Article Text

Abstract
Objective Recently approved direct acting antivirals provide transformative therapies for chronic hepatitis C virus (HCV) infection. The major clinical challenge remains to identify the undiagnosed patients worldwide, many of whom live in low-income and middle-income countries, where access to nucleic acid testing remains limited. The aim of this study was to develop and validate a point-of-care (PoC) assay for the qualitative detection of HCV RNA.
Design We developed a PoC assay for the qualitative detection of HCV RNA on the PCR Genedrive instrument. We validated the Genedrive HCV assay through a case–control study comparing results with those obtained with the Abbott RealTime HCV test.
Results The PoC assay identified all major HCV genotypes, with a limit of detection of 2362 IU/mL (95% CI 1966 to 2788). Using 422 patients chronically infected with HCV and 503 controls negative for anti-HCV and HCV RNA, the Genedrive HCV assay showed 98.6% sensitivity (95% CI 96.9% to 99.5%) and 100% specificity (95% CI 99.3% to 100%) to detect HCV. In addition, melting peak ratiometric analysis demonstrated proof-of-principle for semiquantification of HCV. The test was further validated in a real clinical setting in a resource-limited country.
Conclusion We report a rapid, simple, portable and accurate PoC molecular test for HCV, with sensitivity and specificity that fulfils the recent FIND/WHO Target Product Profile for HCV decentralised testing in low-income and middle-income countries. This Genedrive HCV assay may positively impact the continuum of HCV care from screening to cure by supporting real-time treatment decisions.
Trial registration number NCT02992184.
- Diagnostic Virology
- Chronic Viral Hepatitis
- Hepatitis C
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Recent direct-acting antiviral therapies have provided transformative therapeutic options for chronic hepatitis C virus (HCV) infection, yet the challenge remains to identify the 1% of the world’s population that is chronically infected with HCV.
Current WHO guidelines highlight the lack of field-based platforms for point-of-care (PoC) HCV RNA testing.
The Cepheid Xpert HCV Viral Load assay (Cepheid) is currently the only CE-In Vitro Diagnostic (IVD)-certified assay for decentralised HCV viral load determination, yet according to the WHO, it presents many limitations.
What are the new findings?
We developed a PoC assay for the qualitative detection of HCV RNA on the Genedrive PCR instrument. The PoC assay identified all six major HCV genotypes, with a limit of detection of 2362 IU/mL, 98.6% sensitivity (95% CI 96.9% to 99.5%) and 100% specificity (95% CI 99.3% to 100%) to detect HCV viraemia.
The test was also validated in a real-life clinical setting in a resource-limited country.
The Genedrive instrument is a 600 g portable device that can be battery operated, thus making it highly suitable for decentralised testing in field settings, and requires only 30 µL of sample.
We demonstrated a proof of concept for semiquantification of HCV viral load using melting peak ratiometric analysis.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The Genedrive instrument provides a rapid, simple, portable and accurate PoC molecular test for HCV and demonstrated sensitivity and specificity fulfilling the requirements published by FIND, the Foundation for Innovation in New Diagnostics, and WHO for decentralised testing in low- and middle-income countries. Thus, the Genedrive HCV assay could support decentralisation of HCV testing diagnosis, resulting in improved patient outcome and public health. In particular its portability, rapid results, and ease of use make it ideal for confirming infection cases in population-based screening approaches. This is crucial for ensuring that diagnosis translates into treatment and that patients in low- and middle-income settings undergo screening and treatment initiation during a single medical intervention. This highly sensitive and specific test has recently obtained CE-IVD certification and is therefore positioned to impact the continuum of HCV care from screening to cure through real-time treatment decisions.
