Article Text

Abstract
Objective Helicobacter pylori is the strongest risk factor for gastric cancer; however, the majority of infected individuals do not develop disease. Pathological outcomes are mediated by complex interactions among bacterial, host and environmental constituents, and two dietary factors linked with gastric cancer risk are iron deficiency and high salt. We hypothesised that prolonged adaptation of H. pylori to in vivo carcinogenic microenvironments results in genetic modification important for disease.
Design Whole genome sequencing of genetically related H. pylori strains that differ in virulence and targeted H. pylori sequencing following prolonged exposure of bacteria to in vitro carcinogenic conditions were performed.
Results A total of 180 unique single nucleotide polymorphisms (SNPs) were identified among the collective genomes when compared with a reference H. pylori genome. Importantly, common SNPs were identified in isolates harvested from iron-depleted and high salt carcinogenic microenvironments, including an SNP within fur (FurR88H). To investigate the direct role of low iron and/or high salt, H. pylori was continuously cultured in vitro under low iron or high salt conditions to assess fur genetic variation. Exposure to low iron or high salt selected for the FurR88H variant after only 5 days. To extend these results, fur was sequenced in 339 clinical H. pylori strains. Among the isolates examined, 17% (40/232) of strains isolated from patients with premalignant lesions harboured the FurR88H variant, compared with only 6% (6/107) of strains from patients with non-atrophic gastritis alone (p=0.0034).
Conclusion These results indicate that specific genetic variation arises within H. pylori strains during in vivo adaptation to conditions conducive for gastric carcinogenesis.
- Helicobacter pylori
- gastric cancer
- iron deficiency
- high salt
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Helicobacterpylori strains exhibit remarkable genetic diversity as a species and have a panmictic population structure, reflecting a high level of genetic exchange among strains.
H. pylori is the strongest known risk factor for the development of gastric adenocarcinoma; however, the majority of H. pylori-infected individuals do not develop disease.
Two environmental factors associated with increased gastric cancer risk are high dietary salt intake and iron deficiency. Both high salt and low iron significantly augment the severity of H. pylori-induced gastric inflammation and the development of premalignant and malignant lesions in rodent models of gastric cancer.
What are the new findings?
Whole genome sequencing of H. pylori strains B128 and 7.13 and in vivo-adapted 7.13 output isolates harvested from animals maintained on iron-replete, iron-depleted, regular salt or high salt diets identified a small number of common polymorphisms that occurred within the context of iron deficiency and high salt intake.
Specific cancer-related genetic loci were identified when in vivo-adapted carcinogenic H. pylori strain 7.13 was compared with its progenitor human H. pylori isolate B128, which does not cause cancer.
Environmental-specific genetic loci were identified among H. pylori strains isolated from carcinogenic, iron-deficient or high salt, microenvironments.
A single nucleotide polymorphism in fur was selected for by exposure to low iron or high salt conditions and was more frequently identified in clinical H. pylori strains from patients with premalignant lesions.
How might it impact clinical practice in the foreseeable future?
Key H. pylori genetic loci that are susceptible to genetic alteration under in vivo environmental stress may serve as genetic biomarkers for H. pylori virulence potential and gastric cancer risk.
Introduction
Helicobacter pylori persistently colonises the gastric mucosa of more than 50% of the world’s population. H. pylori strains harbour remarkable genetic diversity as a species, as evidenced by variation of gene order and genetic content, the mosaic structure of genes and sequence diversity within conserved genes.1–3 H. pylori exhibits a panmictic population structure, reflecting a high level of genetic exchange among strains,4 5 and recombination between strains can occur in vivo during naturally occurring mixed infections.3 Analyses of complete genomic sequences from a number of isolates indicate that H. pylori has incorporated genetic material from other organisms,4 5 and that variation occurs among single colony isolates of distinct H. pylori from an individual host.6 7
In addition to being one of the most common bacterial infections worldwide, H. pylori is the strongest known risk factor for the development of gastric adenocarcinoma, and pathological outcomes are mediated by complex interactions among bacterial virulence determinants, host constituents and environmental factors. One H. pylori virulence determinant that increases gastric cancer risk is the cag pathogenicity island. H. pylori strains that harbour the cag island induce more severe gastric injury and further augment the risk for gastric cancer compared with cag-negative strains. The cag island encodes a bacterial type IV secretion system, which translocates the oncoprotein CagA into host cells to induce cellular morphological changes, proliferation and migration.2 8
Two environmental factors associated with increased gastric carcinogenesis are high dietary salt intake and iron deficiency.9 Epidemiological studies have demonstrated a link between salt intake and increased gastric cancer risk,10 11 and high salt diets accelerate the development of gastric cancer in animal models of infection.12–14 Similarly, epidemiological studies have supported the concept that iron deficiency contributes to the risk of gastric cancer,15 16 and a recent study from our group demonstrated that iron depletion significantly increased the development of gastric adenocarcinoma in H. pylori-infected gerbils.17 Therefore, both high salt and low iron augment the severity of H. pylori-induced gastric inflammation and the development of premalignant and malignant lesions in rodent models of gastric cancer.
