Article Text

Abstract
Objective Nicotinamide phosphoribosyltransferase (NAMPT, also referred to as pre-B cell colony-enhancing factor or visfatin) is critically required for the maintenance of cellular nicotinamide adenine dinucleotide (NAD) supply catalysing the rate-limiting step of the NAD salvage pathway. NAMPT is strongly upregulated in inflammation including IBD and counteracts an increased cellular NAD turnover mediated by NAD-depleting enzymes. These constitute an important mechanistic link between inflammatory, metabolic and transcriptional pathways and NAD metabolism.
Design We investigated the impact of NAMPT inhibition by the small-molecule inhibitor FK866 in the dextran sulfate sodium (DSS) model of colitis and the azoxymethane/DSS model of colitis-associated cancer. The impact of NAD depletion on differentiation of mouse and human primary monocytes/macrophages was studied in vitro. Finally, we tested the efficacy of FK866 compared with dexamethasone and infliximab in lamina propria mononuclear cells (LPMNC) isolated from patients with IBD.
Results FK866 ameliorated DSS-induced colitis and suppressed inflammation-associated tumorigenesis in mice. FK866 potently inhibited NAMPT activity as demonstrated by reduced mucosal NAD, resulting in reduced abundances and activities of NAD-dependent enzymes including PARP1, Sirt6 and CD38, reduced nuclear factor kappa B activation, and decreased cellular infiltration by inflammatory monocytes, macrophages and activated T cells. Remarkably, FK866 effectively supressed cytokine release from LPMNCs of patients with IBD. As FK866 was also effective in Rag1−⁄− mice, we mechanistically linked FK866 treatment with altered monocyte/macrophage biology and skewed macrophage polarisation by reducing CD86, CD38, MHC-II and interleukin (IL)-6 and promoting CD206, Egr2 and IL-10.
Conclusion Our data emphasise the importance of NAD immunometabolism for mucosal immunity and highlight FK866-mediated NAMPT blockade as a promising therapeutic approach in acute intestinal inflammation.
- Energy Metabolism
- Colonic Mucosal Metabolism
- Gut Inflammation
- IBD Basic Research
- Inflammatory Bowel Disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Energy Metabolism
- Colonic Mucosal Metabolism
- Gut Inflammation
- IBD Basic Research
- Inflammatory Bowel Disease
Significance of this study
What is already known on this subject?
Nicotinamide phosphoribosyltransferase (NAMPT) catalyses the rate-limiting step of the nicotinamide adenine dinucleotide (NAD) salvage pathway, maintaining NAD supply during inflammation.
NAMPT is strongly upregulated in patients with IBD; however, its functional impact still remains unclear.
What are the new findings?
Blocking NAMPT activity with the small-molecule inhibitor FK866 effectively ameliorates experimental colitis and reduces mucosal activities of NAD-dependent enzymes including PARP1 and Sirt6.
FK866-induced NAD depletion skews monocyte/macrophage biology away from a proinflammatory M1 towards an anti-inflammatory M2 phenotype.
FK886 diminishes cytokine release from human IBD-derived lamina propria mononuclear cells comparably to dexamethasone or infliximab.
How might it impact on clinical practice in the foreseeable future?
Targeting NAD immunometabolism by FK866 constitutes a promising novel induction regimen in IBD that warrants further investigation as FK866 has already been used in clinical trials with reasonable toxicity and with nicotinic acid/vitamin B3 as a potential antidote.
Introduction
IBD, including Crohn’s disease (CD) and UC, constitutes multifaceted disorders with chronic relapsing and/or progressive inflammation of the GI mucosa. IBD results from a disturbed intestinal immune system and microbiota on the basis of genetic susceptibility and poorly defined environmental triggers.1 Furthermore, chronic mucosal inflammation and tissue damage predispose patients with IBD to the development of colorectal cancer, and the risk increases with duration, extent and severity of inflammation.2
Current treatment algorithms in moderate to severe IBD intend the use of corticosteroids, immunosuppressors (eg, azathioprine, 6-mercaptopurine, methotrexate), tumour necrosis factor (TNF) antagonists (eg, infliximab, adalimumab), the α4β7-antagonist vedolizumab and the interleukin (IL)-12/23 antagonist ustekinumab.3–6 However, up to one-third of patients are primary TNF non-responders, and about half of patients who initially responded lose response over time.7 Similarly, despite clinical effectiveness, the overall efficacy of vedolizumab remains incomplete, particularly in patients with prior anti-TNF failure.6 8 Together, these data demonstrate the unprecedented need for alternative treatment options in IBD.
Phenotypic characteristics of IBD comprise a heterogeneous spectrum of patients with distinct genetic, environmental and microbial influences such that mechanisms underlying IBD in an individual are difficult to untie.1 Accordingly, the currently most successful treatment strategies target overarching disease mechanisms irrespective of individual phenotypes. Along these lines, cellular energy metabolism may represent an attractive novel therapeutic target. Inflammation and tumorigenesis are heavily energy-consuming processes, partly because activation, proliferation and recruitment of cells require energy-enriched substrates for the biosynthesis of proteins involved in inflammation and immunity.9–11 Similarly, tumour cells switch their metabolism away from oxidative phosphorylation towards aerobic glycolysis to enhance energy metabolism.12
Nicotinamide adenine dinucleotide (NAD) is a major coenzyme in bioenergetic processes. Besides its well-established function in oxidative phosphorylation and redox reactions, NAD has evolved as a substrate for evolutionarily conserved, NAD-cleaving enzymes such as poly(ADP-ribose) polymerases (PARPs), sirtuins (SIRT) and cADP-ribose synthases such as CD38/CD157.13–15 NAD cleavage by the latter proteins is key to many biological processes such as cell metabolism, inflammatory responses and cell death.13–15 A permanent supply of the dynamic NAD turnover is essential for energy-consuming processes,16 which is accomplished by two distinct biosynthetic pathways, the NAD de novo pathway and the NAD salvage pathway. In mammals, the salvage pathway is the predominant source of NAD due to its high adaptability. In this pathway, nicotinamide (Nam; ie, vitamin B3, dietary or from NAD cleavage) is enzymatically converted into nicotinamide mononucleotide (NMN) by intracellular nicotinamide phosphoribosyltransferase (iNAMPT), which catalyses this first and rate-limiting step of the NAD salvage pathway.17 One of three isoforms of NMN-adenylyltransferase completes the catalysis to NAD. As such, NAMPT is biologically indispensable and has been implicated in a variety of inflammatory disorders such as sepsis, rheumatoid arthritis and diabetes.18–20 Furthermore, NAMPT overexpression has been associated with tumorigenesis.21 We have reported elevated NAMPT concentrations in colonic tissue and serum of patients with IBD and recently, NAMPT has been identified as a marker for severity in paediatric IBD.22 23 The small molecule inhibitor FK866 potently inhibits NAMPT enzymatic activity in a competitive fashion with little toxicity24 and may represent an attractive target in various inflammatory disorders.19 25 26
Based on these observations, we hypothesised that blockage of the NAD salvage pathway by the NAMPT inhibitor FK866 may be effectively limiting intestinal inflammation. Here, we demonstrate that FK866 protects against experimental colitis and colitis−associated tumorigenesis by suppression of activated leukocytes particularly macrophages, inflammatory monocytes and T cells. NAMPT blockage influenced macrophage differentiation processes, suppressing inflammatory characteristics of M1 and promoting anti-inflammatory properties of M2 macrophages. Importantly, FK866 also reduced inflammatory responses of lamina propria mononuclear cells (LPMNC) from colonic biopsies of patients with IBD to a comparable extent as dexamethasone. Thus, we suggest FK866 as a promising novel compound for the treatment of IBD.
