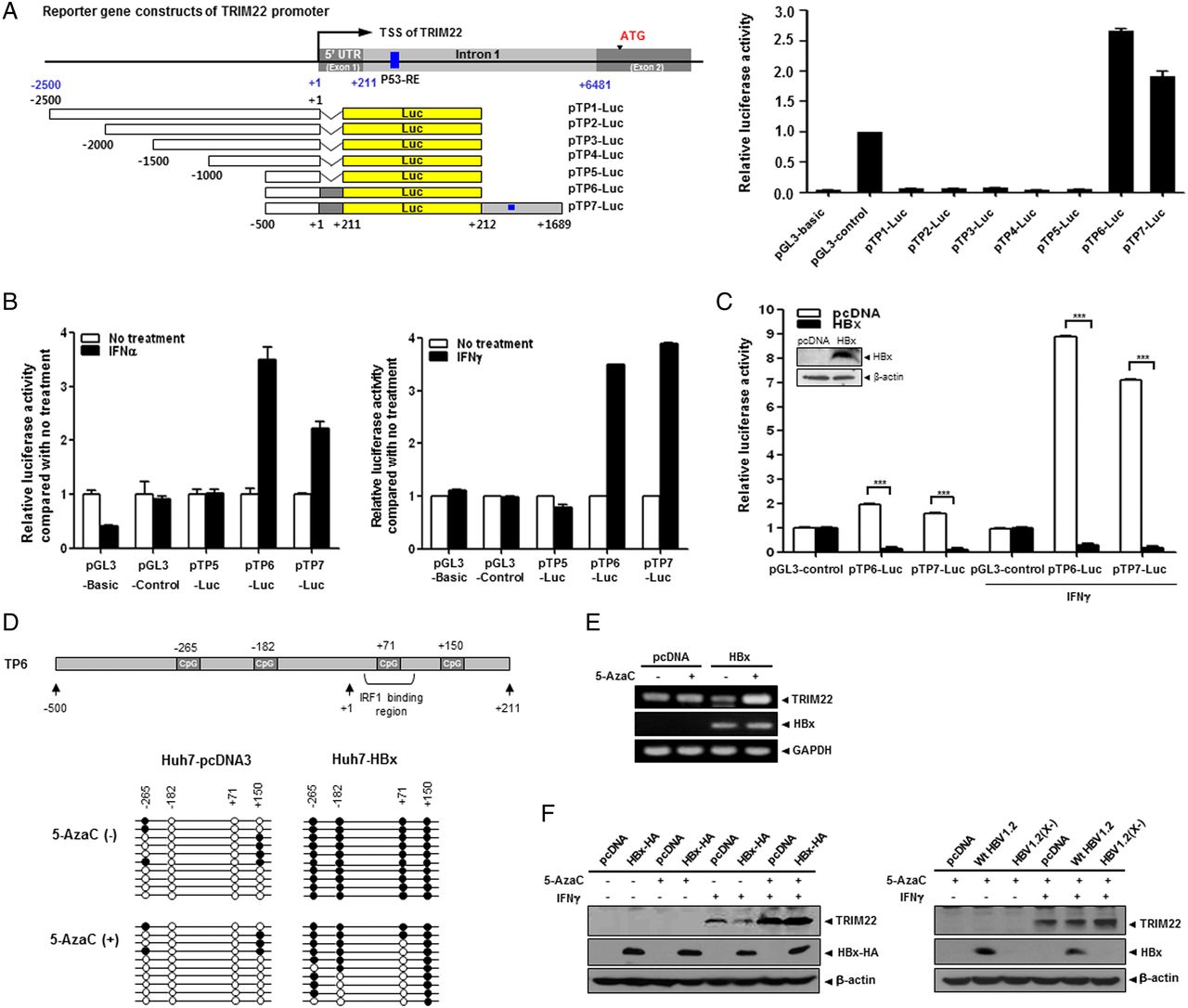
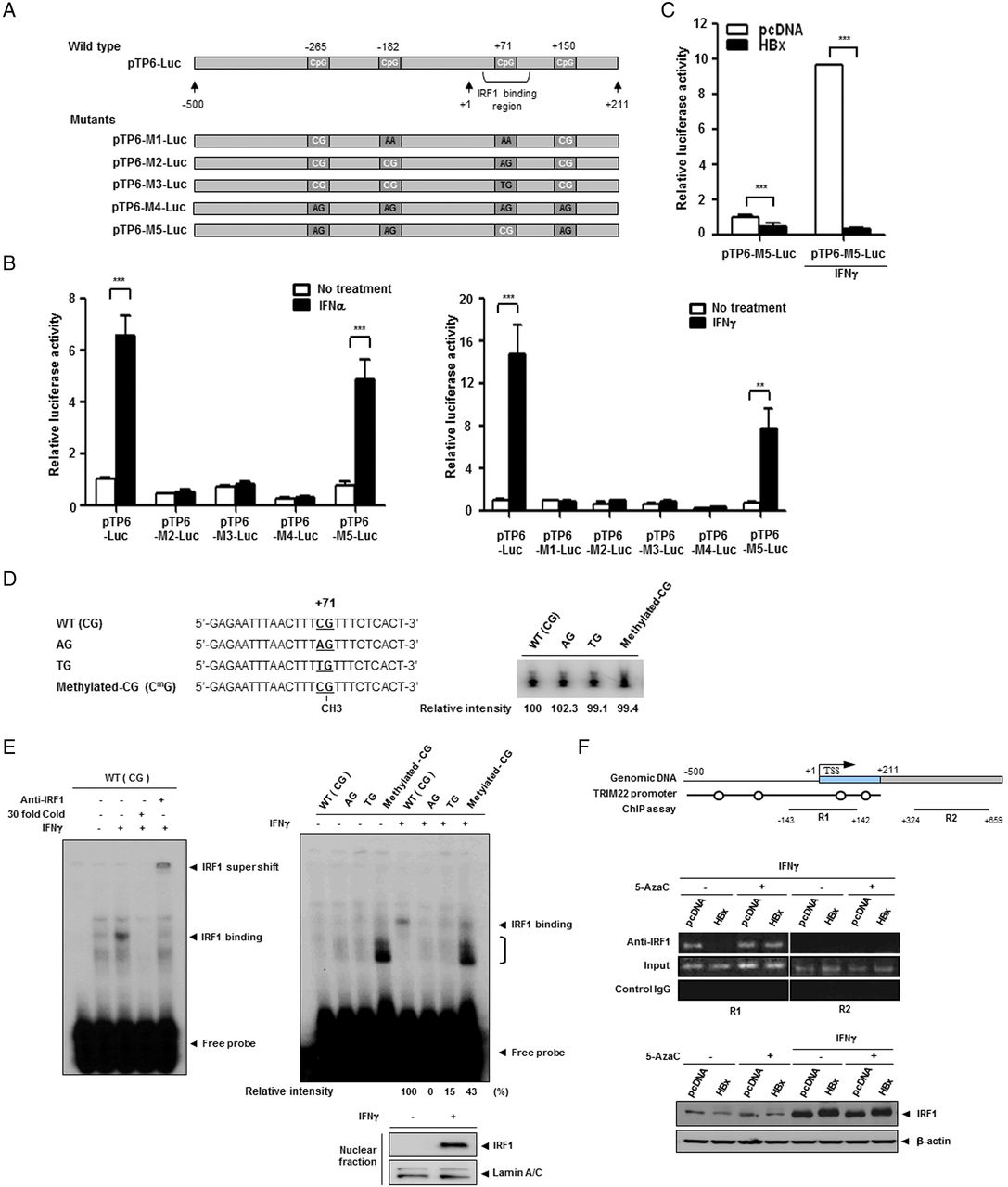
To identify the elements responsible for HBx-mediated suppression of TRIM22, we constructed seven luciferase reporter plasmids, including five serial deletion constructs spanning 2.5 kb upstream of the transcription start site of the TRIM22 gene. Construct TP6 contained 5′-UTR, and construct TP7 contained both the 5′-UTR and the p53 response element in intron 1, which was suggested to be necessary for promoter activity32 (figure 3A). The luciferase assay revealed that TP6 and TP7 have strong basal promoter activity, suggesting that the 5′-UTR (+1 to +211) is essential for TRIM22 transcription (figure 3A). Further analysis demonstrated that the 5′-UTR is also responsible for both TRIM22 induction (figure 3B) and HBx-mediated desensitisation of TRIM22 induction by IFNγ (figure 3C). Interestingly, this region resembles a IFNγ-stimulating element, a region that binds IFN regulatory factor-1 (IRF1).33 Accordingly, we examined whether the HBx-mediated suppression of TRIM22 is regulated by IRF1. IFNγ but not HBV or HBx increased IRF1 expression (see online supplementary figure S4). These findings suggest that the 5′-UTR (+1 to +211) is critical for transcriptional activity, induction by IFNs and HBx-mediated suppression of TRIM22 expression.
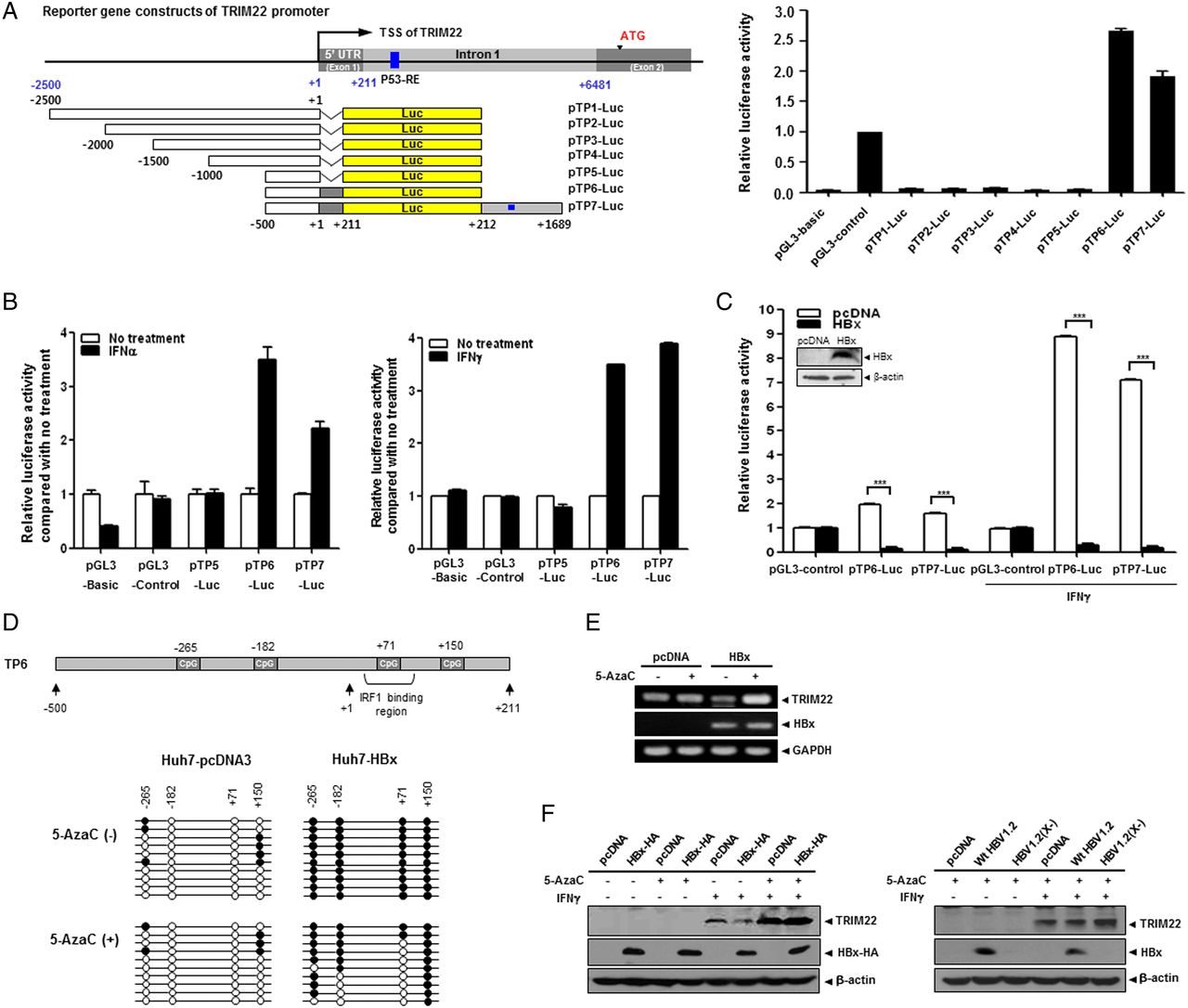
HBV X protein (HBx) suppresses TRIM22 induced by interferons (IFNs) through epigenetic regulation in the 5′-UTR of TRIM22. (A) Schematic representation of the promoter and 5′-UTR of TRIM22. Seven luciferase reporter plasmids (pTP1-Luc to pTP7-Luc) that included the putative TRIM22 promoter were constructed. Basal activity of putative TRIM22 promoters was determined by luciferase reporter