Article Text

Abstract
Background and aims HBV infection represents a major health problem worldwide, but the immunological mechanisms by which HBV causes chronic persistent infection remain only partly understood. Recently, cell subsets with suppressive features have been recognised among monocytes and natural killer (NK) cells. Here we examine the effects of HBV on monocytes and NK cells.
Methods Monocytes and NK cells derived from chronic HBV-infected patients and healthy controls were purified and characterised for phenotype, gene expression and cytokines secretion by flow cytometry, quantitative real-time (qRT)-PCR, ELISA and western blotting. Culture and coculture of monocytes and NK cells were used to determine NK cell activation, using intracellular cytokines staining.
Results In chronic HBV infection, monocytes express higher levels of PD-L1, HLA-E, interleukin (IL)-10 and TGF-β, and NK cells express higher levels of PD-1, CD94 and IL-10, compared with healthy individuals. HBV employs hepatitis B surface antigen (HBsAg) to induce suppressive monocytes with HLA-E, PD-L1, IL-10 and TGF-β expression via the MyD88/NFκB signalling pathway. HBV-treated monocytes induce NK cells to produce IL-10, via PD-L1 and HLA-E signals. Such NK cells inhibit autologous T cell activation.
Conclusions Our findings reveal an immunosuppressive cascade, in which HBV generates suppressive monocytes, which initiate regulatory NK cells differentiation resulting in T cell inhibition.
- regulatory NK cells
- monocytes
- HBV
- PD-L1
- HLA-E
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
In chronic HBV infection, natural killer (NK) cells show a functional impairment with defects in their antiviral function, and the insufficiency of T cell immunity has been clearly demonstrated.
The cell subsets with suppressive features have been recognised among monocytes and NK cells.
What are the new findings?
HBV employs hepatitis B surface antigen to induce suppressive monocytes with HLA-E, PD-L1, interleukin (IL)-10 and TGF-β expression via the MyD88/NFκB signalling pathway.
HBV-induced monocytes educate NK cells to secrete IL-10.
HBV-induced IL-10 producing NK cells through monocytes is mediated by PD-L1/PD-1 and HLA-E/CD94.
HBV-induced regulatory NK (NK-reg) cells inhibit autologous T cell activation.
How might it impact on clinical practice in the foreseeable future?
It contributes to better understand the immunopathogenesis of how HBV evades immunity and maintains persistent infection. Future investigation needs to address how to manipulate the function of these NK-reg cells in the clinical setting.
HBV infection represents a major health problem worldwide. Over 240 million individuals are chronically infected with HBV and are at high risk of developing liver cirrhosis and hepatocellular carcinoma.1 Chronic HBV infection (CHB) occurs as a result of an ineffective antiviral immune response.2 Although the insufficiency of T cell immunity in HBV persistent infection has been clearly demonstrated, the role of innate immune cells involving monocytes and natural killer (NK) cells still remains to be clarified.
The innate immune system provides the first line of defence in antiviral responses and activates adaptive immune responses.3 Monocytes are innate immune cells and play multiple roles in immune function.4 5 Hepatitis B surface antigen (HBsAg) is the most abundant HBV protein in the liver and peripheral blood of patients with CHB.6 High concentration of HBsAg in the bloodstream of patients with CHB could theoretically contribute to the impaired immune response. Several studies have shown that HBsAg can suppress the release of c-induced cytokines by interfering with the Toll-like receptor (TLR) signal pathway in monocytes/macrophages, which may contribute to the establishment of chronic infections.7 8
NK cells are another important component of the innate immune system,9 which respond very rapidly to virus-infected or transformed cells and kill virus-infected or malignant transformed cells without presensitisation and restriction by major histocompatibility antigen.10 NK cells play key roles in innate antiviral immunity and have important functions that shape and influence adaptive immune responses and play immunoregulatory roles.11 Human interleukin (IL)-10-producing regulatory NK cells were showed to express low transforming growth factor (TGF)-β and IL-13 but no Interferon-γ (IFN-γ).12 In patients with breast cancer, circulating NK cells include rare cells producing TGF-β and IL-10, which exert a suppressive influence on innate and adaptive immunity.13 In CHB, NK cells show a functional impairment with defects in their antiviral function,14 15 but the mechanism is only partly understood. The immunosuppressive cytokine environment in CHB, created through high levels of IL-10, may inhibit the ability of NK cells to produce IFN-γ,16 as has already been shown in acutely infected patients.17 This defect persists in patients with CHB receiving antiviral therapy but can be reversed in vitro by specific blockade of IL-10 and TGF-β.18