Several studies have investigated specific mechanisms through which iron deficiency or high salt may modify H. pylori and enhance the risk for gastric cancer. Iron deficiency and high salt-dependent alterations of H. pylori per se include alterations in bacterial cell morphology, gene regulation and protein expression.17–26 Specifically, CagA expression and translocation into gastric epithelial cells are increased in response to both high salt and low iron conditions, and, under iron restriction, function of the cag type IV secretion system is enhanced.17 22 24 All of these modifications likely contribute to the increased risk of H. pylori-induced gastric cancer associated with these dietary alterations.14 17 22 24 26
In this study, we tested the hypothesis that prolonged adaptation of H. pylori to an in vivo carcinogenic gastric microenvironment induced by iron deficiency or high salt would select for genetic variants important for pathogenesis. We previously identified a strain of H. pylori, 7.13, that reproducibly induces gastric cancer in rodent models of gastritis. This strain was derived via in vivo adaptation of a human clinical H. pylori strain, B128, which induces inflammation, but not cancer, in rodent gastric mucosa. Thus, H. pylori strains B128 and 7.13 are closely related genetically, but differ in oncogenic potential.27 Whole genome sequence analyses were performed on H. pylori strains B128 and 7.13, and in vivo-adapted 7.13 output isolates exposed to iron-replete or iron-depleted conditions and indicate that targeted genetic variation in H. pylori arises during in vivo adaptation to conditions conducive for the development of gastric cancer.
Results
H. pylori genetic alterations during in vivo adaptation
We initially conducted whole genome sequence analyses of human progenitor H. pylori strain B128 and its in vivo gerbil-adapted carcinogenic derivative strain 7.13. Sequences were assembled using the genome sequence of H. pylori strain B8, a closely related strain for which its complete genome sequence is available, as the reference strain.28 In addition to these strains, we also performed whole genome sequence analysis using in vivo gerbil-adapted H. pylori isolates exposed to iron-replete or iron-depleted conditions. Three single colony isolates from H. pylori strains B128 and 7.13 and three single colonies from 7.13-infected gerbils maintained on either iron-replete (n=3) or iron-depleted (n=3) diets were used for whole genome sequence analysis (figure 1). In vivo-adapted strains from iron-replete and iron-depleted conditions were also compared with in vivo-adapted strains isolated from regular salt or high salt conditions14 (figure 1).

Experimental design. Helicobacter pylori strain B128 was originally isolated from a human patient.54 Strain B128 (green) was used for orogastric challenge of Mongolian gerbils and was passaged three times through gerbils in a previous study.28 The in vivo-adapted output strain, designated H. pylori strain B8 (orange), was isolated from those gerbils, sequenced and used as the reference strain in this study.19 In an independent experiment, strain B128 (green) was also used for orogastric challenge of a single Mongolian gerbil, and the in vivo-adapted output strain isolated from this gerbil was designated H. pylori strain 7.13 (orange).27 Carcinogenic H. pylori strain 7.13 was then used to infect another cohort of Mongolian gerbils maintained on either iron-replete or iron-depleted diets.17 Single colonies (n=3) from three independent gerbils maintained on iron-replete diet (n=9) and three independent gerbils maintained on iron-depleted diet (n=9) were subjected to whole genome sequence analysis. In vivo-adapted strain from iron-replete (red) and iron-depleted (blue) conditions was also compared with previously described H. pylori single colonies (n=4) harvested from one gerbil maintained on a regular salt (pink) or high salt (light blue) diet.21
Whole genome sequence analysis
The total length of the H. pylori reference strain B8 genome (1673 997 bp) was completely covered, and the guanine-cytosine content was 38.8% for all sequenced strains. Overall sequence read count, average sequence read length, coverage and unmapped reads for H. pylori sequences are shown in online supplementary table S1. On average, the total read count was 645 132 bp, the mean read length was 263 bp, the fold coverage was 102 and the percentage of unmapped reads was 6.6% for all H. pylori strains analysed in this study.
Supplementary file 1
Phylogenetic analysis confirmed that human H. pylori B128 isolates are more closely related to each other than to H. pylori strains adapted to gerbil stomachs (figure 2A). Consistent with in vivo adaptation within the same host, the reference H. pylori strain B8, which is also gerbil adapted, is more closely related to the gerbil-adapted strain 7.13 than to its parental human-adapted strain B128 (figure 2A). Furthermore, H. pylori strains isolated from gerbils under iron-replete conditions are more closely related to each other than to strains isolated from gerbils maintained on iron-depleted diets (figure 2A). These findings demonstrate that in vivo adaptations to different hosts (human vs gerbil) and to different environmental conditions (iron-replete vs iron-depleted) induce H. pylori genetic changes.

(A) Phylogenetic analyses of whole genome sequences from Helicobacter pylori strains B128 (green) and 7.13 (orange), and in vivo-adapted iron-replete (red) and iron-depleted (blue) strains. R1-3 and D1-3 designate independent gerbils maintained on either iron-replete (n=3) or iron-depleted (n=3), respectively, while a–c designate different single colonies isolated from each gerbil. H. pylori strains isolated from humans or gerbils are grouped within the grey boxes, respectively. The scale bar represents amino acid changes per site. (B) Venn diagram20 showing the number of common and unique single nucleotide polymorphisms (SNPs) among human-adapted H. pylori strains B128 (green) and 7.13 (orange), and in vivo-adapted H. pylori strains isolated from gerbils maintained on iron-replete (red) and iron-depleted (blue) diets, all relative to the reference H. pylori strain B8. The number in the parentheses indicates the total number of unique SNPs identified in each strain and present exclusively in that strain, but not any other strain.
