Article Text

Abstract
Objective Alcohol-related pancreatitis is associated with a disproportionately large number of hospitalisations among GI disorders. Despite its clinical importance, genetic susceptibility to alcoholic chronic pancreatitis (CP) is poorly characterised. To identify risk genes for alcoholic CP and to evaluate their relevance in non-alcoholic CP, we performed a genome-wide association study and functional characterisation of a new pancreatitis locus.
Design 1959 European alcoholic CP patients and population-based controls from the KORA, LIFE and INCIPE studies (n=4708) as well as chronic alcoholics from the GESGA consortium (n=1332) were screened with Illumina technology. For replication, three European cohorts comprising 1650 patients with non-alcoholic CP and 6695 controls originating from the same countries were used.
Results We replicated previously reported risk loci CLDN2-MORC4, CTRC, PRSS1-PRSS2 and SPINK1 in alcoholic CP patients. We identified CTRB1-CTRB2 (chymotrypsin B1 and B2) as a new risk locus with lead single-nucleotide polymorphism (SNP) rs8055167 (OR 1.35, 95% CI 1.23 to 1.6). We found that a 16.6 kb inversion in the CTRB1-CTRB2 locus was in linkage disequilibrium with the CP-associated SNPs and was best tagged by rs8048956. The association was replicated in three independent European non-alcoholic CP cohorts of 1650 patients and 6695 controls (OR 1.62, 95% CI 1.42 to 1.86). The inversion changes the expression ratio of the CTRB1 and CTRB2 isoforms and thereby affects protective trypsinogen degradation and ultimately pancreatitis risk.
Conclusion An inversion in the CTRB1-CTRB2 locus modifies risk for alcoholic and non-alcoholic CP indicating that common pathomechanisms are involved in these inflammatory disorders.
- Genome wide association study
- chronic pancreatitis
- genetic rearrangement
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Genetic risk underlying alcoholic chronic pancreatitis (CP) is poorly understood. In contrast, the genetic basis of non-alcoholic CP has been more comprehensively characterised.
Alcohol abuse is a predominant cause of CP; however, only a small percentage of alcohol abusers develop the disease, suggesting that genetic susceptibility may contribute to pathogenesis.
A genome-wide association study (GWAS) reported variants in the PRSS1-PRSS2 and CLDN2-MORC4 loci associated with CP. More recent studies indicate that these variants may have the strongest effect in alcoholic CP.
What are the new findings?
This is the largest European GWAS in patients with alcoholic CP. The results replicate the reported associations with variants in the CLDN2-MORC4, CTRC, PRSS1-PRSS2 and SPINK1 loci.
We identified CTRB1-CTRB2 (chymotrypsin B1 and B2) as a new risk locus for alcoholic CP and found that a 16.6 kb inversion in the CTRB1-CTRB2 locus was in linkage disequilibrium with the associated single-nucleotide polymorphisms. The association was replicated in non-alcoholic CP.
The inversion changes the expression ratio of the CTRB1 and CTRB2 isoforms and thereby affects protective trypsinogen degradation and ultimately pancreatitis risk.
How might it impact on clinical practice in the foreseeable future?
The results indicate that alcoholic and non-alcoholic CP share common pathomechanisms.
As the same variants influence development of alcoholic and non-alcoholic CP, therapeutic approaches should be guided by disease mechanism rather than aetiology.
The identified risk variants explain about 18% of the variance in alcoholic CP and may serve as the basis for risk assessment in the clinical setting.
Introduction
Chronic pancreatitis (CP) is a relapsing, progressive inflammatory disorder of alcoholic (ACP), idiopathic or hereditary aetiology. In about half of non-alcoholic CP (NACP) cases, a genetic background has been identified with mutations in risk genes CTRC (chymotrypsin C), PRSS1 (serine protease 1, cationic trypsinogen) and SPINK1 (serine protease inhibitor Kazal type 1).1 Most changes in these genes increase ectopic intra-pancreatic activation of the digestive serine protease trypsin.2–5 However, the relevance of such a trypsin-dependent mechanism in ACP has remained contentious as association with high-effect CTRC, PRSS1 and SPINK1 variants is absent or considerably weaker than in NACP.5 6 Similarly, rare PRSS1 and CPA1 (carboxypeptidase A1) variants that result in misfolding and endoplasmic reticulum stress are associated with NACP but not with ACP.7 8 The so far unidentified genetic susceptibility in ACP is an intriguing observation as only a small percentage of alcohol abusers develop ACP, suggesting that factors other than alcohol may contribute to disease onset.9 10
Indeed, a recent genome-wide association study (GWAS) revealed association of common variants in the CLDN2-MORC4 and the PRSS1-PRSS2 loci with ACP and NACP.11 These findings were first replicated in a large European cohort where association was strongest in the ACP group followed by similar observations in Japanese and Indian CP cohorts.12–15 Functional studies indicated that the protective PRSS1 promoter variant reduces transcription that should result in lower intra-pancreatic trypsinogen levels.16
Here, we present the largest European multicentre GWAS combining 1959 ACP cases from nine countries using Illumina chips. As controls, chronic alcoholics without CP (n=1332) and population-based controls (n=4708) with data on alcohol consumption were used (total n=6040).