Introduction
Chronic infection with hepatitis C virus (HCV) is a major public health problem, estimated to infect 1.0% of the world’s population (71 million people)1 and to be responsible for 400 000 annual deaths as a result of cirrhosis and liver cancer.2 With the advent of highly potent direct-acting antiviral (DAA) therapy, cure is now possible in >95% of treated individuals achieving a sustained virological response,3–7 thus changing the landscape of clinical HCV management in recent years. Consequently, the WHO published in 2016 a global strategy to eliminate HCV as a public health threat by 2030.8
To achieve this ambitious goal, it is essential to scale up diagnosis and treatment capacities in low-income and middle-income countries, particularly in Africa and Central Asia where HCV is highly endemic.9 However, in these regions, access to HCV diagnostic tools is severely limited.10 The current diagnostic algorithm is based on serological screening of HCV-specific antibodies (two different positive serological tests may be mandatory in some countries), followed by nucleic acid testing to detect presence of HCV RNA.10–12 Although rapid serological diagnostic tests exist for anti-HCV antibodies,10 13 nucleic acid testing for HCV RNA often involves dedicated facilities and highly qualified personnel, even if platforms such as Cepheid, Panther or VERIS/MDx are changing such requirements. In low-income and middle-income countries, HCV RNA tests are currently only available in centralised laboratories, resulting in less than 1% of infected people in these regions being aware of their infection.14 In certain at-risk patient subgroups (eg, migrants and prisoners) with current testing practices, it requires multiple visits to make a diagnosis, and as a result, many positives cases are missed. Therefore, to improve the ‘suboptimal’ cascade from screening to linkage to care and efficient therapy, there is an urgent unmet clinical need for decentralised HCV RNA testing.14 Point-of-care (PoC) molecular technologies have emerged as a viable strategy for improving clinical management of HCV, as they facilitate diagnosis and treatment programmes with the possibility to work in rural, resource-limited settings. In addition to decentralised nucleic acid amplification testing (NAAT), serological rapid diagnostic tests are also contributing to the improvement of diagnosis strategies.
We developed the Genedrive HCV assay for the detection of HCV RNA and conducted analytical and clinical validation studies to assess its diagnostic accuracy. We used plasma and serum samples from a large cohort of treatment-naïve patients with chronic HCV and HCV-negative controls at two centres (Institut Pasteur, Paris, France, and National Health Service (NHS), Nottingham, UK). Samples were assayed using the newly developed PoC assay, and results were compared with the commercial Abbott RealTime HCV Assay. We further validated this new PoC test in a resource-limited setting (Lancet Laboratories, Johannesburg, South Africa).
Methods
Genedrive HCV assay development
The Genedrive HCV assay is a two-step procedure requiring a plasma or serum preparation step, followed by a reverse transcription (RT) reaction to generate the complementary DNA (cDNA) from the target HCV RNA. This cDNA undergoes asymmetric PCR to generate linear amplification of single stranded products, followed by detection using a secondary hybridisation probe and dissociation curve analysis. The Genedrive HCV assay amplifies and detects a 91 base-pair from the 5′ untranslated region of the HCV genome.15 The pan-genotypic HCV target selection was performed through an iterative process comprising 339 sequences from the Los Alamos National Security Database, representing all major HCV genotypes (1a and 1b, 2, 3, 4, 5 and 6) (online supplementary table S1 A). Diagnostic accuracy was shown for all six majors genotypes (online supplementary table S1 B).
Supplemental material
To enable PoC testing, the Plasma Preparation Cartridge contains lyophilised reagents for plasma processing (protease and reagents to prevent thermal-induced coagulation of plasma proteins), as well as the internal positive control (IPC) oligonucleotides and template (pUCV57 plasmid). The IPC reaction targets a DNA sequence located in the backbone of the pUCV57, which shows non-cross reactivity for all HCV genotypes.