Materials and methods
Reagents, cell lines and in vitro experiments
FK866 was kindly provided by RTI International (North Carolina, USA). RAW 264.7 (ATCC) and MODE-K cells (kindly provided by Kaiserlian D) were cultured in RPMI1640 medium or Dulbecco’s modified Eagle’s medium 4.5 g (DMEM) (Sigma) supplemented with 1% Pen/Strep (Biochrom), 10% heat-inactivated fetal calf serum (FCS) and 1% NEAA for DMEM (Life Technologies). Cells were stimulated with ultrapure 100 ng/mL lipopolysaccharide (LPS) or 1 µg/mL flagellin (Invivogen) followed by incubation with or without 200 nM FK866 overnight. Supernatants (SN) were harvested and protein was extracted with NE-PER containing protease and phosphatase inhibitors (Thermo Fisher Scientific) and stored at −80°C until further workup. Additional protocols and procedures are described in online supplementary methods.
Supplementary file 1
Animal studies and colitis induction
Acute colitis was induced in 8-week-old female wild-type (WT) or Rag1tm1Mom/J (Rag1−/− ) mice with 3.5% or 3% dextran sulfate sodium (DSS, MP Biomedicals, MW 36.000–50.000) ad libitum for 5 consecutive days, followed by a tap water period until end of experiments. Control mice received tap water during the study period. Mice were injected intraperitoneally with 10 mg/kg bodyweight FK866 or vehicle control twice daily until termination of experiments. A detailed description of experimental procedures including isolation of LPMNC and flow cytometry is given in the online supplementary methods.
Liquid chromatography tandem-mass spectrometry quantification
Mucosal scrapings were collected from mice, immediately snap frozen and later homogenised by chemical (25% water, 67.5% methanol and 7.5% chloroform) and mechanical (Precellys, Peqlab) lysis. The preanalytical workup included two steps of centrifugation and transfer of target molecules to a chloroform-based carrier. Respective standards were purchased from Sigma Aldrich. Metabolite concentrations were normalised to protein content. Samples were analysed on an Eksigent 425 LC system hyphenated to a TripleTOF 5600+ (AB Sciex).
Ex vivo culture of human LPMNC
Mucosal biopsies from inflamed areas of patients with CD and UC were obtained during colonoscopy at our department according to a protocol approved by the Institutional Ethics committee (AN2015-0132 to HT). LPMNCs were isolated as described in online supplementary methods, incubated with or without FK866, dexamethasone or infliximab for 18 hours and SN assayed for cytokines.
Statistics
All data are expressed as mean±SEM. Statistical significance was assessed using a two-tailed Student’s t-test. p<0.05 was considered significant. For comparisons of more than two groups, one-way analysis of variance with Bonferroni’s post hoc test was used. Data were analysed and illustrated using GraphPad Prism V.6.0 (GraphPad Software).
Results
The NAMPT inhibitor FK866 ameliorates DSS-induced colitis
NAMPT is strongly upregulated in the inflamed mucosa and the serum of patients with IBD.22 23 Accordingly, we observed a continuous NAMPT increase in the course of DSS colitis as demonstrated on mRNA and protein levels (figure 1A). NAMPT immunopositivity on stained colonic sections mapped to infiltrating leukocytes and intestinal epithelial cells (IEC) and was further investigated by flow cytometry (figure 1B). In the non-inflamed colon, NAMPT is highly expressed in Epcam+ epithelial cells, which outnumber NAMPT+-expressing CD45+ LPMNC. Among these, CD11b+ F4/80+ macrophages and CD49b+natural killer (NK)cells represent the major source of NAMPT in the steady state. In contrast, the expression pattern of NAMPT dramatically changed during DSS-induced colitis with a strong increase of NAMPT+-expressing CD45+ LPMNC compared with epithelial cells. Notably, NAMPT expression particularly occurred in CD11b+ F4/80+ macrophages, Ly6G+ neutrophil granulocytes and NAMPT+/Ly6Chi inflammatory monocytes.
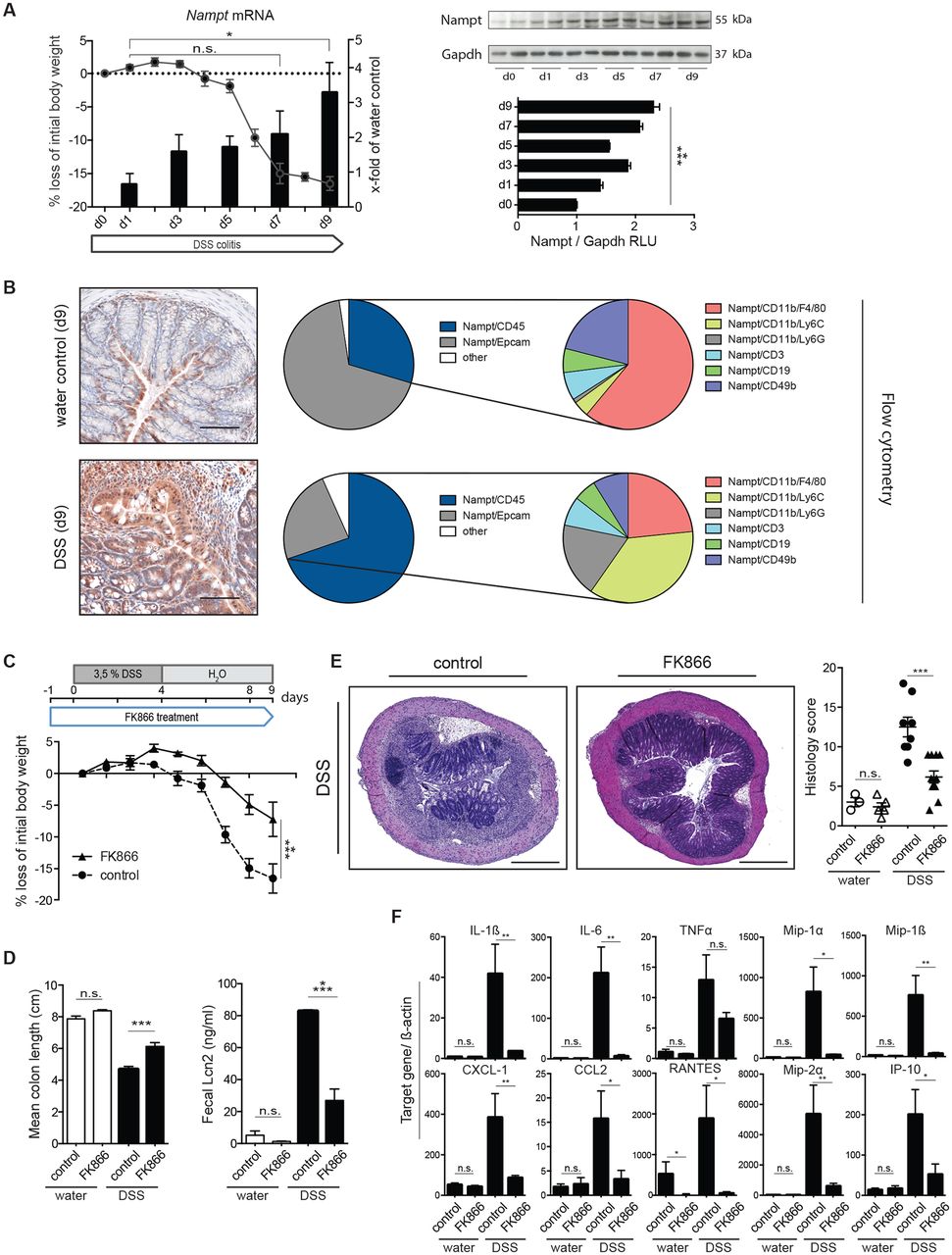