assay. pGL3-basic and pGL3-control (SV40 promoter) plasmids were used as negative and positive controls, respectively. TSS, transcription start site; UTR, untranslated region; p53-RE, p53 response element. (B) Effect of IFNs on the induction of TRIM22 promoter. HepG2 (left panel) and Huh7 (right panel) cells were transfected with reporter plasmids (0.25 μg) and treated with or without IFNα or IFNγ (500 units/mL) for 24 hours, and luciferase activity was determined. (C) HBx suppresses the expression of TRIM22 regardless of IFN treatment. At 24 hours post transfection with reporter plasmids, cells were treated with IFNγ (500 units/mL) for 24 hours, and luciferase activity was measured. The expression of HBx was confirmed by western blotting. Data are mean±SD of at least three independent experiments; ***p<0.001. (D) Upper panel, a schematic outline of the four single CpG sites in the promoter and 5′-UTR of TRIM22. The third CpG belongs to the IFN regulatory factor-1 (IRF1)-binding region. Lower panel, effect of HBx on CpG methylation. The results of bisulfite sequencing with or without 5AzaC treatment are shown. The methylation status of 10 clones from each sample was analysed. Open circles, CpG; filled circles, methyl-CpG. (E and F) Treatment with 5AzaC restores the HBx-mediated suppression of TRIM22. Cells were transfected with 2 μg of the indicated plasmids and were treated with 5AzaC with or without IFNγ treatment. The TRIM22 mRNA (E) and protein (F) levels were determined by semiquantitative RT-PCR and western blot analysis, respectively. 5AzaC (10 μM) was present throughout the cell culture. IFNγ (500 units/mL) was added for 24 hours before harvesting.
HBx represses IFN-induced TRIM22 expression through epigenetic regulation