Methods
Study population
In all participating study centres, the corresponding medical ethical review committees approved the study. CP was defined by a typical clinical course with recurrent attacks or chronic pain and characteristic morphological changes in imaging studies as well as functional impairment with exocrine and/or endocrine insufficiency. CP was considered as ACP if alcohol consumption was >60 g for females and >80 g for males per day over at least 2 years. The cohorts are summarised in table 1 and in online supplementary table S1.
Supplementary file 1
Description of the cohorts included in the analysis
Genotyping, quality control and GWAS statistics
Genotyping was performed on Illumina BeadChip arrays (Illumina, San Diego, California, USA). The ACP samples of the screening cohort and NACP samples of replication cohort 1 were genotyped at the Helmholtz Center Munich (Dr P. Lichtner). The study design is summarised in online supplementary figure S1.
All analyses were performed using PLINK V.1.917 and R (www.R-project.org). Data were filtered to achieve an individual-wise and single-nucleotide polymorphism (SNP)-wise call rate >0.99.18 Genotyped sex was determined in each sample from gonosomal data. Related individuals were excluded (pi-hat >0.185). Heterozygosity outliers defined by heterozygosity more extreme than median ±3 IQRs and ethnic outliers according to Price et al 18 with 6 SD criterion in principal component analysis were not used for further analysis. Hence, the 1959 ACP samples that passed quality control as well as controls (n=6040) derived from different consortia (KORA S3/F3 Illumina Omni data set from Augsburg, Germany; GESGA consortia data set from Mannheim, Germany; INCIPE Illumina Omni data set from Verona, Italy) were included for further analyses.
Imputation was performed with SHAPEIT V.2 and IMPUTE V.2.3.0 applying the 1000Genomes reference phase 1, V. 319 and logistic regression was applied with the first three principal components of the SNP data included as covariates to account for possible population stratification resulting in a standardised overall inflation factor of 1.02.20
Imputation of the 16.6 kb inversion for further analysis
Imputation of a 16.6 kb inversion21 in the CTRB1-CTRB2 locus within the screening cohort was performed using a 5 Mb region on chromosome 16 between 71 310 697 bp and 76 045 524 bp including 475 SNPs overlapping between all cohorts and the reference. As reference panel, quality filtered data of HumanOmniExpress BeadChips from 227 CP patients with successfully genotyped inversion genotypes were used (for details, see online supplementary file 1). To improve accuracy, the IMPUTE2 parameters number of Markov chain Monte Carlo iterations were set to 30 and number of hidden Markov Model states were increased to 200.
Calculation of variance of ACP
Joint variance explained by all identified loci was measured by calculating McFadden’s pseudo-R2. Here, we compared a logistic regression including the three principal components and literature SNPs rs497078, rs17107315, rs10273639, rs12688220 and the inversion as independent variables with a logistic regression model including the first three principal components only.
Replication genotyping
Details of polymerase chain reactions (PCR) and melting curve assays used for analysis of the CTRB1-CTRB2 locus can be found in the online supplementary file 1. In the first replication cohort of the German NACP patients and controls, chip data were used with similar preprocessing as described (n=584 NACP patients; n=4892 population-based controls from LIFE, the latter were genotyped using Affymetrix AXIOM-CEU (Affymetrix, St. Clara, California, USA) genome-wide SNP array22). Thereby, imputation on 1000Genomes reference phase 1, V. 3 was performed for a 5 Mb region on chromosome 16 between 71 310 697 bp and 76 045 524 bp using 285 SNPs fulfilling quality control in all individuals.