FK866 treatment mitigates clinical, morphological and biochemical parameters of DSS colitis. (A) Colonic Nampt mRNA induction as determined by RT-qPCR (black labelling) is shown together with the weight course (grey symbols and labelling) and NAMPT protein expression using immunoblot analyses along with densitometric quantification during DSS colitis. (B) Comparative NAMPT immunohistochemistry of colons with or without DSS on day 9 of experiments is shown (left panel). NAMPT positivity (red) is observed in IEC and inflammatory cells (15× magnification; scale bars 100 µm). Colonic NAMPT expression of Epcam+epithelial cells and CD45+LPMNC including respective immune cell subsets was analysed by flow cytometry in the steady state and on day 7 of colitis (pie charts). (C) Weight course of vehicle control and FK866-treated mice during DSS colitis. The experimental outline is shown above. (D) Mean colon lengths and faecal Lcn2 of respective groups are shown. (E) Representative H&E stainings of colons along with histological severity scores (5× magnification; scale bars 500 µm). (F) mRNA expression of cytokines and chemokines of mucosal scrapings was determined by RT-qPCR and normalised to ß-actin (day 7 of experiments). n=6–10 per group, five independent experiments. Data represent mean±SEM. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Student’s t-test or one-way analysis of variance followed by Bonferroni’s post hoc corrections when more than three groups analysed. CCL, CC-chemokine ligand; CXCL, C-X-C motif ligand; DSS, dextran sulfate sodium; IEC, intestinal epithelial cells; IL, interleukin; IP, induced protein; Lcn2, lipocalin 2; LPMNC, lamina propria mononuclear cells; Mip, macrophage inflammatory protein; NAMPT, nicotinamide phosphoribosyltransferase; RLU, relative light units; TNFα, tumour necrosis factor alpha.
To assess the impact of increased NAMPT activity on intestinal inflammation, we used the competitive NAMPT inhibitor FK866 during DSS colitis in a prophylactic setting as outlined in figure 1C. Notably, FK866-treated WT mice were clinically protected from DSS-induced injury in terms of weight loss (figure 1C) and mean colon length (figure 1D) along with reduced concentrations of faecal lipocalin 2 (Lcn2, figure 1E), a protein that has been implicated in the pathogenesis of IBD and that correlates well with disease activity.27 28 Furthermore, mice exhibited a substantially reduced histological severity of colitis compared with vehicle-treated controls (figure 1E). FK866 treatment substantially impaired the expression of inflammatory and chemotactic cytokines such as IL-1β, IL-6, chemokine (C-X-C motif) ligand 1 (CXCL) 1, CC-chemokine ligand (CCL) 2 and others in response to DSS-induced injury (figure 1F). Importantly, we did not observe any FK866-related toxicity such as weight loss, ruffled fur or hunched posture in WT mice not exposed to DSS. As NAMPT has been implicated in cell cycle regulation and survival,29 we analysed IEC proliferation by BrdU incorporation and cell death by TUNEL labelling. Although there was a trend towards increased IEC apoptosis and cell proliferation in vehicle-treated animals, the observed differences did not reach statistical significance in this short-term treatment model (online supplementary figure 1A,B).
FK866 mitigates DSS-induced colitis in T and B cell-deficient Rag1−/−mice
FK866 has been reported to exert particularly strong anti-inflammatory effects on activated T and B lymphocytes.26 29 To discriminate whether FK866 treatment targets cells of the innate or adaptive immunity in our model, FK866 or vehicle was administered to DSS-exposed mice lacking T and B cells due to deletion of recombination-activating gene 1 (Rag1 −/−). Similar to WT mice, vehicle-treated Rag1 −/− mice developed colitis based on weight loss (figure 2A), histologic severity (figure 2B), colonic shortening (figure 2C), faecal Lcn2 concentrations (figure 2D) and augmented inflammatory cytokine expression (figure 2E) when compared with Rag1 −/− mice in the steady state. Notably, FK866 significantly ameliorated all features of DSS-induced colitis in Rag1 −/− mice (figure 2A−E), demonstrating that NAMPT inhibition effectively suppresses inflammatory innate immune responses in the absence of adaptive immunity.

FK866 treatment is effective independent of T and B cells. (A) Weight course of vehicle control and FK866-treated RAG−/− mice during DSS colitis along with the experimental outline shown above. (B) Representative H&E-stained colonic sections along with histology score of indicated groups (5× magnification; scale bars 500 µm). (C,D) Mean colon lengths and (D) faecal Lcn2 of experimental groups are shown. (E) Colonic scrapings were analysed for mRNA expression of respective cytokines/chemokines on experimental day 7 with or without DSS. n=10 per group, two independent experiments. Data represent mean±SEM. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Student’s t-test or one-way analysis of variance followed by Bonferroni’s post hoc corrections when more than three groups analysed. CCL, CC-chemokine ligand; CXCL, C-X-C motif ligand; DSS, dextran sulfate sodium; IL, interleukin; IP, induced protein; Lcn2, lipocalin 2; Mip, macrophage inflammatory protein; TNFα, tumour necrosis factor alpha.