Previously, we and other groups reported that HBx regulates the expression of several host proteins through epigenetic control, mainly DNA methylation in their regulatory regions.8 ,9 Therefore, we examined whether downregulation of TRIM22 by HBx is associated with epigenetic modification. We first analysed the CpG sites using TP6, which contains the 5′-UTR (+1 to +211). There were only four CpGs scattered from upstream of 500 bp to downstream of 200 bp (figure 3D, upper panel). Interestingly, bisulfite sequencing revealed that each CpG site in the TRIM22 promoter was fully methylated in HBx-expressing cells, unlike in control cells (figure 3D, middle panel). Unexpectedly, the four CpG sites showed different sensitivity to 5′-aza-2′-deoxycytidine (5AzaC), a DNA methylation inhibitor (figure 3D, lower panel). Methylation at the first and last CpG sites was stable, but the third CpG site (+71) was unmethylated after 5AzaC treatment. This suggests that methylation of the third CpG site plays a critical role in the suppression of TRIM22 transcription.
Overexpression of the DNA methyltransferases DNMT1, DNMT3A1 or DNMT3A2 did not affect the expression level of TRIM22 (see online supplementary figure S5A). The expression levels of DNA methyltransferases were not considerably affected by HBx in HepG2 cells (see online supplementary figure S5B).
As shown in figure 3E, F (left panel), the mRNA and protein levels of TRIM22, which had been dramatically repressed by HBx, were restored by 5AzaC treatment. The repression of TRIM22 by genome-driven HBx (figure 2D) was also completely abolished by 5AzaC treatment (figure 3F, right panel). These results suggest that the suppression of TRIM22 by HBx is mediated by epigenetic regulation of the TRIM22 promoter.