The number of unique single nucleotide polymorphisms (SNPs) among all sequenced H. pylori strains, relative to H. pylori reference strain B8, is shown in figure 2B.29 The human-adapted strain B128 had 26 unique SNPs (ie, SNPs present exclusively in strain B128, but not in any other strain) (figure 2B, online supplementary table S2). The gerbil-adapted strain 7.13 only had two unique SNPs (figure 2B, online supplementary table S2), highlighting the similarity among strains adapted to the gerbil host. However, subsequent H. pylori adaptation to iron-replete (29 SNPs) or iron-depleted (49 SNPs) conditions resulted in the greatest number of unique SNPs (figure 2B, online supplementary table S2), reinforcing the importance of genetic adaptation to different environmental conditions.
When all of the sequences were analysed, a total of 180 genetic loci with single and multinucleotide polymorphisms were identified, including 33 within intergenic regions and 147 within coding regions (online supplementary table S3). When each single colony was analysed individually for total polymorphisms, there was variation among the single colonies (online supplementary tables S4 and S5). The H. pylori human isolate B128 had, on average, 98 polymorphisms, 12 insertions and 10 deletions. Of the polymorphisms within coding regions of strain B128, 67% resulted in a non-synonymous change (online supplementary table S4). Among the three single colonies, gerbil-adapted H. pylori strain 7.13 contained, on average, 47 polymorphisms, nine insertions and three deletions. Of the SNPs within coding regions of strain 7.13, 65% resulted in a non-synonymous change (online supplementary table S4). Similarly, among the nine single colonies harvested from three gerbils maintained on iron-depleted diets, there were, on average, 52 polymorphisms, 12 insertions and five deletions, while in vivo-adapted strains isolated from gerbils maintained on iron-replete diets had, on average, 43 polymorphisms, 11 insertions and three deletions. Of the SNPs within coding regions of the iron-depleted and iron-replete strains, 54% and 52% resulted in a non-synonymous change, respectively (online supplementary table S5). Based on the number of variants identified, most of the genetic variation arose through in vivo adaptation to a new gerbil host. However, all H. pylori isolates were found to be unique, including isolates from a single host.
Many of the genetic alterations resulting in deletions or insertions occurred within genetic loci important for H. pylori pathogenesis, including polymorphisms within loci regulating chemotaxis (cheA), adhesion (hopZ, oipA, sabA), survival (katA), iron acquisition or active transport (tonB) and cag type IV secretion system function (cagY) (table 1).
Deletions and insertions in Helicobacter pylori strains B128 and 7.13, and in vivo-adapted strains isolated from gerbils maintained on iron-depleted or iron-replete diets
Among the 180 polymorphisms identified, 72 were located within genes with predicted function and resulted in non-synonymous amino acid changes (table 2). Similar to the insertions and deletions, many of these variants were within genes encoding proteins that function in pathogenesis, including motility (fliI, fliF), adhesion (babA, babB), iron acquisition or active transport (tonB) and cag IV secretion system function (cagY). When we compared unique SNPs within coding regions between in vivo-adapted strains isolated from gerbils maintained on iron-depleted versus iron-replete diets, we identified 35 polymorphisms, distributed among 15 genes, including babB, cagY, pepP, oppD, hcpD, hcpE, prfB, and various outer membrane and hypothetical proteins. Additional SNPs were present in both groups, but were more prevalent in iron-depleted versus iron-replete strains; these included SNPs within babA, babB and fur.
Unique genetic loci with polymorphisms resulting in non-synonymous amino acid changes in Helicobacter pylori strains B128 and 7.13, and in vivo-adapted strains isolated from gerbils maintained on iron-depleted or iron-replete diets
The SNP (FurR88H) located within fur, a gene encoding the ferric uptake regulator, was identified previously in in vivo-adapted H. pylori strains isolated from a gerbil maintained on a high salt diet.30 Therefore, we compared the number of SNPs from in vivo-adapted strains isolated from gerbils maintained on iron-depleted diets with those previously isolated from a gerbil maintained on a high salt diet. Specifically, we sought to identify SNPs present in these isolates that were either not present or present at a lower frequency in strains isolated from control gerbils maintained on iron-replete or regular salt diets. We identified five SNPs distributed among four genes, including HPB8_593, HPB8_716 (cagY), HPB8_1139 and HPB8_1145 (fur) (online supplementary table S6) in iron-depleted and high salt H. pylori in vivo-adapted output strains, suggesting that certain loci are subject to variation following in vivo adaptation to different host microenvironments.