NK cell activation can be triggered by the interaction of the receptor 2B4 with the ligand CD48 present on the surface of peripheral blood mononuclear cell (PBMC)-derived macrophages.19 Blockade of NKp46 and DNAX accessory molecule-1 decreases NK cell cytotoxicity against cytomegalovirus-infected macrophages and lipopolysaccharides (LPS)-stimulated macrophages derived from PBMC.20 Monocytes activated by TLR increase the expression of activation-induced C-type lectin (AICL), which is the ligand of the NK cell activating receptor NKp80, the interaction AICL–NKp80 induces IFN-γ production of NK cells.21 More recently, the secretion of IL-12 by CpG DNA or Bacillus anthracis-infected macrophages enhances the release of IFN-γ by mouse NK cells but depends on cell-to-cell contact.22
In the present study, we found that compared with cells from normal donors, both monocytes and NK cells derived from the chronic HBV-infected patients display suppressive characteristics. We thus hypothesised that HBV induces suppressive monocytes to educate NK cells differentiating into IL-10+ regulatory NK cells. Our findings provide a novel mechanism by which HBV initiates an immunosuppressive cascade involving monocytes, NK cells and T cells; it may contribute to HBV evasion of immunity and the maintenance of persistent infection.
Materials and methods
Human subjects
Thirty-five treatment-naive patients with CHB and 35 healthy individuals were enrolled in this study (online supplementary table 1). These studies were approved by the IRB of Jilin University, The First Hospital (No: 2015215). Written informed consent was obtained from all adult participants, and no children were involved in this study. All experiments were carried out in accordance with the approved guidelines and regulations.
Supplementary file 1
Reagents
HBsAg was purchased from Meridian (Memphis, Tennessee, USA, Catalogue #: R36100), which is purified from human plasma with the purification >95%. ST2825 and BAY 11–7082 were purchased from Medchem Express (Monmouth Junction, New Jersey, USA). . These Abs, rabbit monoclonal anti-IκB-α, mouse monoclonal antiphospho-IκB-α, anti-MyD88 and anti-β-actin, were detected using HRP-conjugated goat or rabbit anti-mouse secondary Abs. All of these Abs were purchased from CST.
Limulus amoebocyte assay for LPS contamination
Endotoxic contamination of HBsAg was assessed using QCL-1000 chromogenic end point assay (Combrex, Cottonwood, Arizona, USA) and found to be less than 1 pg/mL.
Cell culture of HBV (HBVcc)
HepG2.2.15 cells were cultured in tetracycline-free medium to induce virion production; supernatant was collected every other day with culture medium replenishment for 18 days. The pooled supernatant was mixed with polyethylene glycol-8000 powder (final concentration of 10%) and gently rotated at 4°C overnight. HBV particles were then precipitated by centrifugation at 1000 g for 30 min at 4°C and redissolved in serum-free DMEM/F12 medium with 1% vol of the original supernatant samples. The concentrated virus stocks were aliquoted and stored at −80°C.
Cell isolation and purification
PBMCs were freshly isolated from peripheral blood of healthy individuals and patients with CHB by ficoll density gradient separation. Monocytes and NK cells were then purified by magnetic cell sorting with CD14+ microbeads and NK cell isolation kit (Miltenyi Biotec). The purity of cells was equal or greater than 95% as determined by flow cytometry.
Cell culture of monocytes and NK cells
Purified monocytes from healthy HBV-negative and HCV-negative blood donors were cultured with HBVcc or HBsAg, which concentrations are given in the figure legends. To compare the function of monocytes and NK cells between healthy individuals and patients with CHB, monocytes were stimulated with LPS (1 µg/mL) and NK cells by PHA (10 µg/mL) for 16 hours. Cells and supernatants were collected for the detection of phenotype, signalling molecules and cytokines.
Generation of dendritic cells (DCs)
DCs were generated from monocytes with GM-CSF and IL-4 for 5 days following LPS stimulation for another 2 days.