Methylation of single CpG (+71) by HBx is responsible for HBx-mediated desensitisation of TRIM22 induction
Correlation between hypomethylation of the third CpG (+71) (figure 3D) and restoration of TRIM22 expression (figure 3E, F) by 5AzaC treatment elucidates the role of this site in IFN-mediated TRIM22 induction. We generated five TP6 mutant reporter clones containing one CpG mutation each (figure 4A) and examined their basal and IFN-induced promoter activities. The results of luciferase assay showed that all mutants except pTP6-M5 lost their induction activities; pTP6-M5 had substitutions in all CpGs except the third one and maintained its IFNα and IFNγ-induced activity (figure 4B). HBx strongly suppressed pTP6-M5 activity under both basal conditions and upon IFNγ treatment (figure 4C). These results demonstrate that a single CpG (+71) is responsible for the suppression of TRIM22 transcription by HBx.

HBV X protein (HBx) suppresses TRIM22 expression through methylation of a single CpG in the 5′-UTR of TRIM22. (A) Schematic representation of five mutant luciferase reporter clones. Cytosines or guanines of CpG were mutated to adenine or thymine. (B) Induction of mutant promoters by interferons (IFNs). HepG2 cells were transfected with the indicated reporter plasmids (0.5 μg), treated with or without IFNα or IFNγ (500 units/mL) for 24 hours, and luciferase activity was measured. (C) Suppression of the M5 TRIM22 promoter by HBx regardless of IFNγ treatment. Cells were transfected with pTP6-M5-Luc (0.5 μg), and basal and IFNγ-induced promoter activity was measured. Data are mean±SD of at least three independent experiments; ***p<0.001, **p=0.003. (D) Sequences of [32P]-labeled probes used for IFN regulatory factor-1 (IRF1) electrophoretic mobility shift assay (EMSA). The single CpG site (+71) is underlined, and mutations or methylated CpG are indicated. The radioactivity of each [32P]-labeled probe was determined. (E) Verification of IRF1 interaction with WT probe through antibody supershift and competition assay with a cold probe (left panel). (E) EMSA of binding affinity between IFN-induced IRF1 and each probe. Induction of IRF1 by IFNγ (500 units/mL) was confirmed by western blot analysis of nuclear extracts (lower panel). The bracket indicates the complex formed by methylated CpG-binding proteins. Relative intensity was calculated by subtracting the intensity without treatment from that with IFNγ treatment (right panel). (F) Chromatin immunoprecipitation (ChIP) assay of the TRIM22 promoter region with IRF1 antibody and genomic DNA obtained from Huh7 cells stably expressing HBx and control cells. Both cell groups were treated with or without IFNγ (500 units/mL) or 5AzaC (10 μM), and ChIP assay was performed. Circles indicate single CpG sites. R2 was used as a negative control. Induction of IRF1 by IFNγ was confirmed by western blotting (lower panel).
As mentioned before, CpG (+71) is located in the IRF1-binding region (figure 4A).33 Therefore, we examined the possibility that methylation at this site impedes IRF1 binding, which is essential for IFNγ-induced TRIM22 expression. We performed electrophoretic mobility shift assay (EMSA) using four DNA probes with comparable radioactivity (figure 4D). Nuclear IRF1 was detected only in IFNγ-treated cells (figure 4E, bottom panel). IRF1 bound to the wild-type probe harbouring a single CpG (+71), as revealed by cold competition and antibody supershift assays (figure 4E, left panel). Both the AG and TG probes lost their ability to bind IRF1 dramatically (figure 4E), in line with abrogated induction activity of pTP6-M2 and pTP6-M3 (figure 4B). In EMSA using the methylated CG probe, the binding affinity of IRF1 was markedly lower than that of the wild-type probe, in which a strong band was observed (figure 4E, right panel). This result implies that IRF1 binding is impeded when the CpG (+71) site is methylated, and confirms the importance of this site in IRF1 binding and TRIM22 induction.
We further investigated the influence of HBx on CpG (+71). In chromatin immunoprecipitation assay, IFNγ-induced IRF1 interacted with the IRF1-binding site in control cells; this interaction was markedly diminished in HBx-expressing cells (figure 4F). IRF1 expression was not altered by HBx. Treatment with 5AzaC completely restored IRF1 binding in HBx-expressing cells, suggesting that CpG methylation by HBx disturbs the IRF1–DNA interaction and downregulates IFN-induced TRIM22 expression.
Suppression of TRIM22 by HBx is involved in viral escape from IFN-mediated antiviral response
TRIM22 exhibits anti-HBV activity through transcriptional suppression of the HBV core promoter,30 whereas HBx epigenetically desensitises TRIM22 expression. We wondered how HBx affects the anti-HBV activity of IFNs. We chose IFNγ to test its anti-HBV effect, since IFNγ induces TRIM22 more prominently than does IFNα (figure 2C) and efficiently inhibits viral replication and transcription. Southern and western blot analyses showed that the amounts of HBV DNA and proteins (HBs and core) were reduced in response to IFNγ in a dose-dependent manner (figure 5A, left panel). The transcription of HBV genomic and subgenomic RNAs was also significantly reduced by IFNγ (figure 5A, right panel). In contrast, the expression level of TRIM22 was inversely related to HBV replication (figure 5A, left panel), suggesting its role in IFNγ-mediated anti-HBV activity.