Analysis of sequence variation in fur
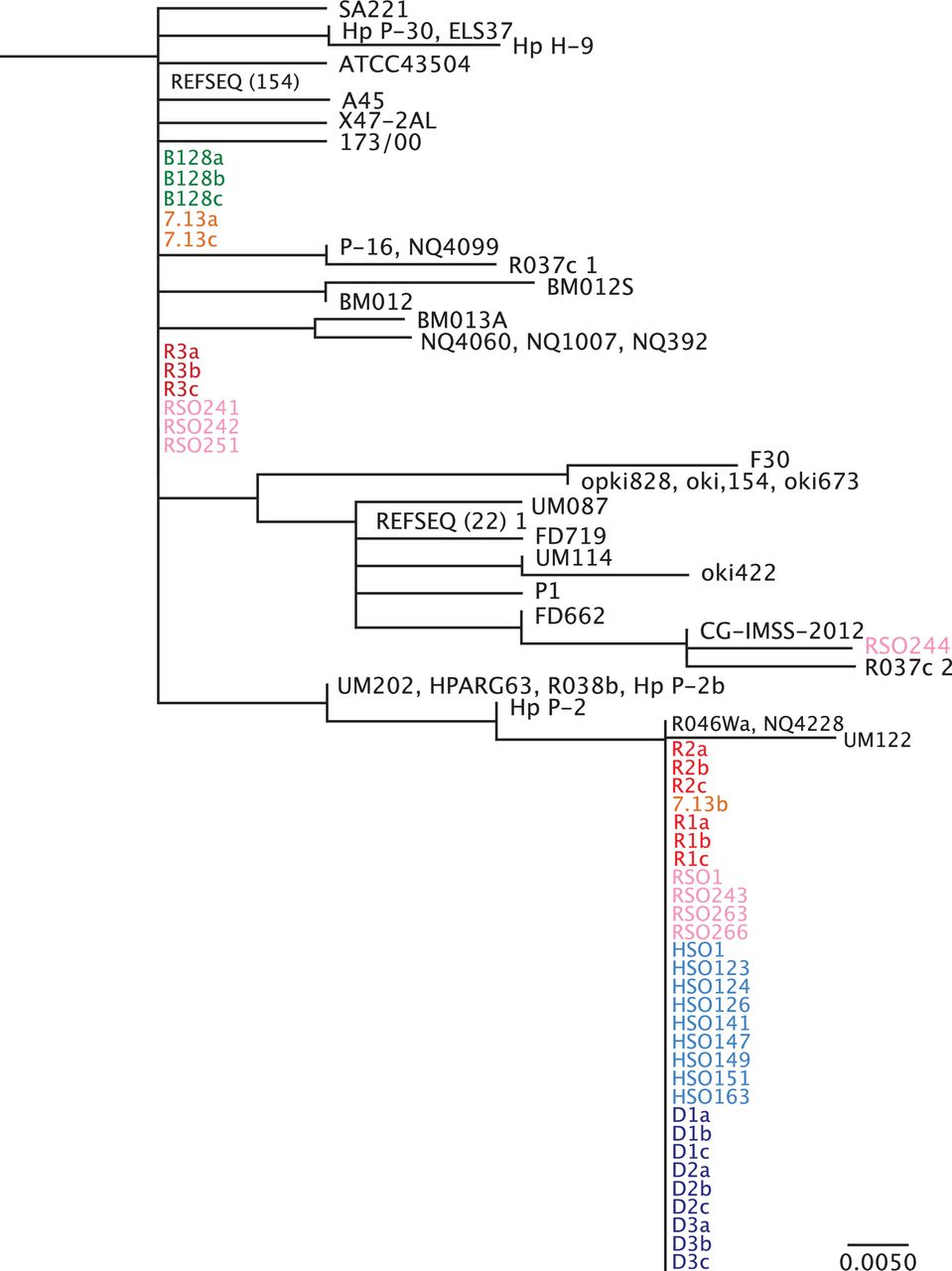
To investigate mechanisms underpinning these changes in greater depth, phylogenetic analysis of Fur in H. pylori strains B128 and 7.13, and in vivo-adapted strains isolated from gerbils maintained on iron-replete, iron-depleted, high salt or regular salt diets was performed (figure 3). In vivo-adapted strains isolated from gerbils maintained on iron-depleted or high salt diets exhibited a high degree of identity within Fur, and segregated based on the FurR88H variant, suggesting that this region may be a susceptible genetic locus subject to variation under these conditions. The presence of the fur SNP in some regular salt21 and iron-replete strains in addition to one 7.13 single colony suggests that this variant is present at a low frequency within H. pylori populations and is favoured under certain conditions such as high salt and low iron.

Phylogenetic analysis of Fur amino acid sequence. Phylogenetic analysis was performed for full-length Fur from all available Helicobacter pylori sequence data within the National Center for Biotechnology Information NCBI database. H. pylori single colonies from strains B128 (green) and 7.13 (orange), and in vivo-adapted strains isolated from gerbils maintained on iron-replete (red) and iron-depleted (blue) diets as well as in vivo-adapted strains isolated from gerbils maintained on regular salt (pink) and high salt (light blue) diets were included in these analyses. R1-3 and D1-3 designate independent gerbils maintained on either iron-replete (n=3) or iron-depleted (n=3), respectively, while a–c designate different single colonies isolated from each gerbil. HSO and RSO designate independent high salt output and regular salt output H. pylori strains, respectively. REFSEQ (#) represents the number of unique H. pylori sequences from different H. pylori strains that cluster under the same phylogeny. Although all Fur sequences from the NCBI database were used, this phylogenetic analysis shows only the branch to which the newly sequenced strains cluster. The scale bar represents amino acid changes per site.