Further replication in two independent NACP cohorts from Germany (Greifswald cohort, 520 patients, 760 controls) and France (Brest cohort, 546 patients, 1043 controls) was carried out by genotyping SNPs with the melting curve assay (see online supplementary table S2).
Measurement of CTRB1 and CTRB2 mRNA expression
Samples of human pancreatic cDNA prepared from pancreatic exocrine fractions discarded after islet isolation were kind gifts from Dr Sohail Husain (Children’s Hospital of Pittsburgh) and Dr Rajinder Dawra (University of Minnesota) or were prepared from discarded surgical specimens at the University of Szeged, Hungary. Details of the methods used are summarised in online supplementary file 1.
Trypsinogen activation and degradation
Human cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2) were produced in Escherichia coli. His-tagged forms of CTRB1 and CTRB2 were expressed in HEK 293 T cells and purified as described previously.23 24 The effect of CTRB1 and CTRB2 on trypsinogen was determined in autoactivation experiments and degradation assays.
Results
Association of ACP with loci CLDN2-MORC4, CTRC, PRSS1-PRSS2 and SPINK1
We robustly confirmed the association of loci CLDN2-MORC4 and PRSS1-PRSS2 reported in a previous GWAS.11 Our lead SNPs at the CLDN2-MORC4 (rs12688091) and the PRSS1-PRSS2 locus (rs2855983) were in linkage disequilibrium (LD) with the previously identified lead SNPs in these loci (see online supplementary table S3). Furthermore, strong association was found with rs545634 in CTRC and with rs146437551 in SPINK1. Both susceptibility genes were previously identified in candidate gene studies of CP.3 5 6 25–29 In line with the literature, our top hits at these loci were in strong LD with the most frequently reported SNPs; CTRC, rs497078 (c.180C>T, p.G60=) and SPINK1, rs17107315 (c.101A>G, p.N34S) (see online supplementary table S3). Association of the lead SNPs at the four risk loci remained essentially unchanged when ACP patients were separately compared with chronic alcoholics and non-alcoholic controls (see online supplementary figure S2).
Association of ACP with an alcohol-dependence locus
We also observed an association when we compared ACP patients versus chronic alcoholics at the known alcohol-dependence locus ADH1B (alcohol dehydrogenase 1B) for rs1229984 (OR 2.49, 95% CI 1.84 to 3.39; p=1.8×10–8). This variant (c.143A>G, p.H48R) alters alcohol metabolism and thereby deters from drinking, resulting in the observed lower frequency among alcoholics (see online supplementary figure S3).30
Novel association of ACP with the CTRB1-CTRB2 locus
We discovered a novel association signal at the CTRB1-CTRB2 (chymotrypsin B1 and B2) locus (figure 1 and table 2 for all top hits) with lead SNP rs8055167 located in intron 1 of CTRB1 (OR 1.35, 95% CI 1.23 to 1.6; p=4.2×10–9). The association was also observed when ACP patients were compared with alcoholics or population-based controls (see online supplementary figure S2). Regional association plots for the top hits and all associations with p<10–5 are summarised in online supplementary figure S4 and online supplementary table S4 and risk estimates for carriers of multiple risk alleles are shown in online supplementary figure S5. Here, the identified risk variants explain about 18% of the variance in ACP.
Supplementary file 2

Genome-wide association analysis of 1959 cases with alcoholic chronic pancreatitis and 6040 controls derived from population studies and a cohort of alcohol-dependent patients. Genome-wide significance-level threshold (p=5×10–8) is represented by the black line. Only single-nucleotide polymorphisms that passed quality control are depicted.
Top associated variants in the overall cohort of European alcoholic chronic pancreatitis patients
Identification of a complex genetic rearrangement in the CTRB1-CTRB2 locus associated with CP
The CTRB1-CTRB2 locus harbours complex genomic rearrangement variants which include a 16.6 kb inversion (see figure 2) and a 584 bp deletion in CTRB2 (not shown).21 First, we excluded the deletion in CTRB2 as an explanation for the association as genotyping of 289 ACP patients revealed only negligible LD (R²=0.12) with our lead SNP rs8055167. Subsequently, in order to examine whether our lead SNP is in LD with the 16.6 kb inversion in the CTRB1-CTRB2 locus, we genotyped 227 ACP patients for the inversion and found stronger LD (R2=0.50). By combining our genotype data on the inversion with genetic data from SNP chips, we successfully imputed the inversion in all 1959 ACP and 6040 control samples (info-score of the inversion =0.81, correlation of imputed inversion vs measured inversion: R²=0.99, see online supplementary figure S6A). The major allele of the inversion conferred risk for ACP with a similar effect size as the lead SNP at this locus (OR 1.36, 95% CI 1.20 to 1.53; p=3.1×10–6) (figure 3). From the ACP-associated SNPs within the CTRB1-CTRB2 locus, rs8048956 tagged the inversion the best (R2=0.987) and it was therefore used as a reporter for the inversion in further analyses (see online supplementary figure S6B).