Suppression of TRIM22 by HBV X protein (HBx) is involved in HBV evasion from interferon (IFN)-mediated anti-HBV response. (A) IFNγ inhibits HBV replication and transcription and involvement of TRIM22 induction. HepG2 cells were transfected with WT HBV1.2 (1 μg) and treated with IFNγ for 48 hours. Genome replication, expression of TRIM22 and viral surface/core proteins (left panel) and transcription of pregenomic RNA (pgRNA)/subgenomic RNAs (right panel) were analysed by southern, western and northern blotting, respectively. Relative level of HBV transcription was calculated using a phosphorimager; data are from at least three independent experiments (***p<0.001). (B) Left panel, effect of TRIM22 knockdown on Enhancer I/II activity. HepG2 cells were transfected with plasmids (pEnhI-II-Luc (0.4 μg), pcD-TRIM22-flag and pβ-gal (0.25 μg)) with or without indicated siRNAs (20 nM). Total amounts of DNA were adjusted with pcDNA3.1. Relative Enhancer I/II activity was measured 48 hours post transfection. Transfection efficiency was normalised by β-gal assay. ***p<0.001; **p=0.001; siU, siRNA control. Right panel, effect of endogenous TRIM22 knockdown on Enhancer I/II activity. HepG2 cells were transfected with pEnhI-II-Luc, pcDNA3.1, pβ-gal and siRNAs, and treated with IFNγ (500 units/mL) for 24 hours before luciferase analysis. **p=0.001; *p=0.004 (right panel). (C) Effect of TRIM22 knockdown on viral transcription and protein expression. Cells were transfected with the indicated plasmids and siRNAs (20 nM), and transcription (left panel) and expression of viral proteins (right panel) were analysed 72 hours post transfection by northern blotting, western blotting and ELISA. siGL3 was used as a negative control. Relative HBV RNA transcription was calculated from data obtained from three independent experiments and quantified using a phosphorimager. Secreted HBV surface antigen was analysed in culture medium. ***p<0.001; *p=0.006. (D) Effect of endogenous TRIM22 knockdown on IFNγ-mediated anti-HBV activity. HepG2 cells were transfected with HBV1.2 and indicated siRNAs (20 nM), and treated with or without IFNγ (500 units/mL) for 24 hours before harvesting. At 72 hours post transfection, the level of viral transcription was analysed by northern blotting. The expression level of the core protein and TRIM22 knockdown were confirmed by western blotting (left panel). The relative levels of total and genomic RNA (3.5 kb) were calculated using phosphorimager data from five independent experiments. ***p<0.001 (right panels). (E–F) Effect of TRIM22 knockdown on IFN-mediated antiviral activity in HepG2-NTCP (E) and HepaRG (F) HBV infection systems. Expression levels of HBeAg/HBsAg and TRIM22 were measured by ELISA and western blotting, respectively. The level of HBV DNA was quantified using real-time PCR. (G) Enhancement of IFN-induced anti-HBV activity by 5AzaC. Cells transfected with HBV1.2 were treated with 5AzaC (10 μM) and/or IFNγ (500 units/mL). HBV transcription and protein expression were analysed by northern and western blotting, respectively. Relative HBV transcription was calculated from the data from three independent experiments. ***p<0.001.
To examine whether IFNγ-induced anti-HBV activity is mediated by TRIM22, we analysed enhancer activity and the levels of HBV RNA, DNA and proteins after TRIM22 knockdown by siRNA. siTRIM22 effectively knocked down overexpressed TRIM22-flag as well as IFN-induced TRIM22; however, it did not affect the expression of HBx (see online supplementary figure S6). Since TRIM22 inhibits the activity of the HBV enhancer and core promoter,30 we constructed a reporter plasmid containing HBV Enhancer I/II and core promoter (pEnhI-II-Luc) to investigate the effect of TRIM22 on promoter activity (figure 5B, upper panel). Anti-HBV activities mediated by TRIM22 overexpression or IFNγ treatment were significantly reduced by siTRIM22 (figure 5B). HBV transcription and expression of viral core and surface proteins initially reduced by TRIM22 or IFNγ were markedly restored by siTRIM22 (figure 5C, D, left panel). This result suggests that IFNγ-induced anti-HBV activity is mediated by TRIM22. In particular, the HBV genomic RNA (3.5 kb), a template for genomic DNA replication and mRNA for the core protein, was further recovered by TRIM22 knockdown (figure 5D, lower right panel). The levels of total viral and subgenomic (2.4/2.1 kb, mRNAs for surface proteins) RNAs were also significantly recovered (figure 5D, upper right panel). We further examined the effect of TRIM22 knockdown on IFN-mediated antiviral activity using the HepG2-NTCP and HepaRG HBV infection systems. Genome replication and the expression of viral proteins were almost completely restored by TRIM22 knockdown in both systems (figure 5E, F). Notably, the basal level of TRIM22 was much higher in HepaRG than in HepG2 cells (figure 5F). These results suggest an important role of TRIM22 under physiological conditions.
Finally, we compared IFNγ-induced anti-HBV activity with and without 5AzaC treatment to see if the HBV evasion mechanism against IFNγ antiviral response relies on HBx-mediated epigenetic suppression of TRIM22. Viral RNAs and HBx initially inhibited by IFNγ were further decreased by 5AzaC treatment, whereas the expression of TRIM22 increased (figure 5G).
Consequently, our data indicate that TRIM22 mediates IFNγ-induced anti-HBV activity and that HBV may escape this activity through epigenetic suppression of TRIM22 by HBx.
HBV suppresses IFN-induced TRIM22 expression through HBx in a mouse model, primary human hepatocytes, and human liver tissues
To check the biological relevance of our findings in vivo, we used an acute HBV infection model described previously.9 Because mice lack theTRIM22 gene, we introduced TRIM22 promoter clones (pTP5-Luc or pTP6-Luc) with indicated plasmids (pcDNA, HBx-HA, HBV1.2(X-) or WTHBV1.2) using hydrodynamic injection to access the role of HBx in IFN-inducible TRIM22 expression (figure 6A).