To extend these findings, we next investigated the direct effects of high salt or low iron per se on genetic variation within fur. H. pylori strain 7.13 was continuously cultured in vitro for 6 days under high salt (1.1% NaCl), in the presence of an iron chelator (100 µM dipyridyl) to simulate iron depletion, or supplementation with iron (100 µM FeCl3). As expected, H. pylori growth under high salt or low iron was significantly attenuated (figure 4A). Sequence analysis revealed that adaptation to high salt and/or iron depletion in vitro resulted in selection of the fur SNP (FurR88H) after only 4 to 5 days, respectively, and this variant was maintained throughout the duration of growth in vitro (figure 4B). Exposure of H. pylori to normal growth conditions or normal growth conditions supplemented with iron did not induce sequence variation within fur. These data indicate that in vitro adaptation to high salt or low iron induces genetic variation in fur in vitro, suggesting that adaptation within the context of environmental stress may be regulated by these dietary changes in vivo.

Analysis of genetic variation within fur following continuous culture in vitro under high salt or low iron conditions. (A) Helicobacter pylori strain 7.13 was grown in normal Brucella broth, modified Brucella broth with iron supplementation (100 µM FeCl3), iron chelation (100 µM dipyridyl) or high salt (1.1% NaCl) for 20 hours and continuously over a 6-day time course. The growth rate or ratio of the final OD600 to the initial starting OD600 is represented as mean values with SE from experiments performed on at least six independent occasions. A one-way analysis of variance with multiple comparisons was used to determine statistical significance. ****p<0.0001. (B) H. pylori strain 7.13 was continuously cultured in vitro under these conditions for 6 days to assess the presence (CAC) or absence (CGC) of the SNP in fur (FurR88H). SNP, single nucleotide polymorphism.
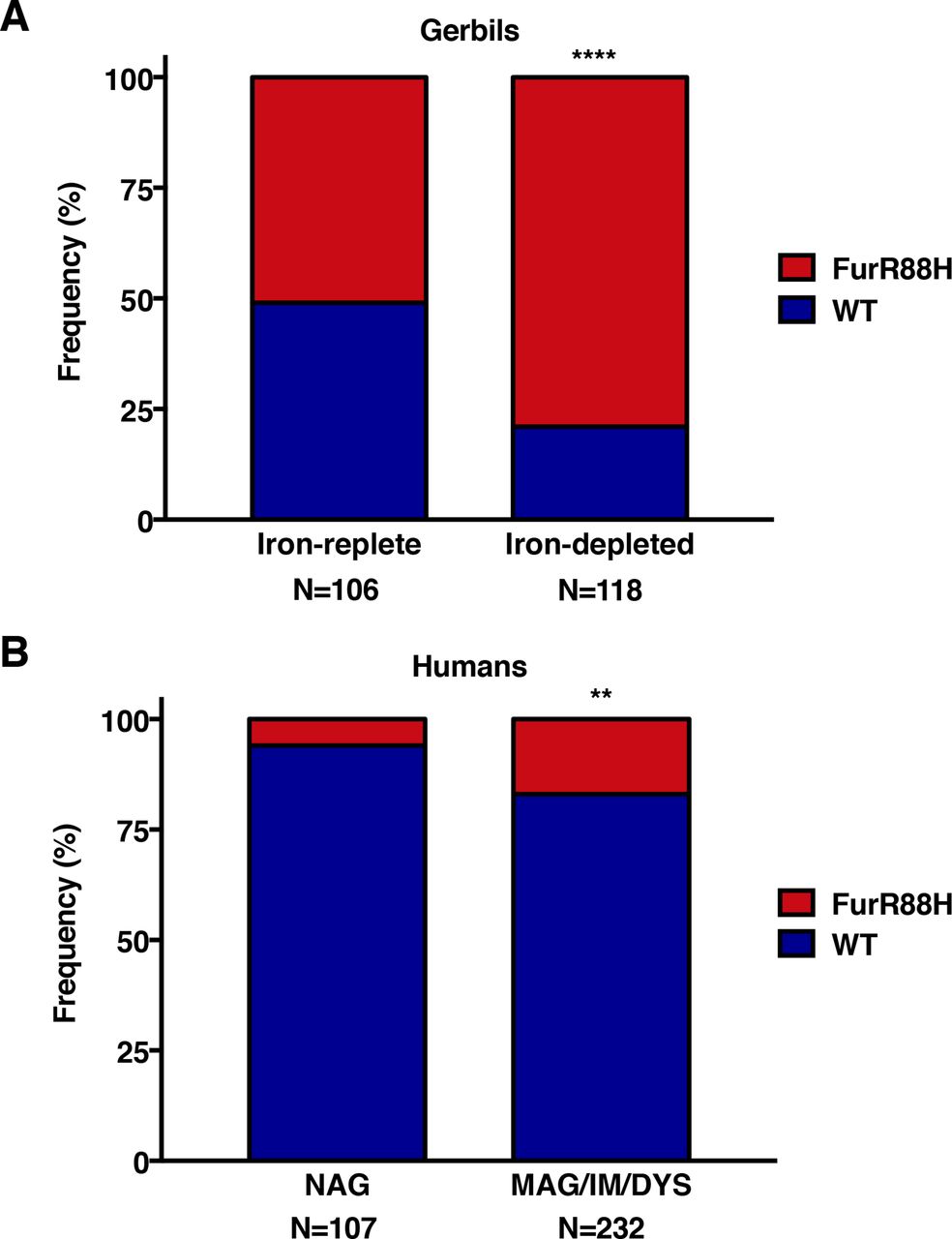
To expand on these in vitro studies and specifically address the relationship between presence of the FurR88H variant and iron deficiency in vivo, we performed sequence analysis on 224 in vivo-adapted H. pylori strains isolated from gerbils maintained on iron-replete or iron-depleted diets. The FurR88H variant was present at a significantly higher frequency among gerbils maintained on iron-depleted diets (93/118, 79%) compared with those maintained on iron-replete diets (54/106, 51%; p<0.0001, figure 5A). These data indicate that in vivo adaptation to environmental iron deficiency leads to genetic adaptation within fur.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of genetic variation within fur in vivo. The frequency of the FurR88H variant was examined among (A) 224 in vivo-adapted Helicobacter pylori strains isolated from gerbils maintained on iron-replete or iron-depleted diets and (B) 339 clinical H. pylori strains isolated from three independent and well-defined patient cohorts within Colombia and the USA.31–33Fischer’s exact test was used to determine statistical significance. **p<0.005 and ****p<0.0001. DYS, dysplasia; IM, intestinal metaplasia; MAG, multifocal atrophic gastritis; NAG, non-atrophic gastritis alone.
Finally, we examined genetic variation in fur among 339 clinical H. pylori strains isolated from three independent and well-defined patient cohorts within Colombia and the USA (online supplementary table S7).31–33 Sequence analysis of fur revealed that 14% (46/339) of these clinical H. pylori strains harboured the same FurR88H variant identified in in vivo gerbil-adapted strains isolated from conditions of high salt or iron deficiency (online supplementary table S7). Among the clinical H. pylori isolates examined, 17% (40/232) of strains from patients with premalignant lesions (multifocal atrophic gastritis, intestinal metaplasia, dysplasia) harboured the FurR88H variant, compared with 6% (6/107) of strains from patients with non-atrophic gastritis alone (p=0.0034, figure 5B). These results demonstrate that this specific fur SNP is likely important for in vivo adaptation to a new host and within subpopulations of humans at increased risk for gastric cancer.