Genedrive HCV assay workflow
Following dilution with nuclease free water (1:2 ratio) of the plasma, 15 µL was added to each of the three channels of the plasma preparation cartridge. The protease incubation step was then performed within the Genedrive instrument, consisting of a 5 min incubation at 37°C followed by 5 min at 95°C, thus rendering the sample non-infectious. During this incubation time, 100 µL of nuclease free water was added to resuspend the RT-PCR reagents. After removal of the cartridge from the device, 30 µL of the RT-PCR suspension was dispensed into each channel, which was resealed with a new lid. The cartridge was placed back in the Genedrive instrument, and the programme resumed. The target RNA was converted to DNA (via RT) and subsequently amplified by asymmetric PCR. Following amplification, a fluorescent probe was used to detect target-specific sequences by monitoring changes in fluorescent signal intensity that occurred during dissociation of the fluorescent probe from its hybridised target sequence (if present) as temperature increased. The Genedrive instrument detects this fluorescence at defined melt temperature positions (Tm) for both the HCV target and for the IPC. A schematic workflow of the Genedrive HCV assay and a representative outcome for a positive and a negative sample are illustrated in online supplementary figures S1 and S2. The melting temperatures (Tm) and peak height values for both the IPC and the HCV peak are also illustrated (online supplementary figure S2). Each of the three channels can result in either one or two separate melt peaks: the IPC peak and the HCV peak (if positive). The calling logic for determining a result and the four possible outcomes from the sample test replicates and IPC are depicted in online supplementary figures S3 and S4. In all cases, a positive/negative outcome on the first test run was taken as a result. Control fail-retest and indeterminate-retest outcomes were retested, and the second outcome was taken as the final result. If that second run returned an indeterminate/control fail, no further retests were performed, and no result was obtained. The instrument runs for 88 min (from 1 hour 40 min to 2 hours including experimental manipulation). The runs were monitored, and the data were saved using the Genedrive Engineering software.
Analytical validation
Analytical specificity was assessed using a group of HCV RNA negative plasma samples, some of which were positive for hepatitis B virus (HBV), HIV-1 or flaviviral samples (dengue, yellow fever, Zika and GBV-C) as determined by serology, PCR or both. These specimens were obtained at either NHS, Nottingham or Instituto de Salud Carlos III & Hospital la Paz, Madrid, as part of routine diagnostic procedures, with clearance for samples surplus to diagnostic use being permitted for assay development and validation.
Analytical sensitivity was assessed for each of six major HCV genotypes using 16 commercial standard plasma panels (Seracare (USA) and Tissue Solutions (UK)). Each sample was diluted to 5000, 3000 and 1000 IU/mL, and at each concentration, 24 replicates were tested to obtain the limit of detection (LoD) using probit analysis. To examine the effect of anticoagulant on the LoD, analytical sensitivity was further assessed using synthetic, stable, homogenous and non-infectious armoured RNA in EDTA and Na-heparin plasma (Assuragen, Quant HCV GT 2b).16
Case–control study
We defined cases as treatment-naïve patients with chronic HCV infection (anti-HCV positive and HCV RNA positive). At the Institut Pasteur (Paris, France), pretreatment heparinised plasma samples were obtained from three approved cohort studies in which consent was obtained for secondary use of samples: Inserm C10-08, Inserm C10-54 and ANRS Cupic CO20. The negative control group consisted of samples from healthy donors, negative for both anti-HCV antibody and HCV RNA. Control heparinised plasma samples were obtained from the CoSImmGEn cohort of the Investigation Clinique et Accès aux Ressources Biologiques platform (Centre de Recherche Translationnelle, Institut Pasteur, Paris) and the Etablissement francais du sang. Nottingham samples were derived from those sent to the diagnostic laboratory for routine investigation of HCV or other blood-borne virus infection that were surplus to requirement and could be used for assessment of an in vitro assay. The daily positive controls were obtained from Seracare (USA) and used at a viral load (VL) of 10 000 IU/mL. The negative controls were from a healthy donor plasma (K2EDTA) internally sourced by genedrive plc. Samples were stored at −80°C, thawed, aliquoted (operators were blinded to infection status) and kept frozen at −80°C prior to use for both Genedrive and Abbott RealTime HCV Assay testing. Additionally, to test a potential freeze cycle effect, matched fresh and frozen plasma from HCV RNA positive and negative individuals were assessed. The study was sponsored and approved by the ANRS (France Recherche Nord&Sud Sida-HIV Hépatites) and registered with clinicaltrials.gov (NCT02992184: PoC-HCV Genedrive Viral Detection Assay Validation Study).
Reference standard test for HCV RNA
VLs were measured by RT real-time PCR using the Abbott RealTime HCV assay, according to manufacturer’s instructions. The Abbott m2000sp and m2000rt instruments were used for automated sample preparation and real-time amplification and detection, respectively. The lower limit of quantification of the assay is 12 IU/mL. HCV genotyping was performed based on NS5b region sequence phylogenetic analysis using the Abbott RealTime HCV Genotype II on the Abbott m2000, following manufacturer’s instructions.