FK866 diminishes recruitment of inflammatory monocytes and activated T cells during DSS colitis
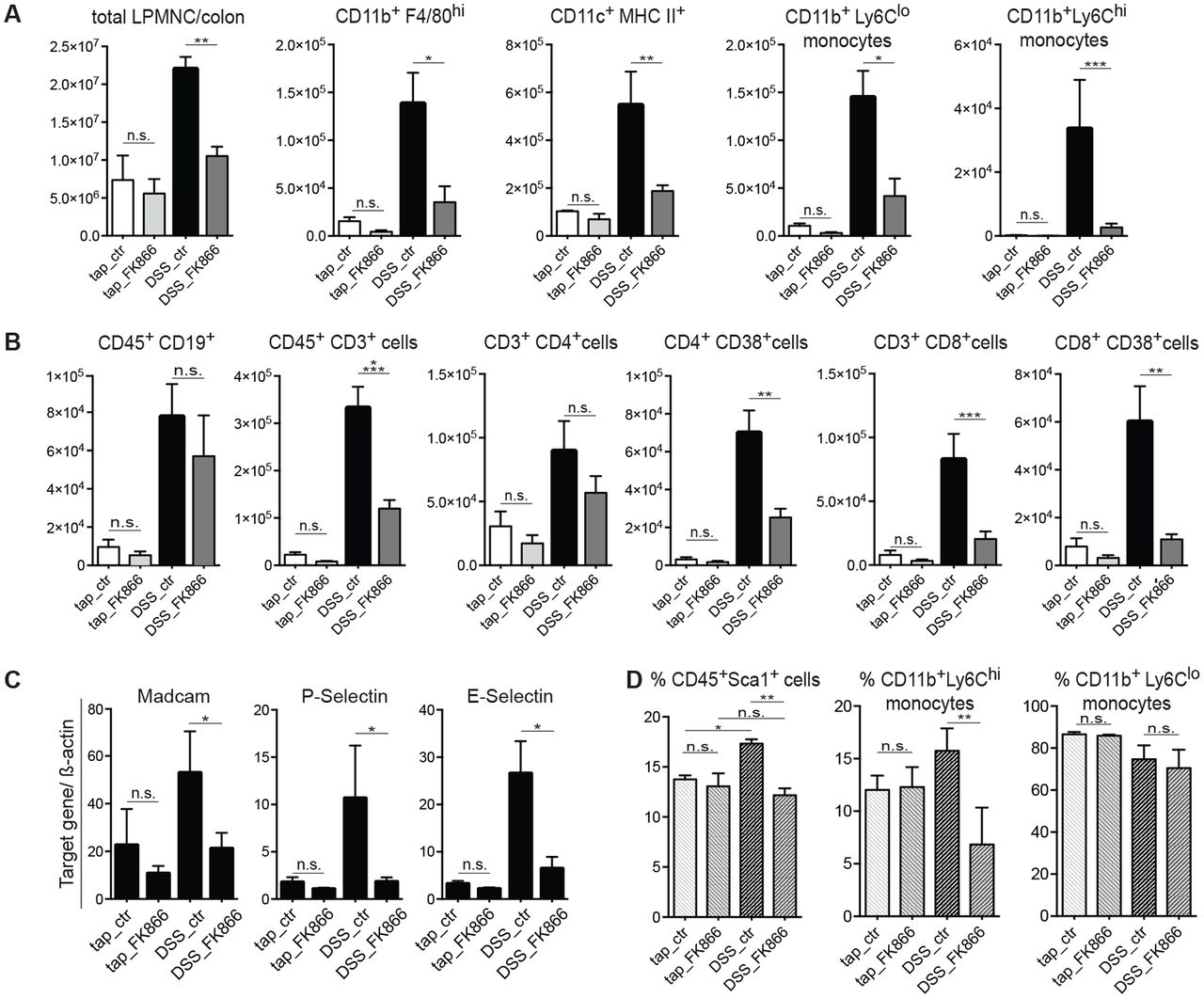
As FK866 appeared to dampen inflammatory responses, we next examined its impact on cellular infiltration consequent to DSS-induced injury. Mouse LPMNCs were isolated and analysed by flow cytometry (gating strategy outlined in online supplementary figure 2A,B). In agreement with histology, total numbers of colonic LPMNC were strongly reduced in FK866-treated animals after DSS exposure (figure 3A).
In regard to innate immune cells, DSS treatment resulted in an increase of CD11b+F4/80hi tissue macrophages, CD11c+MHC-II+-activated dendritic cells, CD11b+Ly6Clo-resident and CD11b+Ly6Chi-inflammatory monocytes. FK866 treatment resulted in a marked reduction of all aforementioned cell populations compared with vehicle-treated controls with the most pronounced effect observed in CD11b+Ly6Chi cells (figure 3A). Notably, these ‘newly recruited’ inflammatory monocytes, in contrast to resident monocytes, have emerged as critical drivers of acute inflammation in the intestine.30 31 Notably, FK866 did not affect numbers of CD11b+Ly6G+Ly6Clo neutrophils (online supplementary figure 2C).
We next determined the cellular composition of the adaptive immune compartment. Here, FK866 treatment was associated with a strong reduction of CD45+CD3+ total T lymphocytes when compared with vehicle-treated mice exposed to DSS (figure 3B). This was mainly attributed to a reduced abundance of CD3+CD8+ cytotoxic T cells and more specifically CD8+CD38+ and CD4+CD38+-activated T cells (figure 3B). No differences were observed between FK866 and vehicle-treated mice in the number of CD3+CD4+ T helper cells, CD4+CD25+Foxp3+ regulatory T cells and CD45+CD19+ B cells (figure 3B and online supplementary figure 2C). Importantly, WT mice treated with FK866 without DSS exposure (ie, in the steady state) did not show any significant differences in the number of cellular subsets compared with vehicle controls (figure 3A,B and online supplementary figure 2C). To assess whether decreased leucocyte infiltration was a consequence of a diminished extravasation or generation in the bone marrow (BM), we analysed the expression of key chemokines and integrins involved in leucocyte recruitment and further profiled BM leukocytes baseline and during DSS-induced colitis. FK866 treatment of DSS-exposed WT mice, but not tap water controls, resulted in a diminished expression of various chemokines including CCL2 (MCP-1), macrophage inflammatory protein (Mip)-1α, Mip-1β, RANTES, CXCL1, interferon gamma (IFNγ)-induced protein 10 (IP-10), mucosal addressin cell adhesion molecule 1, P-selectin and E-selectin (figures 1F and 3C) in colonic scrapings. DSS colitis induced a significant expansion of BM Sca-1+ stem cell-like cells, which were reduced upon FK866 treatment but not in the steady state (figure 3D). Similar to our observations in LPMNC, FK866 treatment was associated with reduced frequencies of Ly6Chi-inflammatory monocytes upon DSS exposure yet Ly6Clo-resident monocytes in the BM remained unaffected (figure 3D and online supplementary figure 2D).