Cell coculture
Purified NK cells and monocytes were cocultured for 16 hours at a 1:1 ratio in complete medium in presence or absence of HBsAg and HBVcc. Brefeldin A (Sigma-Aldrich) was subsequently added at a final concentration of 5 µg/mL. NK cells were stained for intracellular expression of IL-10 and IFN-γ. Mouse IgG was used as an isotype-matched control antibody. To investigate the contact dependence of the interaction, monocytes and NK cells were separated by a membrane (0.4 µm pore size) in transwell plates (Costar, Corning). To investigate the involvement of selected molecules, blocking experiments were performed by adding the following mAbs: α-TGF-β, α-IL-10, α-TNF-α, α-PD-L1, α-PD-1, α-HLA-E and α-CD94. Control experiments were performed using isotype-matched mouse antibodies (all mAbs from R&D Systems). All mAbs were used at a final concentration of 10 µg/mL.
For the regulatory effect of NK cells on non-specific T cell activation, purified T cells were cocultured with autologous NK cells, which were repurified from monocyte/NK cell coculture with or without HBsAg/HBVcc and stimulated with PHA in the presence or absence of anti-IL-10 antibody. For the regulatory effect of NK cells on HBV-specific T cell response, monocytes, NK cells and T cells were purified from patients with CHB. T cells were cocultured with autologous DCs loaded with either HBsAg or hepatitis E virus open reading frame 2 (HEV-ORF2) protein in the presence of NK cells, which were repurified from monocyte/NK cell coculture with or without HBsAg/HBVcc.
Flow cytometry
Freshly isolated PBMCs or cultured monocytes and/or NK cells were resuspended in staining buffer and preincubated with FcR blocking reagent (Miltenyi Biotec) for 15 min at 4°C. Monocytes were stained with V450 Mouse Anti-Human CD14, PE-Cy7 Mouse Anti-Human PD-L1, PE Mouse Anti-Human HLA-E, NK cells stained with APC Mouse Anti-Human CD3, PE Mouse Anti-Human CD56, APC Mouse Anti-Human CD94 and FITC Mouse Anti-Human PD-1. Cells for detection of intracellular cytokines expression were operated by methods as above. TNF-α, TGF-β and IL-10 were used in monocytes, and IFN-γ IL-10 were used in NK cells. All the antibodies were obtained from BD Biosciences. BD LSRII Fortesa instrument was used to perform experiments, and the data acquired were analysed with FlowJo (Treestar software, Ashland, Oregon, USA).
T cell proliferation assay
T cell proliferation was performed as described previously.23 Briefly, before cocultured with autologous DCs and/or NK cells, T cells were labelled with 5 mM CFSE (Molecular Probes, Grand Island, New York, USA) according to the manufacturer’s recommended protocol. After cocultured for 5 days, when clumps were visible, cells were harvested and assessed the proliferation of CD4+ and CD8+ T cells by flow cytometry.
Enzyme-linked immunosorbent assay
The cell culture supernatants were collected after 2 days incubation in each experimental condition. The concentration of TNF-α, IL-10, TGF-β and IFN-γ were measured by ELISA according to the manufacturer’s instructions.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR analysis was performed using standard procedures. RNA extraction, cDNA transcript and real-time PCR experiment were performed according to the manufacturer’s instructions. The sequences of gene-specific and GAPDH primers used for quantitative real-time PCR are listed in online supplementary table 2.
Western blotting analysis
Pure monocytes were incubated with 10 µg/mL HBsAg or in the presence of HBsAb, after 3 hours of stimulation, cells were collected and lysed for western blotting analysis as described previously.23
Statistical analysis
Statistics were performed using Prism statistical software (GraphPad). All analyses were unpaired, one-way, non-parametric Mann-Whitney U tests.