Robustness, repeatability and stability
Daily controls (positive and negative samples) were performed at both sites, and results were as expected (online supplementary table S2). Nine hundred and twenty-five case and control samples were tested across 12 Genedrive instruments, five operators and three different assay batches. Although our study was not specifically designed and powered for this analysis, no effect of the Genedrive instruments, the operator and the assay batch were observed on assay performance. To determine the effect of interfering substances on assay performance, 37 different substances including a wide range of antiviral drugs and several endogenous substances such as bilirubin or haemoglobin were tested using HCV positive plasma at 3× LoD (online supplementary table S3).
Real-life clinical setting validation
A total of 130 plasma and serum samples were tested across three Genedrive instruments and four operators. They were collected as part of a routine HCV RNA diagnostic testing using Abbott RealTime HCV Genotype II on the Abbott m2000, in different African countries: Ghana (9 cases, 3 controls), Kenya (37 cases, 1 control), Mauritius (18 cases), Mozambique (1 case), Nigeria (3 cases), South Africa (33 cases, 6 controls), Tanzania (10 cases, 2 controls), Uganda (1 case), Zambia (1 case) and Zimbabwe (5 cases). Samples were stored at −70°C prior to testing.
Data analysis
The accuracy of the Genedrive HCV assay to detect HCV viraemia was assessed by diagnostic sensitivity and specificity, and positive and negative likelihood ratios. Factors associated with the lack of result or false-negative results were identified using Wilcoxon rank-sum test for continuous and Fisher’s exact test for categorical variables. All the variables found to be significantly associated with the lack of result in the crude analysis (P<0.05) were further assessed in the multivariable logistic regression. The sample size of the case–control study was determined based on the Foundation for Innovative New Diagnostics (FIND)/WHO Target Product Profile indicating the minimally acceptable sensitivity and specificity as 95% and 98%, respectively.17 18 Four hundred and nine cases were required to demonstrate that sensitivity is at least higher than 95% at a two-sided significance level of 5% with a power of 90% when the true sensitivity was 98%. Similarly, 495 controls were needed to show that the specificity is at least higher than 98% with a power of 80%, when the true specificity was 99.5%.
To assess the performance of the Genedrive HCV assay to quantify/semiquantify HCV RNA levels, we split the HCV RNA-positive cases into a derivation (Paris site) and validation set (Nottingham site). The correlation between HCV/IPC peak ratio (calculated as the mean value of ratios of HCV peak and IPC peak at each channel) and VL was evaluated using Pearson’s correlation coefficient. To predict VL, a conversion factor from the HCV/IPC peak ratio to HCV RNA levels (log10 IU/mL) was developed by fitting a linear regression model in the Paris cohort. The conversion factor was then applied to the HCV/IPC peak ratio of the Nottingham cohort, and the agreement between the observed HCV RNA levels using RT-PCR and the estimated HCV RNA levels from the Genedrive was assessed using Bland and Altman plots. All analyses were performed using STATA, V.13.0. The study was reported in accordance with the standards for reporting of diagnostic accuracy checklists.19
Results
Analytical sensitivity
The Genedrive HCV assay detected all major HCV genotypes, with a LoD ranging from 1406 IU/mL (genotype 6) to 3203 IU/mL (genotype 5) (online supplementary table S4). The LoD of the 16 standard plasma samples was 2362 IU/mL (95% CI 1966 to 2758). Using synthetic armoured RNA, the LoD in plasma derived from K2 EDTA collected blood (1918 IU/mL, 95% CI 1789 to 2205) was significantly lower (P<0.0001) than the LoD in Na-heparin derived plasma (2359 IU/mL, 95% CI 2147 to 2826).
Analytical specificity
Analytical specificity was examined for 17 different pathogens including samples infected with HIV-1, HBV and various Flaviviridae (including: dengue, Zika, yellow fever and GBV-C) (online supplementary table S5). The Genedrive HCV assay was shown to be non-cross-reactive for any of the aforementioned samples with the sole exception of one positive out of two replicate runs on a GBV-C sample.