HBV suppresses interferons (IFNs)-inducible TRIM22 via HBV X protein (HBx) in a mouse model. (A) The experimental scheme. (B) Induction of IFNs in mouse liver by hydrodynamic injection of plasmids. At 48 hours after hydrodynamic injection, total RNA was extracted from three mouse liver tissues. The mRNA levels of mouse IFNs (mIFNα, mIFNβ and mIFNγ) were analysed by semiquantitative RT-PCR. (C) Effect of HBx on TRIM22 promoter activity in liver. Reporter plasmids were hydrodynamically injected into mouse liver, and luciferase activity was analysed in liver lysates. Relative luciferase activity was measured in nine independent experiments and normalised using β-gal assay. ***p<0.001. (D) Effect of genome-driven HBx on TP6 activity in mouse liver (n=5 per group). ***p<0.001. (E) Experimental scheme of 5AzaC (5 mg/kg) injection into the mouse tail vein. (F) Effect of 5AzaC on HBx-mediated suppression of TRIM22 promoter activity. Three mice per group were used for luciferase analysis. ***p<0.001; NS, not significant.
The mouse model of acute HBV infection was validated by observing HBV replication and viral protein expression (HBx-HA and HBsAg) in mouse hepatocytes (see online supplementary figure S7). Since HBx is critical for HBV replication,34 the replication of HBV1.2(X-) in the liver was marginal compared with that of WT HBV1.2 (see online supplementary figure S7A). However, there was no significant difference in the expression levels of surface proteins (see online supplementary figure S7B). Introduction of foreign DNA strongly induced type I and type II IFNs (IFNα, IFNβ and IFNγ) (figure 6B). Luciferase assays showed that only the TRIM22 promoter containing the 5′-UTR (+1 to +211) (pTP6-Luc) was strongly activated in response to IFNs. TRIM22 induction in mouse liver tissues was dramatically suppressed by HBx (figure 6C). Furthermore, replication-competent WT HBV1.2, which is capable of expressing genome-driven HBx, strongly reduced TRIM22 promoter activity in vivo compared with the effect of HBV1.2(X-) (figure 6D). The latter observation suggests that suppression of TRIM22 by HBx occurs during the natural course of HBV infection. In addition, we tested epigenetic regulation of HBx in vivo by examining whether 5AzaC can abolish the repression of the TRIM22 promoter by HBx (figure 6E). Tail vein injection of 5AzaC strongly restored the activity of the pTP6 promoter (figure 6F), indicating that the 5′-UTR of TRIM22 is also critical and responsible for HBx-mediated suppression of TRIM22 expression in vivo.
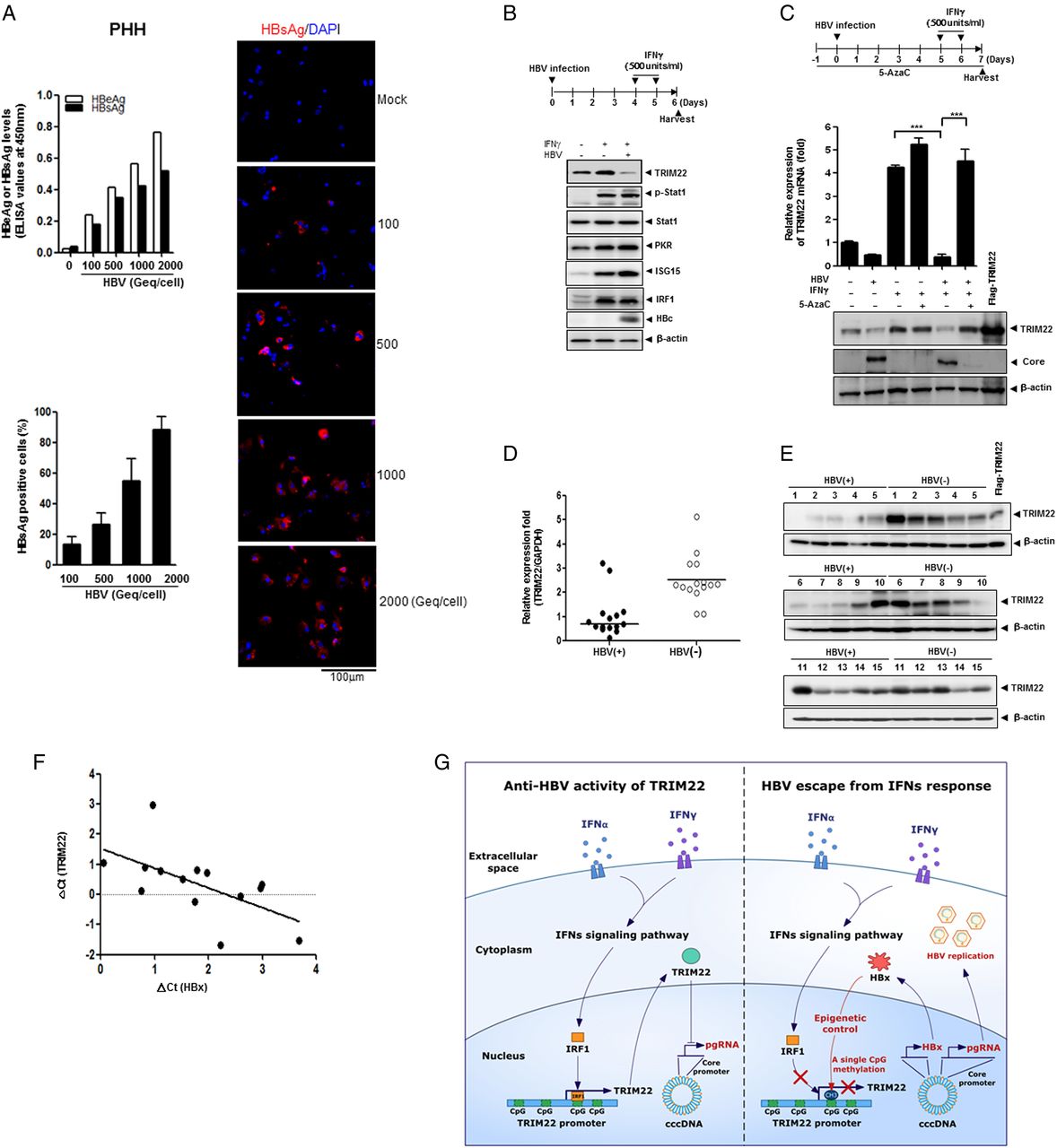
We performed similar experiments in primary human hepatocytes (PHHs) to check the physiological relevance of our findings. To determine the infection efficiency in PHHs, the cells were infected with various amounts of HBV (100–2000 viral genome equivalents per cell) and were analysed 7 days after infection by measuring the levels of secreted HBeAg and HBsAg.35 The percentage of HBV infection was approximately 90% (figure 7A). A slightly lower infection rate (∼70%) was observed in HepaRG cells (see online supplementary figure S8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HBV suppresses TRIM22 induced by interferons (IFNs) in primary human hepatocytes (PHHs) and human liver tissues. (A) Efficiency of HBV infection in PHHs. The level of secreted HBeAg and HBsAg were measured by ELISA at 7 days post infection. HBsAg-positive cells (%) were quantitated using the confocal images. Data are mean±SD of at least five regions. (B) Effect of HBV infection on expression levels of TRIM22 and other IFN-stimulated genes (ISGs) in PHHs. At 6 days post infection, the indicated proteins were detected by western