NAMPT blockage results in a diminished recruitment of inflammatory/activated cells upon intestinal inflammation. (A) Total numbers of colonic infiltrates were quantified by automated cell counting (left). The absolute numbers of CD11b+F4/80hi tissue macrophages, CD11c+MHC-II+-activated dendritic cells, CD11b+Ly6Cloresident monocytes and CD11b+Ly6Chi inflammatory monocytes per colon with or without FK866 during tap water or DSS colitis were analysed by flow cytometry on day 7. (B) On day 7 of colitis, the absolute numbers of CD19+ B cells, CD3+, CD4+ and CD4+CD38+, and CD8+ and CD8+CD38+ T cells per colon of respective groups were analysed by flow cytometry. (C) The mRNA expression of common adhesion molecules was determined by RT-qPCR in mucosal scrapings (n=6–8, two independent experiments). (D) The frequency of Sca1+ stem cell-like cells, CD11b+Ly6Chi inflammatory and CD11b+Ly6Clo resident monocytes of CD45+-gated BM cells was analysed by flow cytometry (n=4–6, two independent experiments). n=4–8 per group, six independent experiments if not otherwise stated. Data represent mean±SEM. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; one-way analysis of variance followed by Bonferroni’s post hoc corrections when more than three groups analysed. BM, bone marrow; DSS, dextran sulfate sodium; LPMNC, lamina propria mononuclear cells; NAMPT, nicotinamide phosphoribosyltransferase.
FK866 impairs mucosal NAD recycling resulting in a dampened PARP/SIRT-mediated inflammatory response and alters macrophage polarisation
Nampt expression correlates well with intracellular NAD levels29 and inhibition by FK866 results in cytosolic NAD depletion particularly in inflammation.25 32 We used a liquid chromatography tandem-mass spectrometry approach to study the effects of FK866 on abundances of NAD salvage pathway metabolites (ie, Nam, NMN, NAD) and ATP concentrations in colonic scrapings (workflow outlined in figure 4A). FK866 did not alter the concentrations of NAD, Nam and NMN in the steady state yet strongly reduced its abundances in the context of DSS-induced inflammation (figure 4A). Since NAD/nicotinamide adenine dinucleotide hydride is important for the mitochondrial synthesis of ATP, we hypothesised that FK866 might exert its anti-inflammatory capacity by ATP energy depletion. However, FK866 did not affect mucosal ATP concentrations baseline or during DSS treatment (figure 4A).
NAD serves as substrate for NAD-cleaving enzymes such as PARPs and SIRT, which have been implicated in ubiquitous biological processes and immune regulation. Therefore, we investigated the effect of FK866 on the NAD-consuming enzymes PARP1, the main isoform of the PARP family and SIRT in the context of DSS colitis. Indeed, we noted increased PARP1 abundance in mucosal scrapings of WT mice upon DSS exposure (figure 4B). While FK866 had no effect on PARP1 abundances in the steady state, FK866 strongly reduced PARP1 in DSS-exposed mice (figure 4B). This finding was corroborated by a pharmacodynamic assay, measuring poly(ADP-ribose) (PAR) formation in lysates of colonic scrapings. PAR activity was reduced more than 50% in FK866-treated DSS mice compared with vehicle-treated DSS controls (figure 4C). Increased PARP1 immunoreactivity in DSS-treated mice localised to infiltrating leukocytes and IEC as demonstrated by immunohistochemistry (figure 4D). Similarly, FK866 reduced mucosal abundance of various SIRT (online supplementary figure 3A) during intestinal inflammation with the most pronounced effect on Sirt6 (figure 4B,D). Again this was paralleled by a markedly decreased enzymatic function as measured by deacetylase activity (figure 4C).
As SIRT and PARPs have been linked to the regulation of inflammation, we next investigated whether FK866 directly modulated inflammatory responses. Indeed, FK866 minimised the release of proinflammatory cytokines such as IL-1β, IL-6, TNFα and Lcn2 after LPS treatment of RAW 264.7 cells (online supplementary figure 3B). In line with our in vivo data, we observed a strong reduction in nuclear expression of PARP1 and Sirt6 in FK866-treated, LPS-stimulated RAW cells. PARP1 and Sirt6 have been shown to interfere with nuclear factor kappa B (NF-κB) signalling.33 34 Notably, FK866 treatment strongly reduced NF-κB phosphorylation consequent to LPS treatment (online supplementary figure 3C). Notably, we did not observe such effects on the abundance of PARP1 and Sirt6 in flagellin-stimulated MODE-K (online supplementary figure 3D) or LPS-challenged CMT.93 IEC (data not shown).
These observations together with the distinct monocyte/macrophage infiltration in the DSS model prompted us to investigate the impact of FK866 on the differentiation and polarisation of human and murine primary monocytes/macrophages. Addition of FK866 during polarisation of bone marrow-derived macrophages (BMDM) had negligible effects on the expression of typical M1 (LPS/IFNγ polarised) or M2 (IL-4 polarised) markers such as suppressor of cytokine signalling 3 (Socs3) or IL-10. However, blockade of the NAMPT enzymatic activity during the entire differentiation process followed by polarisation diminished proinflammatory activity of M1 macrophages including reduced expression of surface CD86 and MHC-II, Socs3 mRNA and IL-6 release. By contrast, in a M2 set-up, FK866 promoted anti-inflammatory M2 properties including increased expression of surface CD206, arginase 1 (Arg1) and IL-10 mRNA, and IL-10 release (figure 4E and online supplementary figure 4A). Importantly, these observations could be corroborated by in vivo experiments, showing that FK866 had a marked effect on the M1/M2 ratio suppressing M1 and favouring M2-like macrophages during intestinal inflammation (figure 4F and online supplementary figure 4B).
These results indicate that FK866’s anti-inflammatory capacity involves a reduction in the activity of NAD-dependent enzymes including PARP1 and Sirt6 thereby suppressing NF-κB-dependent immune responses and skewing macrophage differentiation and polarisation towards M2-like macrophages.
FK866 treatment protects mice from colitis-associated cancer
Colitis-associated cancer (CAC) represents an important complication of chronic intestinal inflammation.2 Whereas anti-inflammatory strategies reduce tumour multiplicity and progression, M2 macrophages have rather been implicated in tumour progression.35 To assess the net effect of FK866 on CAC, we next performed an azoxymethane (AOM)/DSS model administering FK866 or vehicle during DSS cycles and beyond (figure 5A). Long-term administration of FK866 revealed no adverse effects in terms of weight loss (figure 5A) or haematotoxicity compared with vehicle-treated controls (data not shown). On the contrary, FK866 treatment rather protected animals from AOM/DSS-induced mortality by trend, although this was not statistically significant (figure 5B). Accordingly, FK866-treated AOM/DSS-exposed mice also showed reduced tissue injury as reflected by reduced colonic shortening (figure 5C), and decreased IEC death as assessed by the TUNEL method (figure 5D). Together, this resulted in marked reductions in tumour numbers and tumour areas (figure 5E).