Clinical validation study (case–control study)
A summary of all samples tested in all study sites is illustrated in table 1. The characteristics of case and control subjects including age, sex, median HCV RNA values and HCV genotype are summarised in table 2. For the first part of the study performed in Western laboratories (Institut Pasteur, Paris, and Queen’s Medical Centre, Nottingham), a total of 422 cases and 503 negative controls were included, and the flow chart of the study samples is illustrated in figure 1. Results were obtained at the first attempt for 97.2% of the samples (899/925). Consequently, 26 samples were retested, of which 61.5% (16/26) returned a result with the second test. The 10 samples (4 cases/6 controls) that failed such retesting were excluded from subsequent analysis for sensitivity and specificity. Multivariable analysis identified low HCV RNA levels as the sole independent factor significantly associated (P<0.001) with the lack of a result at the first attempt in the HCV RNA positive case samples (online supplementary table S6). The median HCV RNA level (IQR) was 5.9 log10 IU/mL (5.4–6.3) in cases with results and 2.9 log10 IU/mL (2.6–3.5) in cases without results. No factor was identified as associated with a lack of result in the control samples.
Study sample characteristics
Characteristics of cases and control samples

Flow chart of the study participants.
Diagnostic performance of the HCV Genedrive assay
The diagnostic sensitivity and specificity of the Genedrive HCV assay to detect HCV RNA in Western laboratories was 98.6% (412/418; 95% CI 96.9% to 99.5%) and 100% (497/497; 95% CI 99.3% to 100%), respectively (table 3). Apart from one sample with a VL of 2508 IU/mL, all of the false-negative samples were below 1000 IU/mL. Low HCV RNA level was the only factor significantly associated (P<0.0001) with false-negative results; age, sex, study site, sample type or viral genotype were not found to influence the accuracy of Genedrive HCV assay (online supplementary table S7). No differences in sensitivity were observed between the two sites, which tested samples with difference anticoagulants. To examine for potential effects of a freeze/thaw cycle on the assay, the diagnostic performance was further assessed on 100 freshly collected (non-frozen) plasma samples. After excluding four samples (two cases/two controls) that did not give a result, the sensitivity and specificity was 98.0% (48/49; 95% CI 89.1% to 99.9%) and 100% (47/47; 95% CI 92.5% to 100%), respectively (table 3), with one false negative sample that had a VL of 230 IU/mL. This demonstrates that the accuracy of the assay was not affected by a freezing step.
Diagnostic performance of Genedrive using frozen and fresh samples
HCV VL semiquantification
In the derivation data set (Paris cohort), the mean of the HCV/IPC peak ratio was closely correlated with VL as measured by the Abbott platform (Pearson’s correlation coefficient: r=0.76, P<0.0001) (figure 2A). By fitting a linear regression, we obtained a conversion factor from the mean of the HCV/IPC peak ratio to the HCV RNA levels. The estimated log10 VL was equal to 3.5+0.4* HCV/IPC peak ratio. This conversion factor was then applied to the mean HCV/IPC peak ratio in the validation set, for which historical VL measurements were used. There was a linear correlation between the VL measured by the Abbott RealTime platform and the value estimated using the Genedrive results (Pearson’s correlation coefficient: r=0.72, P<0.0001) (figure 2B). The agreement between the observed and estimated VL was assessed using Bland and Altman plots; mean difference was −0.06 log10 IU/mL (SD: ±0.72; 95% limits of agreement: −1.47, 1.33) (figure 2C). Low HCV VL was associated with non-agreement (supplementary table S8). These results support the potential use of the Genedrive HCV assay to semiquantify VL.