Results
Phenotypic and functional comparison of monocytes and NK cells between patients with CHB and healthy controls
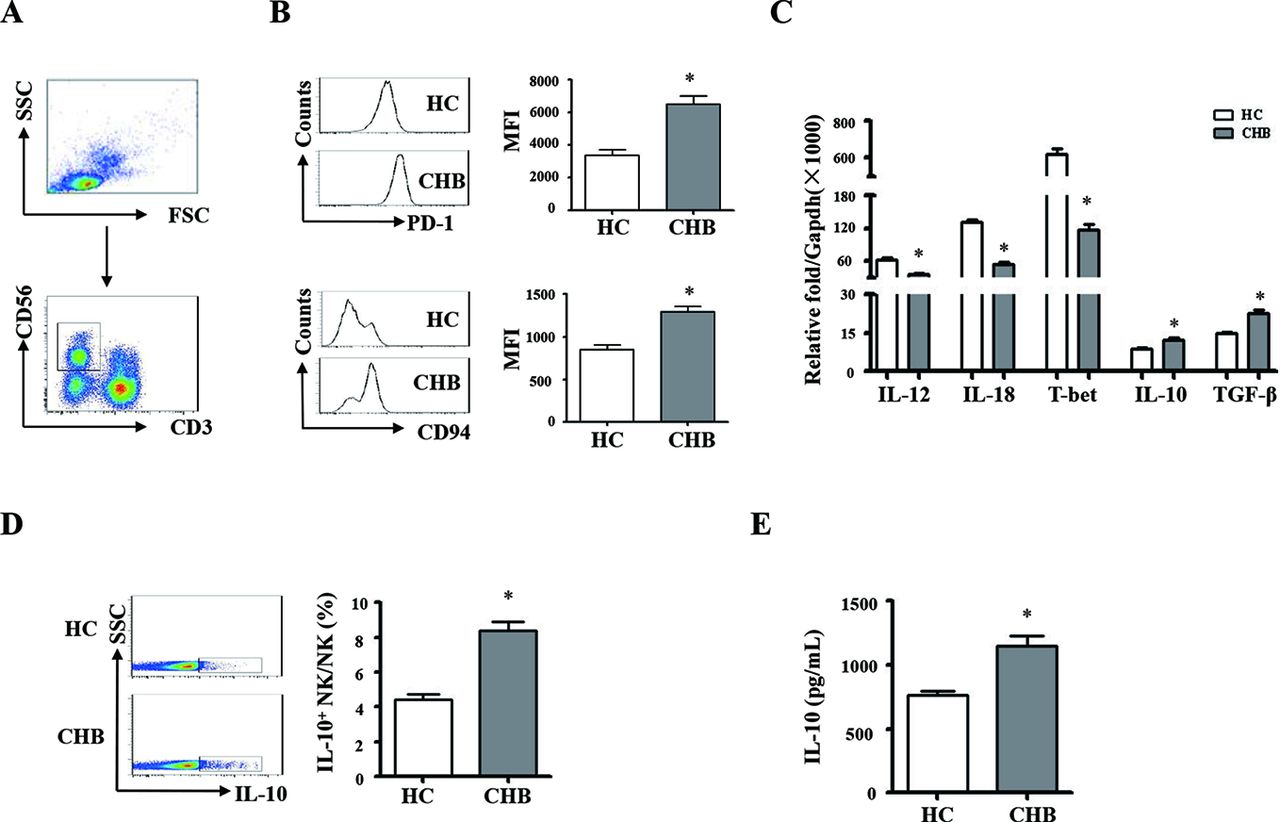
We compared the phenotypic and functional difference of monocytes and NK cells between the patients with CHB and healthy controls. The gating strategies of monocytes and NK cells from PBMCs were showed in figures 1A and 2A; we observed that the expression levels of PD-L1 and HLA-E on monocytes from patients with CHB are much higher than that of healthy controls (figure 1B, p<0.05). The expression levels of PD-1 and CD94 on NK cells from patients with CHB are significantly higher than healthy controls (figure 2B, p<0.05). The mRNA expression levels of TNF-α, TGF-β and IL-10 are significantly higher in pure monocytes from patients with CHB than that of in healthy donors (figure 1C, p<0.05). Compared with healthy individuals, the expression levels of TGF-β and IL-10 were much higher, and the expression levels of IL-12, IL-18 and T-bet were much lower in pure NK cells from the patients with CHB (figure 2C, p<0.05).