Schematic illustration of the CTRB1-CTRB2 locus with the 16.6 kb inversion. The inversion breakpoints lie within the region indicated by the dashed lines. The genomic reference sequence corresponds to the minor allele. The locations of the lead single-nucleotide polymorphism (SNP) rs8055167 and the best tagging SNP rs8048956 are denoted by the empty and black diamond symbols, respectively. Genomic distances are not scaled. Although not shown here, in this locus CTRB2 also harbours a 584 bp deletion variant (allele frequency ~7% in German controls) that eliminates exon 6. CTRB1, chymotrypsin B1 gene; CTRB2, chymotrypsin B2 gene; CTRB1* and CTRB2*, hybrid CTRB1 and CTRB2 genes created by the inversion; E, exon.
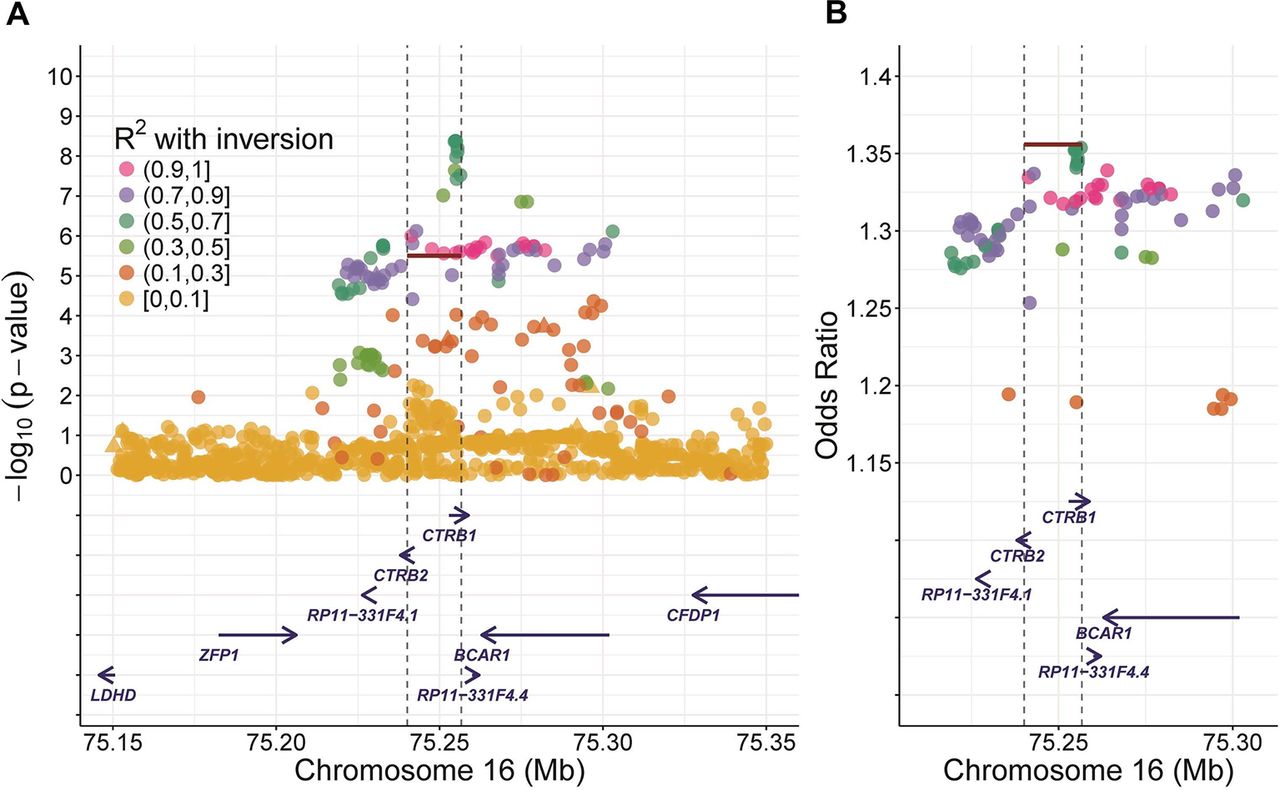
Regional association plots for the CTRB1-CTRB2 locus inversion. (A) p Values (–log10) are displayed against single-nucleotide polymorphism (SNP) genomic position (genome build hg19). The inversion is represented by the red line, triangles are genotyped SNPs, circles are imputed SNPs. For calculations, all alcoholic chronic pancreatitis patients and all controls were included. (B) The OR-based regional association plot indicates that the association is driven by the inversion. Here ORs are represented against the genomic position.