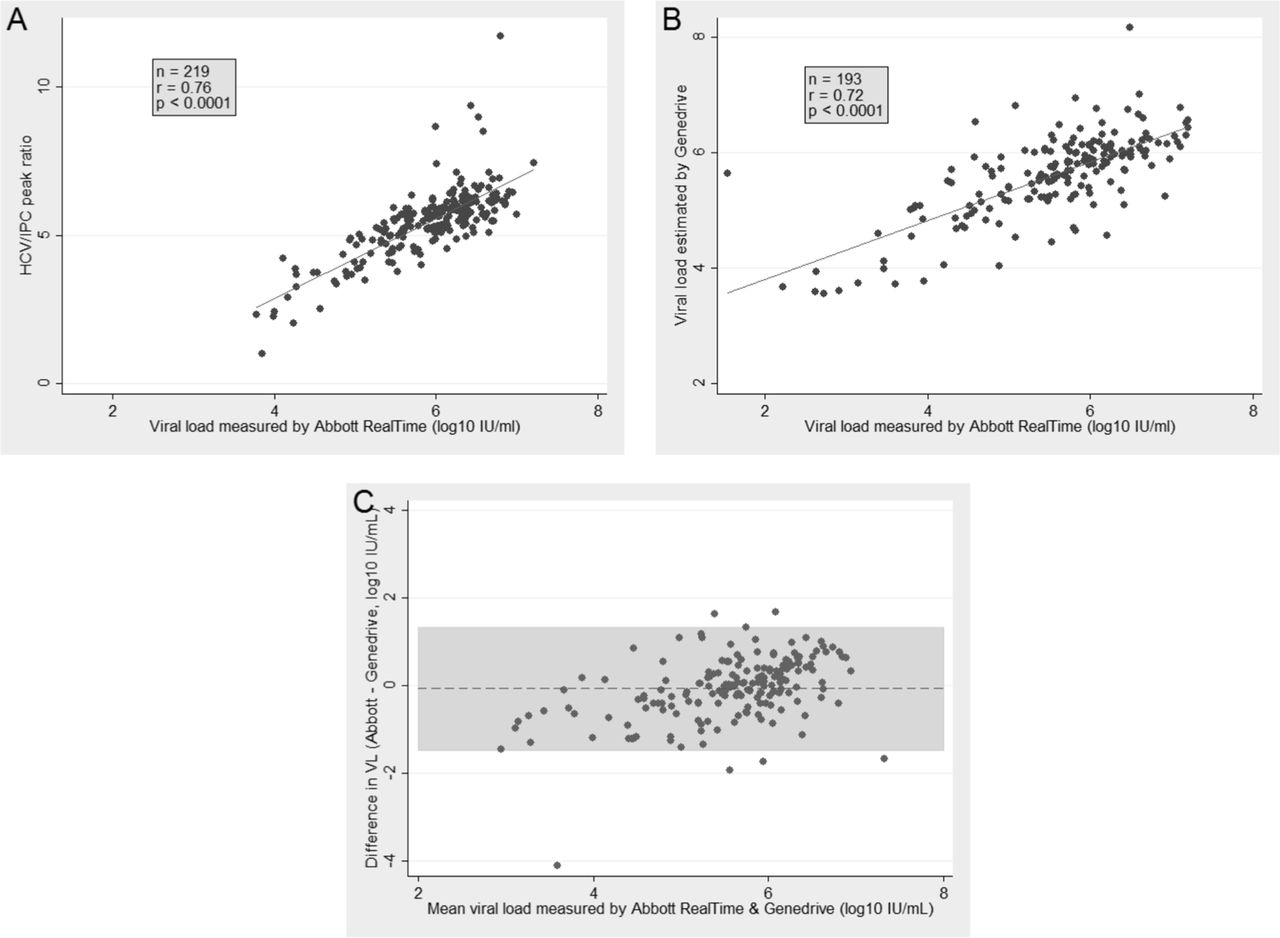
{kind=link}
{kind=link}
Comparison between the HCV/IPC peak ratio (Genedrive) and HCV RNA levels (Abbott RealTime). (A) Correlation between the HCV/IPC peak ratio and the viral load measured by Abbott RealTime in the Paris cohort (derivation set). (B) Correlation between the viral load estimated by Genedrive and viral load measured by Abbott RealTime in Nottingham cohort (validation set). (C) Agreement between the viral loads estimated by Genedrive and viral loads observed by Abbott RealTime in Nottingham cohort (validation set).