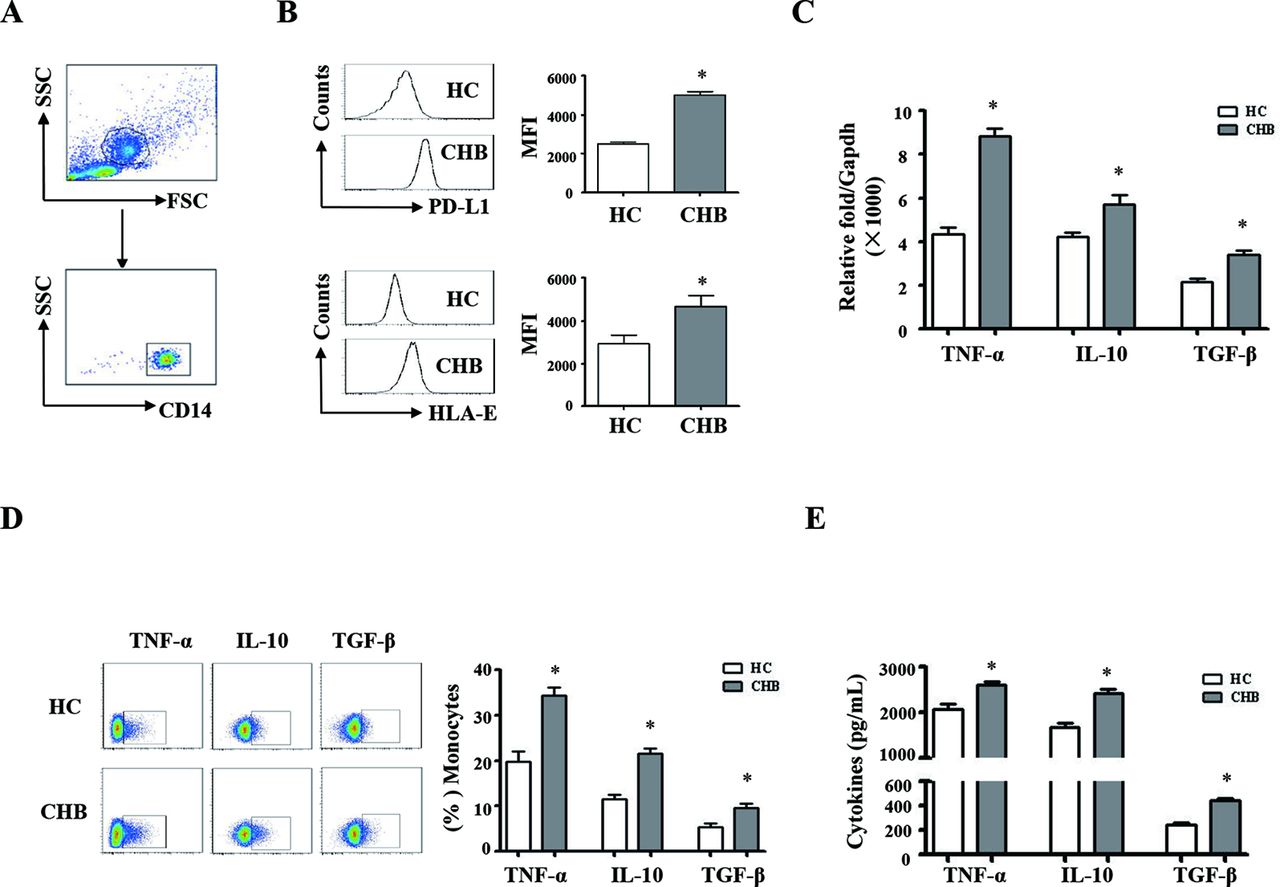
Phenotypic and functional difference of monocytes between chronic HBV-infected patients and HCs. Peripheral blood mononuclear cells (PBMCs) were isolated from health individuals (n=35) and chronic hepatitis B patients (n=35). CD14 staining was used to identify monocytes. (A) The gating strategies of monocytes from PBMCs. (B) The expression levels of HLA-E and PD-L1 on monocytes from chronic HBV-infected patients and HCs were analysed by flow cytometry. (C) TNF-α, TGF-β and IL-10 mRNA expression of pure monocytes were determined by qRT-PCR. (D and E) Monocytes from patients with CHB and HCs were purified and stimulated with LPS for 16 hours. The expression and secretion of TNF-α, TGF-β and IL-10 were detected by intracellular cytokine staining and ELISA, respectively. A representative experiment from 35 independent experiments is shown. The error bars represent SEM. *p<0.05. CD14, Cluster of Differentiation 14; CHB, chronic HBV infection; FSC, forward scatter; HCs, healthy controls; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin; qRT-MFI, mean fluorescence intensity; PCR, quantitative real-time PCR; SSC, side scatter; TGF-β, transforming growth factor-β.

Phenotypic and functional difference of NK cells between chronic HBV-infected patients and HCs. PBMCs were isolated from health individuals and patients with chronic hepatitis B. CD3 and CD56 were used for identify NK cells. (A) The gating strategies of NK cells from PBMCs. (B) The expression levels of PD-1 and CD94 on NK cells were analysed by flow cytometry. (C) NK cells were purified, and messenger RNA expression levels of TGF-β, IL-10, IL-12, IL-18 and T-bet were determined by qRT-PCR. (D and E) NK cells from chronic HBV-infected patients and HCs were purified and stimulated with PHA for 16 hours. The expression and secretion of IL-10 were detected by intracellular cytokine staining and ELISA, respectively. A representative experiment from 35 independent experiments is shown. The error bars represent SEM. *p<0.05. CD94, cluster of differentiation 94; CHB, chronic HBV infection; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HCs, healthy controls; IL, interleukin; MFI, mean fluorescence intensity; NK, natural killer; PBMCs, peripheral blood mononuclear cells; PD-1, programmed death1-ligand; qRT-PCR, quantitative real-time PCR; TGF-β, transforming growth factor-β.