blotting. (C) Effect of 5AzaC on HBV-mediated suppression of TRIM22. At 7 days post infection, the relative expression level of TRIM22 was measured by real-time PCR and the expression of TRIM22 was determined by western blotting. Data are mean±SD from at least three independent experiments. ***p=0.001. (D) TRIM22 expression levels in HBV-positive and HBV-negative liver tissues were evaluated by real-time PCR. (E) The level of the TRIM22 protein in liver tissues was determined by western blotting. (F) TRIM22 and HBV X protein (HBx) expression levels in HBV-positive tissues were measured by real-time PCR. Respective delta Ct values were subjected to Pearson correlation analysis. (G) A schematic model illustrating the epigenetic regulation of TRIM22 by HBV for viral evasion from IFN-mediated antiviral activity. IRF1, IFN regulatory factor-1.
To test whether HBV infection suppresses the expression of TRIM22 through epigenetic regulation in PHHs, we examined the expression level of TRIM22 using real-time PCR and western blot analysis after 5AzaC treatment. Notably, PHHs produced a basal level of TRIM22, which was marginal in HepG2 cells (figure 2). HBV infection strongly repressed the expression of IFNγ-induced TRIM22, but not that of other ISGs such as STAT1, p-STAT1, PKR, ISG15 or IRF1 in PHHs (figure 7B). Both mRNA and protein levels of TRIM22 were dramatically restored by 5AzaC treatment, indicating that the suppression of TRIM22 by HBV is mediated by epigenetic regulation in PHHs (figure 7C). To assess the clinical implications of our finding, we analysed HBV-positive and HBV-negative human liver tissues by real-time PCR to determine the TRIM22 levels (baseline characteristics of included patients are provided in online supplementary table S3). The TRIM22 mRNA level was significantly lower in HBV-positive tissues (figure 7D). The level of the TRIM22 protein was also significantly decreased in HBV-positive tissues (figure 7E). The level of the TRIM22 transcript was inversely correlated with that of HBx mRNA in HBV-positive liver tissues (figure 7F) (Pearson r=−0.6551, p<0.05).
Discussion
The well-developed evasion strategies of some viruses allow them to survive despite the IFN response. Viruses such as HIV and influenza virus possess various mechanisms to outmanoeuvre the IFN response.36 ,37 Hepatitis viruses including HCV, HDV and HEV also impair the IFN response.38–40 Since pegylated-IFN treatment for chronic HBV shows a poor response,18 ,19 it has been speculated that HBV has an IFN evasion mechanism, but its strategy to circumvent the IFN response is not fully understood.
In this study, we demonstrated the ability of HBV to counteract the induction of TRIM22, a mediator of the IFN-induced antiviral response. Proteome-wide analysis identified the ISG candidates suppressed by HBx during viral escape (figure 1). We found that HBV significantly desensitises the IFN-mediated induction of TRIM22 through methylation of a single CpG site in the 5′-UTR in vitro and in vivo. This possibly leads to HBV escape from IFN and antiviral activity of TRIM22 (figure 7G). Because HBx suppresses TRIM22 induced by either IFNα or IFNγ (figure 2C, D), it is likely that HBx suppresses the antiviral responses induced by both type I and type II IFNs simultaneously. These results also suggest why the response to treatment with pegylated-IFNα is not satisfactory. TRIM22 specifically suppressed viral enhancer and core promoter activity (figure 5B),30 and TRIM22 knockdown substantially restored viral RNAs inhibited by IFNγ (figure 5D–F). These results suggest that the therapeutic efficacy of IFNs could be improved by blocking TRIM22 suppression by HBx (figure 5G).