Replication of the association in NACP cohorts
To evaluate whether rs8048956 tagging the inversion also associates with NACP, we analysed three independent cohorts of patients (n=1650) and controls (n=6695) from Germany and France (table 1 and online supplementary table S1). In all three cohorts, we found a significant association and the combined effect across all cohorts was OR 1.62 (95% CI 1.42 to 1.86); p=1.64×10–12 (see online supplementary figure S7A). On the other hand, association of the GWAS lead SNP rs8055167, which had a lower LD with the inversion, was less pronounced, supporting the pathogenic relevance of the inversion in NACP (see online supplementary figure S7B).
Functional characterisation of the genetic rearrangement in the CTRB1-CTRB2 locus
To investigate whether the inversion affects protein translation and secretion, CTRB1 and CTRB2 were expressed in HEK 293 T cells with the two different 5′ untranslated regions and signal peptides of the major and minor alleles. Chymotrypsin levels in the conditioned media of cells transfected with these constructs were essentially identical, indicating that the inversion has no impact on protein translation and secretion. To assess the link between the inversion and expression of CTRB1-CTRB2, we quantified the relative mRNA expression ratio of the two chymotrypsins using cDNA samples obtained from human pancreatic tissue or acinar cells with different inversion genotypes. We found that the major risk allele was associated with higher relative CTRB1 expression while heterozygous carriers with one minor allele expressed higher levels of CTRB2 (figure 4A). In accordance with our findings, data from the GTExPortal (gtexportal.org) indicated that the major allele of rs8048956 tagging the inversion was associated with increased CTRB1 and decreased CTRB2 mRNA expression relative to the minor allele. The association of the lead SNP rs8055167 with CTRB1-CTRB2 expression was similar but smaller, supporting a causal role of the inversion in CP (see online supplementary figure S8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression and effect of CTRB1 and CTRB2 on trypsinogen activation and degradation. (A) Expression of CTRB1 and CTRB2 mRNA in the pancreas of subjects with different inversion genotypes. Expression ratios of CTRB1 and CTRB2 in pancreatic cDNA samples were determined by real-time polymerase chain reaction using the standard curve method. Results were displayed as box plots showing minimum, first quartile (25%), median, third quartile (75%), maximum and the individual values (black dots). Note that subjects carrying two copies of the major risk allele exhibit a significantly higher CTRB1/CTRB2 expression ratio compared with heterozygous individuals with one major and one minor allele. Significance was calculated with unpaired t-test. (B) Effect of CTRB1 and CTRB2 on the autoactivation of human anionic trypsinogen (PRSS2). Trypsinogen (2 µM) was incubated with 10 nM initial trypsin and 200 nM of the indicated chymotrypsin in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 0.05% Tween 20 (final concentrations) at 37°C in 100 µL final volume. At the indicated times, aliquots (2 µL) were withdrawn and trypsin activity was determined using 150 µM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate. Rate of substrate cleavage is given in mOD/min units measured at 405 nm. Note the lower trypsin activity that develops in the presence of chymotrypsins indicating trypsinogen degradation during activation. Under similar conditions, CTRB1 and CTRB2 had a similar but much smaller effect on the autoactivation of human cationic trypsinogen (PRSS1). (C) Degradation of PRSS2 by CTRB1 and CTRB2. Trypsinogen (1 µM) was incubated with 200 nM of the indicated chymotrypsin and 20 nM SPINK1 trypsin inhibitor in 0.1 M Tris-HCl (pH 8.0) and 25 mM NaCl at 37°C. Reactions were stopped at the indicated times by precipitation of 150 µL aliquots with 10% trichloroacetic acid. Samples were analysed by SDS-PAGE and Coomassie Blue staining. Note the disappearance of the intact trypsinogen band in the CTRB2 incubate. Some of the lower bands correspond to the two chains of autolysed CTRB2. Although not shown, CTRB1 or CTRB2 did not degrade cationic trypsinogen (PRSS1) to a detectable extent. CTRB1, chymotrypsin B1; CTRB2, chymotrypsin B2; PRSS2, anionic trypsinogen.