Real-life clinical setting validation
In order to further assess the diagnostic accuracy of the Genedrive HCV assay outside Western laboratories, we extended our study to a real-life clinical setting. Samples tested in Johannesburg (South Africa) are summarised in tables 1 and 2. A total of 118 cases and 12 negative controls were included, and the flow chart of the study samples is illustrated in figure 1. Results were obtained at the first attempt for 95.4% of the samples (124/130). Consequently, six samples were retested, of which 33.3% (2/6) returned a result with the second test. The four samples (four cases) that failed such retesting were excluded from subsequent analysis for sensitivity and specificity. All the samples that required retesting were positive for HCV RNA. As summarised in table 3, the Genedrive HCV assay demonstrated 100% sensitivity and 100% specificity when compared with the Abbot m2000 in real-life conditions.
Discussion
We developed and validated the Genedrive HCV assay for the qualitative detection of HCV RNA. The Genedrive instrument is a handheld rapid thermocycler that permits NAAT on plasma or serum samples, which fulfils the target product profile for HCV diagnosis drafted by FIND/WHO (diagnostic specificity >98% and sensitivity >95%).17 18 Diagnostic accuracy was shown for all six majors genotypes, although genotypes 2, 5 and 6 had a limited presence in the samples tested (online supplementary table S1 B).
Although DAAs for HCV cure are now available, the challenge for HCV infection, and especially for HCV elimination programmes, is the screening of undiagnosed HCV-infected subjects and the linkage to care, namely the availability of DAA at reasonable costs. The majority of infected individuals are unaware of their infection and as such do not benefit from this unprecedented medical advance. HCV infection is often first identified by serological testing, followed by diagnostic confirmation of HCV nucleic acid in circulation.20–22 At present, HCV RNA testing is only available in centralised medical settings resulting in less than 1% of infected people in low-income and middle-income countries being aware of their infection.14 Therefore, there is an urgent unmet clinical need for decentralised HCV testing that can provide rapid diagnosis that may result in improved patient outcome and public health as shown for other diseases.23
Initial assay validation was conducted to establish specificity for HCV with clinical validation performed in a two-site retrospective clinical study to determine sensitivity and specificity. In our treatment-naïve case subjects, the lack of a result and false-negative results were only observed in samples with RNA viral levels below the LoD of the Genedrive HCV assay. It is unlikely that this analytical sensitivity would impact diagnostic sensitivity, as the vast majority of people with pretreatment chronic HCV infection have high VL (>5 log IU/mL). This is supported by a study of 2400 treatment-naïve patients with chronic HCV, where it was shown that <2% of patients had viraemia below 4 log10 IU/mL.24 Furthermore, recent WHO guidelines explicitly acknowledged that ‘a limit of detection of 3000 IU/mL or lower would be acceptable and would identify 95% of those with viremic infection’.10
Linking diagnosis to treatment decisions is now the major clinical challenge: in Europe, the percentage of cured individuals from initial viraemic infections is only 4.1%.25 In the best cases, the treatment rate is around 5% (eg, France and Germany) and does not exceed 1% in resource-limited countries.26 27 Furthermore, it has been well described how HCV prevalence negatively correlates with treatment rate,28 29 as illustrated by countries such as Egypt and Mongolia, which have very high HCV prevalence and treatment rates between 0.1% and 1.2%. These data show how both initial screening and linkage to care after diagnosis are still major challenges, one of the main consequences being the low number of individuals that are prescribed and adhere to treatment.30 This is partially due to a lack of PoC diagnostic tests, as well as the requirement for multiple clinical visits (eg, diagnosis by serology followed by NAAT). Devices like Genedrive could have a major impact in improving the chronic HCV care continuum, as diagnosis through HCV RNA detection could be performed in a wide range of field settings and no intermediate visits would be required. Thus, screening in resource-limited settings could be easy, effective and rapid and, consequently, treatment could be started as soon as a positive result has been obtained.