Discussion
Bacterial population structures differ based on the taxa under investigation. Some species, such as Mycobacterium tuberculosis 34 and Yersinia pestis,35 are of such recent origin that little sequence diversity has accumulated. In such largely clonal species, only a few coding sequences are variably present among different isolates.36–38 However, bacteria with high sequence diversity and frequent homologous recombination, such as H. pylori, harbour different population dynamics.39
H. pylori has co-evolved with humans since their origin, and such lifelong evolution is associated with selection of host-specific variants within particular genes necessary for survival and adaptation to the human host.8 Survival and persistence within the human host depends on the ability of H. pylori to rapidly respond to changing environmental conditions and circumvent host defense mechanisms initiated during colonisation.8 Rearrangement of genomic DNA allows a variety of pathogens to alter expression of surface antigens and evade host immunity, and H. pylori has one of the highest rates of genetic recombination of any known bacterial species,40 suggesting that this process confers a selective advantage in colonisation and survival. Indeed, H. pylori mutants defective in homologous recombination are spontaneously cleared,41 reinforcing the tenet that genetic recombination and variation are critical for H. pylori survival within the human host.
In addition to high rates of recombination, H. pylori displays exceptional genetic variability and intraspecies diversity.4 40 H. pylori has a highly plastic genome, reflecting elevated rates of recombination and development of point mutations, which promotes bacterial divergence and intraspecies diversity within a population.42 43 H. pylori exists within its gastric niche as a bacterial population in a continuous state of genetic variation, which allows rapid adaptation to changing environmental conditions in its current host as well as an ability to successfully colonise a new host.2 44 45 In this study, we hypothesised that prolonged adaptation of H. pylori to an in vivo carcinogenic gastric environment induced by iron deficiency would facilitate genetic changes involved in survival, persistence and increased virulence. Indeed, we demonstrated that H. pylori strains isolated from gerbils maintained on iron-replete diets are more closely related to each other than to strains isolated from gerbils maintained on iron-depleted diets, suggesting that specific variants are advantageous for survival and persistence in altered microenvironments. In addition, the current analysis confirmed that H. pylori B128 isolates recovered from a human stomach are more closely related to each other than to H. pylori strains adapted to gerbil stomachs, and that there are specific genetic variations that arise following adaptation to a new host. The diversity among 7.13 single colony isolates suggests that passage of B128 through a new gerbil host resulted in a more diverse population of isolates poised for further adaptation in vivo. As a result of frequent recombination and genetic variation, almost all H. pylori isolates from unrelated hosts and differing environments possess unique sequences.4 39
Further and consistent with other reports,6 7 we have demonstrated that variability exists among single colony isolates from each individual H. pylori strain. As a result of this diversity, H. pylori has been previously termed a quasispecies, a population of genetic variants comprising a single strain that allows for rapid selection and adaptation to the changing gastric microenvironments of the host.45–47 Recent changes in DNA sequencing technology have afforded the opportunity to assess the genetic diversity of H. pylori populations in much greater depth. We have now demonstrated that polymorphisms exist within single colonies, suggesting that even greater levels of within-host population diversity exist than previously demonstrated. Furthermore, we identified numerous genetic variants that arise under the selective pressure of iron deficiency, and some of these variants were also identified in H. pylori isolates exposed to high salt.