Protective degradation of anionic trypsinogen by CTRB2
When autoactivation of human anionic trypsinogen was followed at pH 8.0 in 1 mM calcium in the presence of CTRB1 or CTRB2, final trypsin levels were reduced by both chymotrypsins with a more prominent effect observed with CTRB2 (figure 4B). A similar but less pronounced effect was seen on the autoactivation of human cationic trypsinogen (PRSS1) (not shown). The stronger effect of CTRB2 was also confirmed in trypsinogen degradation experiments. SDS-PAGE analysis with Coomassie Blue staining revealed more rapid protective degradation of anionic trypsinogen (PRSS2) by CTRB2 relative to CTRB1 (figure 4C). Cationic trypsinogen (PRSS1) was not degraded by either chymotrypsin to a detectable extent (not shown).
Discussion
Chronic pancreatitis is a disease of significant morbidity and suffering associated with a disproportionately large number of hospitalisations among gastrointestinal disorders. In this largest study ever conducted addressing the genetic basis of CP, we identified association with a complex genetic rearrangement in the CTRB1-CTRB2 locus. This locus contains the two highly similar chymotrypsin genes transcribed in opposite directions. Both genes comprise seven exons and the nucleotide sequences are 97% identical with complete identity from exons 2–6. Recently, Pang et al 21 described a 16.6 kb inversion that exchanges the promoter region, exon 1 and intron 1 between CTRB1 and CTRB2. The major, ancestral allele was arbitrarily designated as the inverted allele (figure 2). At the protein level, the inversion switches the signal peptides of CTRB1 and CTRB2, but the secreted mature proenzymes are unchanged. Furthermore, Pang et al 21 also reported a 584 bp deletion within the major allele leading to early termination at the protein level. Although a functional consequence of the deletion or inversion was anticipated, no disease association was reported so far.
The first novel association signal we discovered at the CTRB1-CTRB2 locus was the lead SNP rs8055167 located in intron 1 of CTRB1. This association was maintained when ACP patients were compared with alcoholics or population-based controls (see online supplementary figure S2). To understand whether the reported larger-scale genetic rearrangements might explain the association of the CTRB1-CTRB2 locus with ACP, we analysed LD with our top SNPs. Here, an association of the 584 bp deletion was ruled out as an explanation for the association signal. However, the 16.6 kb inversion within the locus conferred risk with a similar effect size and was best tagged by rs8048956 of the top SNPs. We also found this association in three independent European cohorts with non-alcohol-related CP using the best tagging SNP.
Functional analysis indicated that the inversion results in a reversal of the isoform expression ratio at the mRNA level which is expected to translate to a similar reversal at the protein level. Our previous studies demonstrated that primary cleavage specificity and catalytic efficiency of CTRB1 and CTRB2 are different, with CTRB2 being more active on most substrates.31 Consequently, a change in the isoform expression ratio likely causes altered chymotrypsin activity in the pancreas.
In NACP, increased intra-pancreatic activation of trypsinogen (PRSS1) due to the failure of protective inhibition (SPINK1) and/or degradation (CTRC) is an important mechanism for disease development. The strong association of common CTRC, PRSS1-PRSS2 and SPINK1 locus variants in ACP indicates that this mechanism is also relevant for ACP. Therefore, we hypothesised that the altered CTRB1-CTRB2 activity profile might influence intra-pancreatic trypsinogen activation. Here, in autoactivation experiments with anionic trypsinogen, final trypsin levels were reduced by both chymotrypsins with more prominent degradation seen with CTRB2 (see figure 4). This effect for cationic trypsinogen was similar but less pronounced (not shown). Taken together, the functional studies indicate that in carriers of the major CTRB1-CTRB2 risk allele impaired trypsinogen (PRSS2) degradation due to the altered CTRB1/CTRB2 isoform ratio explains the observed association with CP. The relatively small effect of the CTRB1-CTRB2 inversion on CP risk is consistent with the lesser role of PRSS2 in CP (see ref. 32).