While several NAAT PoC assays are in development, such as Alere q (Alere), EOSCAPE (Wave 80 Biosciences), PanNAT platform (Micronics), Truelab PCR (Molbio Diagnostics) and RT CPA (Ustar Biotechnologies), the Cepheid Xpert HCV Viral Load assay (Cepheid) is currently the only CE-IVD certified assay for decentralised HCV VL determination. The Xpert HCV test has been reported to have good performance but also presents important limitations.31–33 The conventional Xpert HCV assay requires a large volume of sample (1 mL plasma) and an electrical power supply, meaning that basic laboratory infrastructure is needed,34 35 although a recent study carried out by Grebeley and colleagues validated the use of 100 µL of capillary blood.33 Furthermore, the Cepheid Xpert cartridge contains guanidinium thiocyanate as the lysis reagent, which is highly toxic and therefore needs special precautions when handling and should be disposed of by incineration.36 In contrast, the Genedrive assay does not contain any highly toxic chemicals and only requires 30 µL of sample providing the potential for blood pin prick testing, although a requirement for serum/plasma remains an important limitation. Genedrive is a 600 g portable device that can be battery operated, thus making it highly suitable for decentralised testing and use in field settings. While the Genedrive HCV assay is not fully automated, it is a user-friendly device and requires less than a day’s training to operate. While the Xpert HCV assay is quantitative, the Genedrive HCV assay has potential semiquantitative properties as demonstrated herein. Nevertheless, the clinical significance of quantifying HCV VL is less important in the era of highly potent DAAs at least for diagnosing active HCV infection. The aim of this study was to develop and assess the diagnostic accuracy of the Genedrive HCV assay for the qualitative detection of HCV RNA. Thus, we compared Genedrive directly to the Abbott RealTime HCV test, a well-established PCR test that offers HCV VL and genotype testing and is considered the current gold standard. However, it would also be of interest to perform a direct comparison with Cepheid Xpert, to test for equivalence or superiority between the two platforms.
Finally, a crucial factor when assessing the feasibility of PoC technologies is their cost, both for platform and assays. However, direct comparisons of costs to other platforms are challenging as there are geographic differences and unknowns such as subsidised or negotiated pricing, distributor margins and import duty. Furthermore, additional costs related to the platform should be considered, including installation, maintenance, training and lifespan of the equipment and tests. While the final Genedrive costs remain to be defined, the instrument is approximately $5000, which is considerably lower than other molecular systems, and each Genedrive HCV test will be available for $30–$40, depending on the country or region in question, which is equivalent to other products on the market.
The results obtained in Western laboratory-based settings are highly encouraging; however, the accuracy of PoC tests can be often reduced when used in real clinical settings.34 35 To address this point, we performed a validation study in South Africa with samples obtained from various African countries. In this real-life clinical setting, where there is often a large time between sample collection and testing—as illustrated by the high levels of hemolyses (27/130 samples)—the performance of the Genedrive HCV assay was equally specific and sensitive. Although we demonstrated the efficacy of the Genedrive test in real-life clinical conditions, including different temperatures, different time delays from sample withdrawal and processing and different operators, further validation of the whole system for infield use in decentralised settings is necessary to achieve its full potential of supporting wide spread screening campaigns.
Here, we provide proof of concept in a real-life clinical setting that the Genedrive HCV assay has great potential to provide an affordable and robust instrument for decentralised HCV NAAT testing. This highly sensitive and specific test has recently obtained CE-IVD certification and is positioned to enable real-time treatment management of patients with chronic HCV in any clinical setting. Therefore, the next step with the Genedrive HCV assay requires prospective validation in real-life decentralised settings in low-income and middle-income countries.
References
Footnotes
MLA and DD contributed equally.
AL and YS contributed equally.
Contributors AL, T-PB, ARR, J-FM, PC, AM, CPM, WLI, AG and RV analysed clinical samples. AL, YS and SA analysed the data. SA, RF, EH, ML and GM developed the assay. YS, EM, AF, SP, GM, MLA and DD designed the study. AF, SP, GM, MLA and DD secured funding. AL, YS and DD wrote the manuscript. All authors approved the final version and revised it critically before submission.
Funding The overall study was funded by the European Commission FP7 (n° 601851). Work performed in Nottingham and South Africa received additional financial support from genedrive plc.
Competing interests SA, RF, EH, ML and GM are full time employees of genedrive plc. The remaining authors declare that they have nothing to disclose regarding funding or conflict of interest with respect to this manuscript.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval CPP IDF VI (Paris, France)
Provenance and peer review Not commissioned; externally peer reviewed.