Pure monocytes from the patients with CHB and healthy controls were stimulated with LPS for 16 hours. Intracellular and secreted TNF-α, TGF-β and IL-10 were detected by staining (IC) and ELISA, respectively. The results were shown in figure 1D,E. Monocytes from patients with CHB expressed and secreted much more TNF-α, IL-10 and TGF-β, compared with healthy controls (p<0.05). Pure NK cells from patients with CHB and healthy controls were stimulated with PHA. IL-10 synthesis and secretion of NK cells were detected by IC staining and ELISA, respectively (figure 2D,E). NK cells from patients with CHB expressed and secreted much more IL-10 than healthy controls (p<0.05). Taken together, these results suggested that monocytes and NK cells derived from patients with CHB display suppressive characteristics.
HBV employs HBsAg to induce immunosuppressive monocytes
The immunosuppressive effects of HBV or the encoded proteins, HBeAg and HBsAg, have been documented previously.24–26 In order to test whether HBV induces suppressive monocytes, pure monocytes from healthy donors were cultured with increasing concentrations of HBsAg (0.1, 1.0 and 10 µg/mL) or HBVcc (105, 106, 107 copies/mL). HLA-E and PD-L1 expression on monocytes were analysed by flow cytometry. As expected, both HBsAg and HBVcc caused a dose-dependent increase expression of HLA-E (figure 3A) and PD-L1 (figure 3B). Both HBsAg and HBVcc induced a dose-dependent increase in secretion of TNF-α (figure 3C), IL-10 (figure 3D) and TGF-β (figure 3E). 107 copies/mL of HBVcc and 10 µg/mL HBsAg were used in most experiments. We also observed that neutralisation of HBsAg by HBsAb inhibits HBsAg-induced and HBVcc-induced PD-L1 and HLA-E expression (figure 3F,G, p<0.05), and TNF-α, IL-10 and TGF-β productions (figure 3H, p<0.05), but hepatitis B early antigen (HBeAg) and HBcAg antibody could not neutralise the HBV-induced monocytes suppressive phenotype and cytokines production in online supplementary figure 1. These results indicate that HBV employs HBsAg to induce monocytes to express both inflammatory cytokines and immunosuppressive molecules.