A number of studies have shown that epigenetic modulation of host genes is involved in viral evasion from host immune responses.41 We and others have reported that HBx induces epigenetic changes in host genomes.8 ,9 Here, we found a host epigenetic target involved in HBV escape from host immune responses. Epigenetic control is also involved in basal induction of TRIM22 by IFNs because the expression level of IFNγ-induced TRIM22 was increased by 5AzaC treatment (figure 3F, left panel, lane 5 vs 7). Importantly, the epigenetic modulation of TRIM22 was mediated by methylation of a single CpG in the 5′-UTR, not CpG islands in the promoter. The control of gene expression by a single CpG is not common; however, such regulation has been discovered in both host cells and viruses,42 ,43 implying that methylation at a specific single CpG could be sufficient for adaptation to environmental change.
The level of HBV replication in IRF1 knockout mice was higher than that in wild type,12 suggesting that some factors regulated by IFN-mediated IRF1 may be involved in HBV inhibition. We identified TRIM22 as an IRF1-regulated antiviral protein and established that its 5′-UTR is essential for both IFN-mediated induction and HBx-mediated suppression of TRIM22 (figure 3A–C). Although HBx induced methylation at four CpG sites (figure 3D), only methylation at a specific single CpG (+71) decreased IRF1 binding to the 5′-UTR of TRIM22 (figure 4) and subsequent IFN-induced TRIM22 expression. However, the expression of IRF1 was not changed by HBx (see online supplementary figure S4). Demethylation of the third CpG upon 5AzaC treatment restored IFNγ-mediated TRIM22 expression in HBx-expressing cells (figure 3E, F). Our study suggests that single-nucleotide polymorphism at this critical CpG site in TRIM22 might cause different clinical outcomes in individual patients infected with HBV. A clinical study might be warranted to evaluate the association between the level of TRIM22 and virological response to IFN.
Although HBV mainly infects hepatocytes, it has also been reported to infect immune cells such as human peripheral blood lymphocytes (PBMCs) including T lymphocytes.44 ,45 Coinfection with HIV and HBV has a worse prognosis in patients than infection with HBV alone.46 It is well known that TRIM22 has potent anti-HIV activity, and the level of TRIM22 in immune cells is higher than that in other cells.25 Therefore, we assume that downregulation of TRIM22 by HBx in PBMCs could also lead to HIV evasion from IFN-mediated immune responses in coinfected patients. Indeed, this assumption is supported by previous reports of large amounts of HBx detected in PBMCs,44 ,45 and by the ability of HBx to induce HIV replication and transcription and thereby contribute to a faster progression to AIDS.47
In the case of HBV, the evasion strategies against the IFN response through HBV polymerase and HBx have been observed in hepatocytes and mouse models. HBV polymerase and HBx can counteract pattern recognition receptor signalling through binding to DDX3 and IPS-1, and thereby downregulate IFN production in hepatocytes.22 ,23 In addition, HBV effectively suppresses IFN signalling by impairing STAT activation through inhibition of STAT nuclear translocation.21 Since TRIM22 is induced by both type I and type II IFNs, the suppression of TRIM22 by HBx could have a role in HBV escape from a broad range of host IFNs involved in the defence system. It is worth noting that the steady-state protein level of TRIM22 is higher in HepaRG and PHHs compared with HepG2 cells (figures 5E–F and 7B, C). This high level of TRIM22 in PHHs may serve as a restriction factor for HBV spread at early stage of infection. Although the details of the mechanisms of HBV evasion from the IFN-mediated antiviral response are still largely unclear, our finding will help to understand how HBV acts like a ‘stealth’ virus.48
The liver contains non-parenchymal cells including Kupffer cells, sinusoidal endothelial cells and lymphocytes, and is an organ with predominant innate immunity.49 Thus, cytokines (including IFNs) secreted from non-parenchymal cells may contribute to the paracrine anti-HBV response in hepatocytes, although HBV can block the IFN response in hepatocytes. Interestingly, it was recently reported that IFNα-induced anti-HBV activity is transferred by cell-to-cell transmission via exosomes from non-parenchymal cells.50 Accordingly, the strategy of HBV to directly reduce the expression of ISGs such as TRIM22 and therefore to downregulate the IFN response seems to be effective for HBV survival, because HBV cannot infect and regulate non-parenchymal cells effectively.
It has been reported that the induction of ISGs is limited and not detectable in mouse and chimpanzee during the initial phase of HBV infection, unlike infections with other viruses such as HCV.48 Nevertheless, the detailed mechanism of the direct inhibition of IFN-induced antiviral proteins by HBV has not yet been clearly elucidated. Here, we found that TRIM22 induction by IFNs is downregulated by HBx via epigenetic control of a single CpG methylation site in the 5′-UTR of TRIM22. Functional analysis of other proteins from Cluster C (figure 1) would also yield information helpful for understanding HBV evasion from the IFN response.
In conclusion, we showed that epigenetic regulation of TRIM22 by HBx is involved in HBV evasion from the IFN-mediated antiviral response. Our findings will extend the understanding of the pathophysiology of HBV infection and provide a potential way to improve the therapeutic effect of IFNs for HBV clearance.
Experimental procedures
Materials and methods are described in online supplementary information.
References
Footnotes
K-HL and E-SP contributed equally.
Contributors Study conception and design: K-HL, E-SP and K-HK. Acquisition of data: K-HL, E-SP, KCC, YKP, DHK, SHA, SHP, HSK, ARL, SP, HS and JW. Analysis and interpretation of data: K-HL, E-SP, KPK, JSY, BLS and KHK. Material support: K-HK, CWK, J-HL, N-JY, K-WL and K-SS. Obtained funding: K-HK and E-SP. Drafted the manuscript: K-HL, E-SP and K-HK.
Funding This study was supported by the National Research Foundation (NRF) grant funded by the Korean government (No. 2013R1A2A2A01068194, No. 2014M3A9A8064633, NRF-2016R1A5A2012284 and 2016R1A2B4007531). This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute , funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI14C-1529-020014).
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.