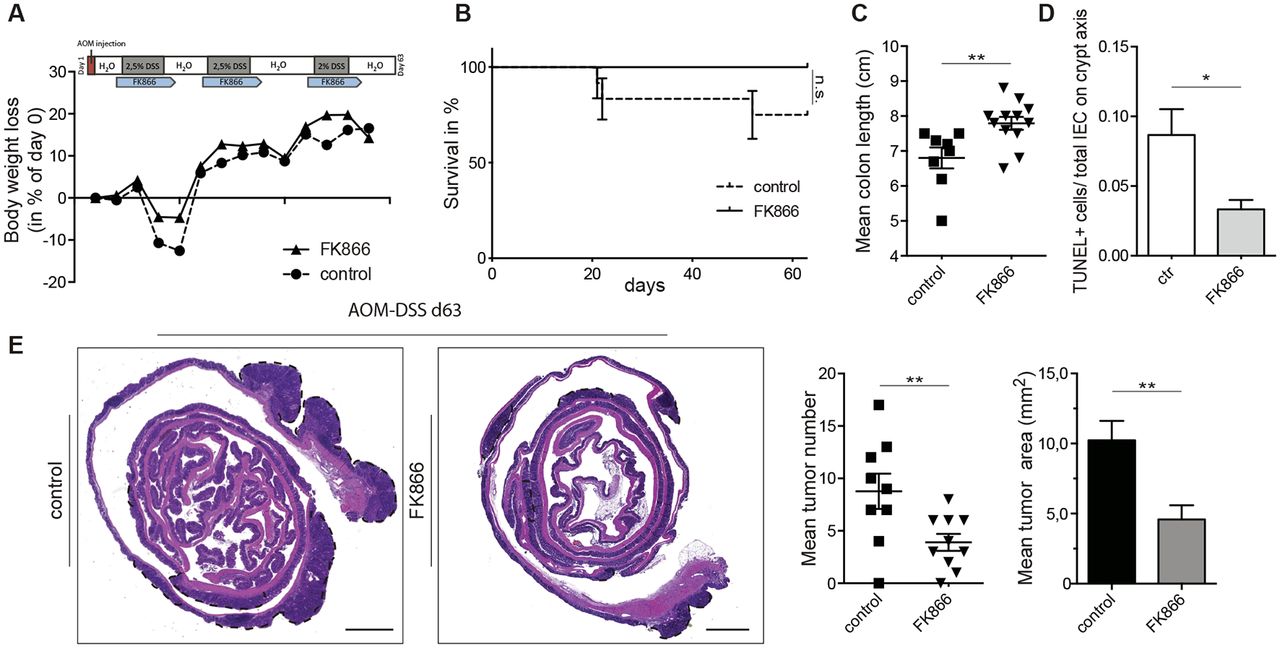
FK866-treated mice are protected in the AOM/DSS model of colitis-associated cancer. (A) The experimental outline along with the weight course is shown. (B,C) Clinical parameters including (B) survival and (C) mean colon lengths of FK866 compared with vehicle-treated mice during the AOM/DSS model. (D) Cell death was evaluated by the TUNEL method. Three crypts/mouse were analysed for TUNEL+cells and expressed as ratio/total IEC count on the crypt axis. (E) The mean tumour numbers and tumour areas of FK866 or vehicle-treated animals were assessed in H&E-stained Swiss roll sections. Dashed, black lines indicate tumour area (5× magnification; scale bars 500 µm). n=12 per group, two independent experiments. Data represent mean±SEM. *p<0.05; **p<0.01; Student’s t-test. AOM, azoxymethane; DSS, dextran sulfate sodium; IEC, intestinal epithelial cell.
FK866 reverses established disease and suppresses cytokine release from LPMNC of patients with IBD
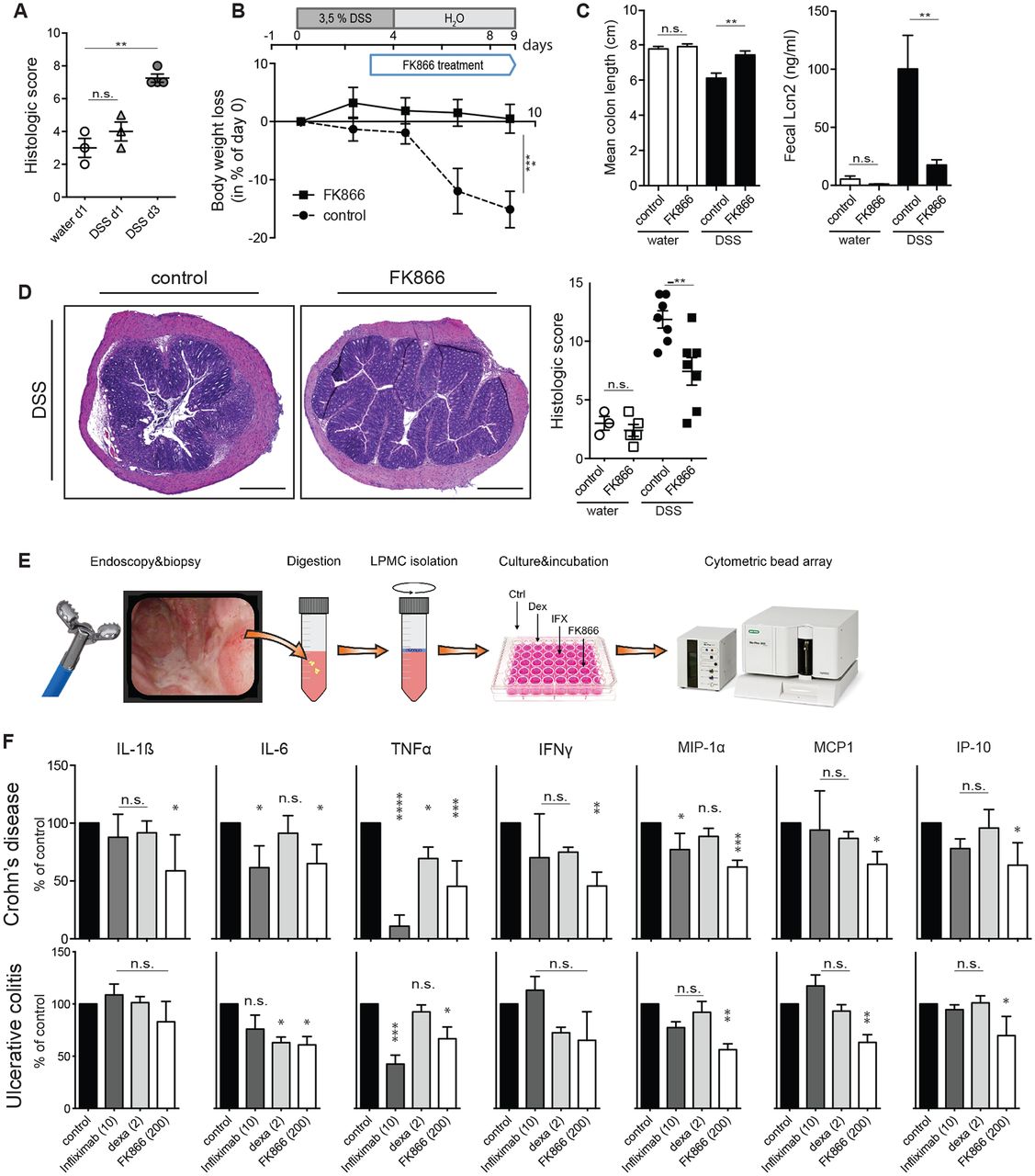
NAMPT inhibition ameliorated colitis and CAC in different mouse models, suggesting that such a treatment strategy may be beneficial in human IBD. To test whether FK866 also influences established intestinal inflammation (see figure 6A), FK866 was administered therapeutically starting 3 days after induction of DSS colitis (outlined in figure 6B). Strikingly, also in a therapeutic setting FK866 effectively ameliorated DSS-induced weight loss (figure 6B), colonic shortening and faecal Lcn2 concentrations (figure 6C) and histological inflammation (figure 6D).