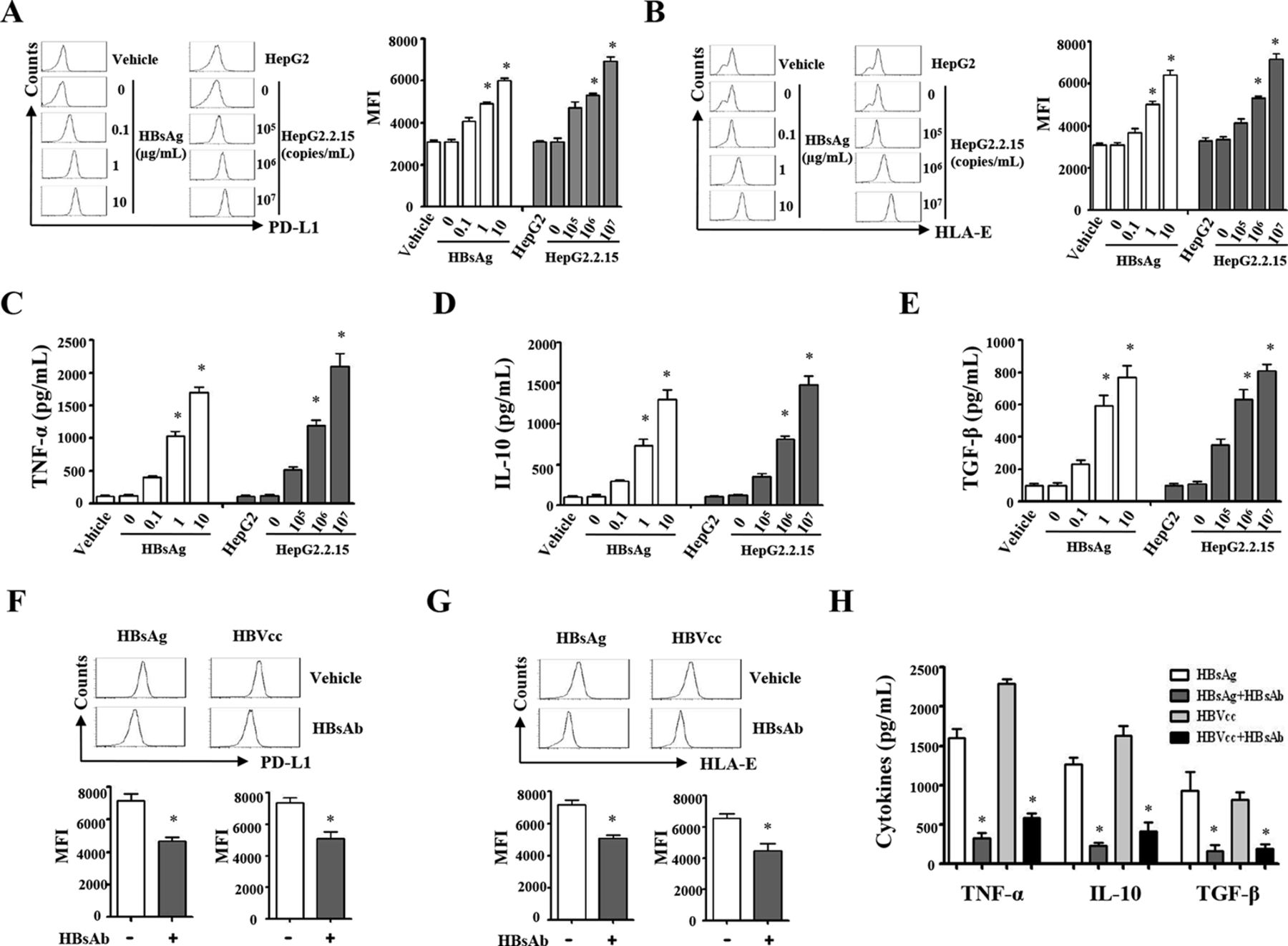
Exposure to HBsAg and HBVcc, monocytes display suppressive phenotypes. Purified monocytes from healthy donors (n=5) were stimulated with HBVcc or HBsAg for 24 hours. Cells and the supernatants were collected for FACS and cytokines detection. (A and B) HBsAg and HBVcc induce a dose-dependent increase expression of PD-L1 and HLA-E. (C–E) HBsAg and HBVcc induce a dose-dependent increase secretion of TNF-α, TGF-β and IL-10. (F and G) Neutralisation of HBsAb inhibits HBsAg-induced and HBVcc-induced the expression of PD-L1 and HLA-E and cytokine production. A representative experiment from five independent experiments is shown. The error bars represent SEM. *p<0.05. FACS, fluorescence activated cell sorter; HBsAb, hepatitis B surface antibody; HBsAg, hepatitis B surface antigen; HBVcc, cell culture of HBV; HLA-E, human leukocyte antigen-E; IL, interleukin; MFI, mean fluorescence intensity; PD-L1, programmed death1-ligand; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-alpha.
HBsAg induced immunosuppressive monocytes via the MyD88/NFκB pathway
We next investigated how HBsAg induces immunosuppressive monocytes. The results of qRT-PCR showed that HBsAg increases the mRNA expression levels of MyD88 and NFκB, compared with medium control (figure 4A, p<0.05). The results of western blotting were shown in figure 4B, HBsAg significantly increased IκB-α phosphorylation (p<0.05) and MyD88 expression (p<0.05). The neutralisation of HBsAg by HBsAb inhibited HBsAg-induced mRNA expression of MyD88 and NFκB, as well as iκB-α phosphorylation and MyD88 expression. ST2825 and BAY 11–7082 were used as inhibitors of MyD88 and NFκB,27 28 respectively. To further test whether HBsAg induced immunosuppressive monocytes via MyD88 and NFκB signalling, ST2825 and BAY 11–7082 were added to monocytes cultured with HBsAg. We observed that the inhibitors of MyD88 and NFκB suppressed the expression levels of HBV-induced PD-L1 and HLA-E (figure 4C, p<0.05). Using ELISA we also found that the inhibitors of MyD88 and NFκB suppressed the secretion levels of HBV-induced TNF-α, IL-10 and TGF-β (figure 4D, p<0.05).