In conclusion, our GWAS identified CTRB1-CTRB2 as a new risk locus for ACP and NACP. The association within the CTRB1-CTRB2 locus was linked to a 16.6 kb inversion that altered CTRB1/CTRB2 expression, thereby affecting protective trypsinogen degradation. Furthermore, we confirmed association of ACP with the CLDN2-MORC4, CTRC, PRSS1-PRSS2 and SPINK1 loci. Taken together, the identified risk variants explained about 18% of the variance in ACP. Our study clearly represents a significant and transformative advance in understanding the genetic basis of CP. The results underpin the prominent influence of commonly occurring variants in ACP and demonstrate that similar disease mechanisms drive both ACP and NACP. Thus, development of therapeutic approaches should be guided by disease mechanism rather than aetiology in CP.
Accession codes
The results for all imputed variants and individual-level data are available from the authors on request. The genomic reference sequence used was NT_010498.16. Reference sequences for mRNA were NM_001906.4 (CTRB1) and NM_001025200.3 (CTRB2). All reference sequences correspond to the minor inversion allele.
Acknowledgments
The authors thank all study participants for providing clinical data and blood samples. They also thank Vera Sahin-Tóth (Boston), Knut Krohn, Kathleen Schön and Birgit Oelzner (IZKF core unit DNA technologies, Leipzig) for excellent technical assistance. They are grateful to Jeff MacDonald and Stephen Scherer for providing details for the data of Pang et al. They thank Stephan Buch and Jochen Hampe (Medical Department 1, University Hospital Dresden, TU Dresden, Dresden, Germany) for initial data collection and fruitful discussions.
References
Footnotes
JPHD, HW, MS and MS-T contributed equally to the supervision of the work.
JR and HK contributed equally.
Contributors JR, PK, JPHD, HW, MS, MS-T and HK conceived, designed and directed the study. JR, EH, FUW, PL, CR and CZ performed genotyping. JR, HK and MS-T drafted and revised the manuscript with substantial help from FXR, JPHD, HW, PK, EH and MS. MS and HK performed bioinformatics work. JR , EH , FUW, HL, PL, CR, JMC, EM, HW, MS, MS-T and HK designed, performed and interpreted genetic analyses. EH and MS-T carried out functional analysis. All other co-authors recruited study subjects, collected clinical data and/or provided genomic DNA samples. All authors approved the final manuscript and contributed critical revisions to its intellectual content.
Funding This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grants RO 3929/1–1, RO 3929/2-1 and RO3929/5-1 (to JR), Wi 2036/2-2 and Wi 2036/2-3 (to HW) and SFB 1052 (to MB, MS, AT, PK), SFB 1052 C01, B01, B03; SPP 1629 TO 718/2-1 (to AT) by a grant of the Colora Stiftung gGmbH (to JR), the Else Kröner-Fresenius-Foundation (EKFS) (to HW), NIH grants R01DK058088, R01DK082412; and R01DK095753 (to MS-T), a grant from the National Pancreas Foundation (to EH), the Institut National de la Santé et de la Recherche Médicale (INSERM; to CF), the Programme Hospitalier de Recherche Clinique (PHRC R 08-04; to CF), the French Association des Pancréatites Chroniques Héréditaires (to CF), the Council of Scientific and Industrial Research (CSIR) (to CF), by grants of the European Regional Development Fund (ERDF) V-630-F-150-2012/133 and V630-S-150-2012/132 (to FUW), by grants 310030_138747 and 310030_169196 from the Swiss National Funds (to FS) and by grants RTICC from Instituto de Salud Carlos III (RD12/0036/0034) and SAF 2015–70857 from Ministerio de Economía y Competitividad (Madrid, Spain) (co-funded by the ERDF-EU) (to FXR) and Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III, Spain (Grant #PI1501573) (to NM). SPP was supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERDF) and by funds of the Free State of Saxony within the framework of the excellence initiative (project numbers 713-241202, 14505/2470, 14575/2470). The KORA study was initiated and financed by the Helmholtz Zentrum München – German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research (BMBF) and by the State of Bavaria. Furthermore, KORA research was financed by a grant from the BMBF to the German Center for Diabetes Research (DZD) and a grant from the Ministry of Innovation, Science, Research and Technology of the state North Rhine-Westphalia (Düsseldorf, Germany). It was supported within the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethic Committee of the University of Leipzig.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All chip data are available upon request.