Among the various SNPs that resulted in non-synonymous variants, the majority arose within genetic loci prone to variation during adaptation to a new host or environmental condition. Many of these included outer membrane proteins that are particularly susceptible to selective pressure for survival and persistence. Adhesins that underwent genetic modification included babA and babB, which have been shown to undergo specific recombination in other models of H. pylori infection.48–50 Another genetic locus, cagY, which encodes an essential component of the cag type IV secretion system, has been shown to be particularly susceptible to variation in vivo.51 52 Another targeted locus was fur, which encodes a transcriptional regulator that controls expression of a large repertoire of genes and is an essential factor for survival and persistence of H. pylori in the stomach. Fur controls bacterial iron homeostasis, and also plays a central role in bacterial metabolism, energy production and immune regulation.53 The specific SNP in fur (FurR88H) identified following adaptation to low iron conditions was also identified in another study of in vivo adaptation to high salt.30 In this study, the fur SNP was directly linked to increased expression of genes controlling iron acquisition and homeostasis as well as increased resistance to high salt conditions and oxidative stress.30 Due to the role of Fur in regulating iron homeostasis, it is likely that this SNP also promotes increased survival and persistence of H. pylori under low iron conditions. Collectively, our data suggest that the accumulation of polymorphisms allows H. pylori to rapidly adapt to changing microenvironments to promote survival and persistence within the host.
Methods
Mongolian gerbils
Male Mongolian gerbils (Charles River Laboratories) were housed in the Vanderbilt University Animal Care Facilities and maintained on iron-depleted (TestDiet 5TWD), iron-replete (TestDiet 5STQ),17 regular salt or high salt (Purina 5001).14 Gerbils were orogastrically challenged with carcinogenic H. pylori strain 7.13. H. pylori strain 7.13 was originally isolated as a single colony following in vivo adaptation of the human H. pylori strain B128 to the Mongolian gerbil model and was then subcultured following minimal passage for single colony purification (figure 1).27 Single colony purified strain 7.13 was isolated from a frozen stock, expanded on trypticase soy agar plates with 5% sheep blood (BD Biosciences) and then grown in Brucella broth (BD Biosciences) supplemented with 10% FBS (Atlanta Biologicals) for 16 hours at 37°C with 5% CO2 prior to gerbil challenge.17 Gerbils were then euthanised 12–16 weeks postchallenge. The Vanderbilt University Institutional Animal Care and Use Committee (IACUC) approved all experiments and procedures.
H. pylori culture
H. pylori strain B128 was originally isolated from a single gastric biopsy of a human patient with gastric ulceration.54 B128 was subsequently used for orogastric challenge of a gerbil, and the in vivo-adapted output strain isolated from this gerbil was designated H. pylori strain 7.13.27 H. pylori strain 7.13 reproducibly induces gastric cancer in rodent models,14 17 27 while B128 only induces gastritis, but not cancer in these models.27 Three single colonies of B128 and 7.13 were isolated and expanded. In a more recent study, carcinogenic strain 7.13 was then used to infect another cohort of gerbils maintained on either iron-replete or iron-depleted diets.17 In vivo-adapted single colonies were recovered from gerbils maintained on iron-replete (n=3) and iron-depleted (n=3) diets, and single colonies were isolated and expanded.
In this study, we analysed in vivo-adapted output strains that were cultured from these gerbils. Specifically, we analysed three single colonies cultured from three gerbils maintained on an iron-replete (n=9) or iron-depleted (n=9) diets, and these were compared with single colonies cultured from one gerbil maintained on regular salt (n=4) or high salt (n=4). All H. pylori isolates were minimally passaged on trypticase soy agar plates with 5% sheep blood (BD Biosciences) and in Brucella broth (BD Biosciences) supplemented with 10% FBS (Atlanta Biologicals) for 16 hours at 37°C with 5% CO2.
H. pylori in vitro adaptation to high salt or low iron conditions
H. pylori strain 7.13 was grown in modified Brucella broth containing 1% Bacto tryptone (BD Biosciences), 1% Bacto Proteose Peptone No. 3 (BD Biosciences), 0.2% yeast extract (Difco), 0.1% dextrose (Fisher Scientific) with high sodium chloride (1.1% NaCl, Sigma), dipyridyl (100 µM 2,2-bipyridyl, Sigma) or addition of iron (100 µM FeCl3, Sigma) approximately 20 hours at 37°C with 5% CO2 and continuously over a 6-day time course. Approximately 5×108 colony forming units (CFUs) were harvested at each time point during mid-log phase for DNA extraction and used to inoculate a fresh broth culture for continuous culture in vitro.