Therapeutic FK866 treatment is effective on established colitis and modulates proinflammatory cytokine secretion in human LPMNC from patients with IBD. (A) Histological severity score of mice treated with DSS at 0, 1 and 3 days. (B) FK866 or vehicle treatment was started 3 days after onset of DSS experiments and animals were assessed for clinical parameters including (B) weight loss, (C) colon length and faecal Lcn2 on experimental day 9. (D) Representative H&E-stained colons and respective colitis scores of indicated groups are depicted. (5× magnification; scale bars 500 µm). (E) A schematic flow chart of LPMNC experiments is outlined. (F) Biopsy-derived LPMNCs from patients with CD (upper panel) or UC (lower panel) were incubated with or without infliximab (10 µg/mL), dexamethasone (2 nM) or FK866 (200 nM) and assayed for cytokine release after 18 hours. Vehicle-stimulated cells were set as 100% and data are expressed as % decrease or increase (n=8–10 per group). n=8 per group if not otherwise stated, two independent experiments. Data represent mean±SEM. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; Student’s t-test or one-way analysis of variance followed by Bonferroni’s post hoc corrections when more than three groups analysed. CD, Crohn’s disease; DSS, dextran sulfate sodium; IFNγ, interferon gamma; IL, interleukin; IP, induced protein; Lcn2, lipocalin 2; LPMNC, lamina propria mononuclear cells; MIP, macrophage inflammatory protein; TNFα, tumour necrosis factor alpha.
To test FK866’s anti-inflammatory potential in human IBD, LPMNCs from inflamed tissue of patients with IBD were incubated with or without FK866, infliximab or dexamethasone and SNs assayed for the release of inflammatory cytokines (workflow outlined in figure 6E). Figure 6F shows that FK866 most potently reduced the production of IL-1β, IL-6, TNFα, CCL2 (=MCP1), Mip-1α and IP-10 compared with vehicle treatment of patient-derived LPMNC from both entities of IBD. Importantly, FK866 appeared advantageous in its in vitro capacity to suppress inflammatory responses compared with dexamethasone and infliximab.
Based on our observations in BMDM, we generated M1 and M2 macrophages from human CD14+ monocytes. Comparably to BMDMs, FK866 reduced proinflammatory M1 characteristics such as IL-6 production and CD86+ expression and promoted M2 features including increased expression of the M2 markers IL-10 and Arg1 compared with control macrophages (online supplementary figure 4).
As such, our data support the notion that FK866 may be useful in a therapeutic setting in patients with human IBD.
Discussion
Here, we provide evidence that inhibition of NAMPT, which is strongly induced in both experimental (figure 1A) and human IBD,22 23 may represent a novel therapeutic approach for active CD and UC. We demonstrate that NAMPT blockade by the small-molecule inhibitor FK866 protects against inflammation in the DSS model of colitis both in a preventive and therapeutic manner. Furthermore, FK866 treatment protects from CAC in the AOM/DSS model. Importantly, FK866 also effectively suppressed cytokine synthesis in LPMNC isolated from patients with IBD suggesting that FK866 could reflect an attractive treatment approach especially in case of acute and/or severe intestinal inflammation.
NAMPT is expressed by various cell types and has been implicated in distinct biological processes including cellular energy metabolism, circadian clock rhythm and importantly immunity.36 Originally described as pre-B cell colony-enhancing factor, secreted NAMPT exerts cytokine-like functions such as growth enhancement of pre-B colony-forming units and stimulation of mononuclear cells as well as promoting macrophage survival.22 37 38 In contrast, intracellular NAMPT represents the rate-limiting enzyme of the NAD salvage pathway that prevents NAD depletion caused by NAD catabolic enzymes such as SIRT, PARPs or CD38, the activity of which is boosted in inflammation.36 As expected and in line with previous reports,25 26 39 NAMPT blockade resulted in a strong depletion of NAD and its precursors Nam and NMN in colonic mucosal scrapings of DSS-treated animals. Of note, FK866 treatment did not affect Nam, NMN and NAD abundances in the steady state. Since mitochondrial NAD plays a critical role in the Krebs cycle and thus cellular energy metabolism,40 shortage of intracellular NAD therefore might affect ATP supply. Interestingly, the modulation of the mitochondrial F1F0-ATPase by LYC-30937-EC protects mice in the TNBS model of colitis.41 Notably, in our short-term treatment protocol FK866 did not alter ATP concentrations in the intestinal mucosa. A possible explanation could be that NAD-catabolic enzymes such as SIRT are active in specific cellular compartments. Fitting into such a concept, iNAMPT has been shown to translocate cell cycle or stress dependently into specific subcompartments such as the nucleus.42 Although we cannot demonstrate compartment-specific NAD concentrations, it appears plausible that mitochondrial NAD concentrations remain unaffected by FK866 treatment.
Several NAD-catabolising enzymes including PARP1, SIRT6 and CD38 have been implicated in human and mouse inflammatory processes. Mice deficient in PARP1 were protected from inflammation and mortality in the DSS colitis model.43 Furthermore, PARP1 inhibitors demonstrated anti-inflammatory and antiproliferative effects in various experimental models including peritonitis, septic shock and ovarian cancer.44 To date, no data on the expression and distribution of PARP1 in human IBD exist, although indirect data support a putative role for PARP1 activation. Inflammatory processes are characterised by the increased formation of reactive oxygen species and reactive nitrogen species such as nitric oxide, both of which have been linked to CD and UC.45 46 These highly reactive molecules are known to induce DNA damage that further result in a consequent PARP-1 activation.47 Accordingly, Sirt6 regulated TNFα production in dendritic cells in a NAD and NAMPT-dependent manner39 and Sirt1 represents an important regulator of monocyte/macrophage polarisation towards M1.48 CD38, a transmembrane glycoprotein that catalyses the production of cyclic ADP-ribose from NAD, has been implicated in the activation and proliferation of T cells.49 Strikingly, FK866-induced NAD depletion uniformly reduced expressions and enzymatic activities of PARP1, Sirt6 and CD38 in the intestinal mucosa of FK866-treated animals (figures 3B and 4B,D), suggesting a relevant impact of FK866 on these pathways in colonic inflammation. However, despite a potent effect on NAMPT activity as demonstrated by substantially reduced NAD levels in colonic tissue, we cannot exclude indirect or off-target immunomodulatory effects independent from NAD activity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FK866-induced NAD depletion results in reduced PARP1 and Sirt6 activity and skews macrophages towards a less inflammatory phenotype. (A) An experimental outline of the LC-MS/MS approach is shown. Mucosal metabolite concentrations of the NAD salvage pathway along with ATP concentrations of indicated groups during tap water or DSS exposure were determined and normalised to total protein contents. (B,C) Representative PARP1 and Sirt6 immunoblots from mucosal scrapings of indicated groups along with densitometry, and (C) poly(ADP-ribosylation) (PAR) assay or sirtuin deacetylase activity (DAA). (D) Comparative PARP1 and Sirt6 immunohistochemistry of colons with or without FK866 during tap water or DSS exposure on day 7 of experiments is shown (20× magnification; scale bars 50 µm). (E) BMDMs were differentiated for 8 days followed by polarisation with M1 (LPS/IFNγ) or M2 (IL-4) stimuli for 24 hours. Cells were incubated with or without (=ctr) 5 nM FK866 from day 0 (=FK866_d0) or from day 8 (=FK866_d8). On day 9, cell viability was assessed by a LUNA automated cell counter and cells analysed for typical M1 (MHC-II, CD86) and M2 (CD206) surface markers by flow cytometry. n=2–4 per group. mRNA expression of typical M1 (Socs3, Nos2) and M2 (IL-10, Arg1) markers was analysed by quantitative RT-PCR and normalised to HPRT and SNs were assayed for IL-6 and IL-10 by ELISA on day 9. (F) The M1/M2 ratio in the colon of mice with or without FK866 in the steady state and during DSS colitis on day 7 is shown. The frequencies of colonic F4/80+ CD38+-M1 macrophages of FK866 or vehicle-treated mice are depicted in the overlays. n=2–8 per group if not otherwise stated, three independent experiments. Data represent mean±SEM. *p<0.05; **p<0.01; ***p<0.001; Student’s t-test or one-way analysis of variance followed by Bonferroni’s post hoc corrections when more than three groups analysed. BMDM, bone marrow-derived macrophages; DSS, dextran sulfate sodium; HPRT, hypoxanthine-guanine phosphoribosyltransferase; IFNγ, interferon gamma; IL, interleukin; LC-MS/MS, liquid chromatography tandem-mass spectrometry; LPS, lipopolysaccharide; MHC, major histocompatibility complex; NAD, nicotinamide adenine; dinucleotide; NMN, nicotinamide mononucleotide; PAR, poly(ADP-ribose); PARP, poly(ADP-ribose) polymerase; RLU, relative light units; SN, supernatant.
A marked infiltration by innate and adaptive immune cells along with increased concentrations of proinflammatory cytokines represents a hallmark of IBD.50 Obviously, FK866 treatment strongly decreased mucosal immune cell infiltration as well as cytokine and chemokine release. Published data are not entirely congruent in terms of relevant FK866-sensitive target cell populations. While an effect on mononuclear cells was relevant in collagen-induced arthritis, suppression of T lymphocytes was suggested as the predominant effect in experimental autoimmune encephalomyelitis.19 26 This might be context dependent and explained by an increased susceptibility of activated cells and tissues to FK866 that demonstrate high NAD turnover and therefore strongly rely on high NAMPT expression and an effective NAD salvage pathway.26 29 As FK866 was highly effective in Rag1-deficient mice, which lack T and B cells, we suggest that effects on cells of the innate immune system were particularly relevant in DSS colitis. Particularly, flow cytometry of LPMNC of FK866-treated WT mice demonstrated strong reductions in the numbers of CD11b+Ly6Chi inflammatory monocytes and also CD3+CD8+CD38+ activated/cytotoxic T cells. Furthermore, the analysis of BM cells revealed additional FK866-mediated extraintestinal effects. Notably, FK866 did not affect the mucosal cellular composition in the steady state.
Immunometabolism in macrophages is critically interwoven with their activation state and function. Metabolic adaptation upon inflammation is key for macrophage plasticity with distinct metabolic programmes in M1 and M2 macrophages.51 Our data are in line with previous reports suggesting that NAMPT is critically required for the activation of M1 macrophages while negligible for M2 macrophages, and NAMPT inhibition even skews macrophages towards an M2 phenotype.52
In regard to tumour biology, M1 macrophages display tumoricidal activity while M2 macrophages have been implicated in tumour progression.35 Noteworthy, various tumour entities including colorectal cancer overexpress NAMPT and FK866 was originally conceived as antitumour compound inducing apoptosis in NAMPT-dependent tumour cells.24 Bruzzone and colleagues demonstrated that FK866 is also capable of inducing apoptosis in activated T lymphocytes.26 Interestingly, FK866 rather protected epithelial cells from apoptosis even after prolonged application in the AOM/DSS model, which seems to be attributable to FK866’s anti-inflammatory potential. Along these lines, we propose that FK866’s anti-inflammatory potential is the predominant contributor for the observed antiproliferative effects in the AOM/DSS model although we cannot exclude additional antitumour effects.
To corroborate our data on the efficacy of FK866 in models of experimental colitis, we finally performed ex vivo experiments using freshly prepared LPMNC from patients with IBD. In support of a relevant translational aspect, we could demonstrate that FK866 significantly reduced chemokine and cytokine release, many of those which are macrophage/monocyte derived. Remarkably, the observed suppression was in the range or even superior to well-established anti-inflammatory compounds such as dexamethasone and infliximab.
In conclusion, we suggest that FK866 dampens immune responses and suppresses the emergence of intestinal inflammation likely by reduced PARP1/SIRT/CD38 activity consequent to NAD shortage. FK866 might thereby act preferably on cells and tissues that rely on the NAD salvage pathway due to the activity of NAD-cleaving enzymes such as SIRT, PARPs or CD38.26 29 Our work highlights the importance of NAMPT and the NAD salvage pathway as a driving force in intestinal inflammation that can be effectively targeted by FK866. FK866 has already been used in clinical trials and the availability of an antidote with nicotinic acid or Nam appears advantageous.32 53 Based on these findings and due to the efficacy of FK866 in the suppression of inflammation in LPMNC derived from patients with IBD, we propose that blocking the Nam-dependent NAD biosynthetic pathway represents an attractive novel strategy for induction therapy in IBD that warrants further investigations.
References
Footnotes
Contributors Conceptualisation: RRG, HT, ARM. Experimental procedures: RRG, VK, SM, CG. Statistical analysis: RRG, VK. Investigation: RRG, VK, KA, VW, AP, NHK. Resources, materials and technical support: AP, PM, GB, HO. Figure preparation: RRG, VK, ARM. Writing of original draft: RRG, TEA, HT, ARM. Writing of revised draft: RRG, TEA, HT, ARM. Funding acquisition: HT, ARM.
Funding This study was supported by the Tyrolean Research Foundation (TWF-0404/1480 to ARM) and the Christian Doppler Research Foundation. The financial support by the Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology and Development is gratefully acknowledged. HT is supported by the excellence initiative (Competence Centers for Excellent Technologies—COMET) of the Austrian Research Promotion Agency FFG: Research Center of Excellencein Vascular Ageing Tyrol, VASCage (K-Project No. 843536) funded by the BMVIT, BMWFW, the Wirtschaftsagentur Wien and the Standortagentur Tirol.
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Ethics approval Institutional Ethics Committee (AN2015-0132 to HT)
Provenance and peer review Not commissioned; externally peer reviewed.