H. pylori whole genome sequence analysis
H. pylori cultures were harvested, and bacterial DNA was extracted using the Wizard Genomic Purification Kit (Promega). H. pylori DNA was sheared using Covaris, yielding an average fragment length of 550 bp. Libraries were prepared using Truseq Nano LT Kit (Illumina) with unique indexes. Postquantitation was performed using the Kapa Library Quantitation Kit (Kapa Biosystems). Libraries were sequenced on the MiSeq Sequencer using the 600V3 Kit (Illumina). For analysis, raw sequence reads were quality trimmed using CLCbio Genomics Workbench V. 8.0 (Qiagen). Alignment files were exported from the CLCbio Workbench in BAM file formats and were then imported into Visual Genomics Analysis Studio, software developed at the Institute for Immunology and Infectious Diseases at Murdoch University, and Geneious V. 6.1.8 (Biomatters) for further analysis. Analyses were performed using a 25% cut-off, which required at least 25% of the reads from at least one single colony to contain the polymorphism, to allow detection of intrapopulation variance. All sequence reads were aligned with the genome sequence of H. pylori strain B8 (GenBank accession number NC_014256.1), a closely related strain whose complete genome sequence is available.28 B8 was chosen as the reference strain because H. pylori strains 7.13 and B8 each originated from the same parental H. pylori strain B128 and each was generated by experimentally infecting gerbils with strain B128. Although B128 (GenBank accession number ABSY0000000) and 7.13 (GenBank accession number NZ_LAQK00000000) have been previously sequenced, these strains were also sequenced in this study to directly compare these sequences within the newly derived strains using the same sequencing technology. All sequences have been deposited to GenBank (BioProject PRJNA362878). Sequence data for H. pylori strains isolated from gerbils on a high salt diet or a regular diet21 were analysed using the same criteria. Specifically, we sought to identify SNPs present in isolates from gerbils on iron-depleted or high salt diets, but not present or present at a lower abundance in isolates from gerbils maintained on iron-replete or regular salt diets.
DNA extraction, PCR and sequence analysis
Approximately 5×108 CFUs were pelleted by centrifugation and stored at −20°C. DNA was extracted from H. pylori using the PureLink Pro 96 Genomic DNA Purification Kit (Invitrogen). DNA was then used as template to amplify a 500 bp product using fur-specific primers: 5′TCCGCCAAAAAGACAAAAAC3′ and 5′GGGCAAGACTTTCACTTGGA3′. PCR products were prepared for sequence analysis using ExoSAP-IT Express PCR Product Cleanup Kit (Affymetrix), quantified using spectrophotometric nucleic acid quantification (BioTek) and sequenced (GeneWiz). Geneious V. 6.1.8 (Biomatters) was used for subsequent analysis.
H. pylori clinical isolates
Clinical H. pylori strains were isolated from three independent and well-defined patient cohorts within Colombia and the USA.31–33Online supplementary table S7 includes information on the clinical H. pylori strains, including strain identification number, patient country of origin, age, sex and histological diagnosis.
Statistical analysis
Mean values with SE are shown from experiments performed on at least six independent occasions. One-way analysis of variance with multiple comparisons and Fisher’s exact tests were used to determine statistical significance. p<0.05 was considered statistically significant.
Study approval
All animal and human studies were conducted in accordance with the Declaration of Helsinki principles and have been approved by Vanderbilt University IACUC and Institutional Review Board, respectively.
Acknowledgments
We would like to acknowledge the following funding sources: R01CA190612 (KTW), R01DK053620 (KTW), P01CA028842 (KTW), R01AI039657 (TLC), R01AI118932 (TLC), Department of Veterans Affairs 2I01B×0 00 627 (TLC), R01CA077955 (RMP), R01DK058587 (RMP), P30DK058404 (RMP), P01CA116087 (RMP, TLC, KTW).
References
Footnotes
Contributors JMN contributed to the study design, performed experiments, gathered and analysed data, and drafted the manuscript. AC generated and analysed whole genome sequences. JTL generated and provided sequence data from Helicobacter pylori strains harvested from gerbils maintained on regular or high salt diets. JRG assisted with all animal experiments and isolation of in vivo-adapted H. pylori strains. MBP provided all patient information from the two Colombian patient cohorts and clinical H. pylori isolates from these subjects. MW and SL generated and analysed whole genome sequences. ACB and KTW provided feedback on the manuscript. TLC contributed to the study design and provided sequence data from H. pylori strains harvested from gerbils maintained on regular or high salt diets. SM contributed to the study design and the whole genome sequence analysis. DAI contributed to the study design, analysed sequence data, and helped with drafting and editing the manuscript. RMP contributed to the study design and helped with drafting and editing the manuscript.
Competing interests None declared.
Patient consent Obtained
Ethics approval Institutional Review Board, Vanderbilt University Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.