HBsAg induce the suppressive monocytes via MyD88/NFκB pathway. Purified monocytes from healthy donors (n=10) were stimulated with HBsAg for 16 hours. (A) HBsAg-induced messenger RNA expression of MyD88 and NFκB were determined by qRT-PCR. (B) HBsAg-induced MyD88 and NFκB expression were detected by western blotting. (C) BAY 11-7082 (NFκB inhibitor) and ST2825 (MYD88 inhibitor) inhibit HBsAg-induced HLA-E and PD-L1 expression. (D) BAY 11-7082 (NFκB inhibitor) and ST2825 (MYD88 inhibitor) inhibit HBsAg-induced cytokine production. The error bars represent SEM. *p<0.05. HBsAg, hepatitis B surface antigen; IL, interleukin; qRT-PCR, quantitative real-time PCR.
HBV-induced monocytes educate NK cells to secrete IL-10
To further investigate HBV-induced monocytes interactions with NK cells, freshly isolated human PBMCs, PBMCs depleted of CD14+ cells, pure NK cells and monocytes were cultured or cocultured with HBsAg or HBVcc. Cells were stained with CD3 and CD56 for NK cell gating, and IL-10 and IFN-γ expression levels of NK cells were determined by intracellular cytokines staining. The effects of the HBsAg or HBVcc on IL-10 and IFN-γ expression by NK cells are represented in figure 5A, and the statistics were analysed and shown in figure 5B. The IFN-γ and IL-10 expression of NK cells from monocytes-depleted PBMCs were significantly lower than that of NK cells from PBMCs (p<0.05). Pure NK cells failed to respond to both HBsAg and HBVcc, and NK cells cocultured with monocytes responded to HBsAg and HBVcc, expressed much higher level of IL-10 (9.5%±1.3% vs 2.8±1.1%, p<0.05) and a lower level of IFN-γ (3.8%±0.8% vs 6.3±1.0%, p<0.05) than that of separated NK cells and monocytes cocultures in a transwell. These results indicate that both HBsAg and HBVcc induce IFN-γ expression of NK cells and regulatory NK cells (NK-reg) cell differentiation manifested as induced IL-10 expression but that this effect requires monocytes. In NK cell/monocyte direct coculture, HBV induced NK cells to produce much more IL-10 than IFN-γ. However, when NK cells and monocytes were cocultured in a transwell, HBV induced NK cells to produce more IFN-γ than IL-10. This argues that different signals are mediated by direct contact and cytokines, such that cell contact was responsible for NK cells producing IL-10, while cytokine signals promoted NK cells to produce IFN-γ.

HBV-induced monocytes as educator of IL-10 producing NK cells. Freshly isolated human PBMCs, PBMCs depleted of CD14+ cells, NK cells and monocytes from healthy control were cultured or cocultured with HBsAg (10 μg/mL) or HBVcc (107 copies/mL) for 24 hours. NK cells were identified by CD3 and CD56 staining, and the expression of IL-10 and IFN-γ were examined by intracellular cytokine staining. (A) A representative experiment from five independent experiments is shown. (B) Statistical analysis for the expression of IL-10 and IFN-γ on NK cells. The error bars represent SEM. *p<0.05. HBsAg, hepatitis B surface antigen; HBVcc, cell culture of HBV; IL, interleukin; NK, natural killer; PBMCs, peripheral blood mononuclear cells.
HBV-induced IL-10 producing NK cells through monocytes is mediated by PD-L1/PD-1 and HLA-E/CD94
To directly investigate the monocytes signals that educate NK cells to produce IL-10, antibodies of PD-L1, PD-1, HLA-E, CD94, IL-10, TNF-α and TGF-β were added to monocyte/NK cell cocultures. A representative experiment was shown in figure 6A, and the statistical analysis was shown in figure 6B. We observed that the blockade of PD-L1/PD-1 and HLA-E/CD94 significantly inhibited the IL-10 expression of NK cells but increased the IFN-γ expression of NK cells (p<0.05). The neutralisation of IL-10, TNF-α and TGF-β had no significant effect on NK cell activation (online supplementary figure 2, p>0.05). These results suggest that HBV-induced PD-L1, HLA-E on monocytes was interacting with their ligands PD-1 and CD94 on NK cells to educate IL-10+ regulatory NK cell differentiation.
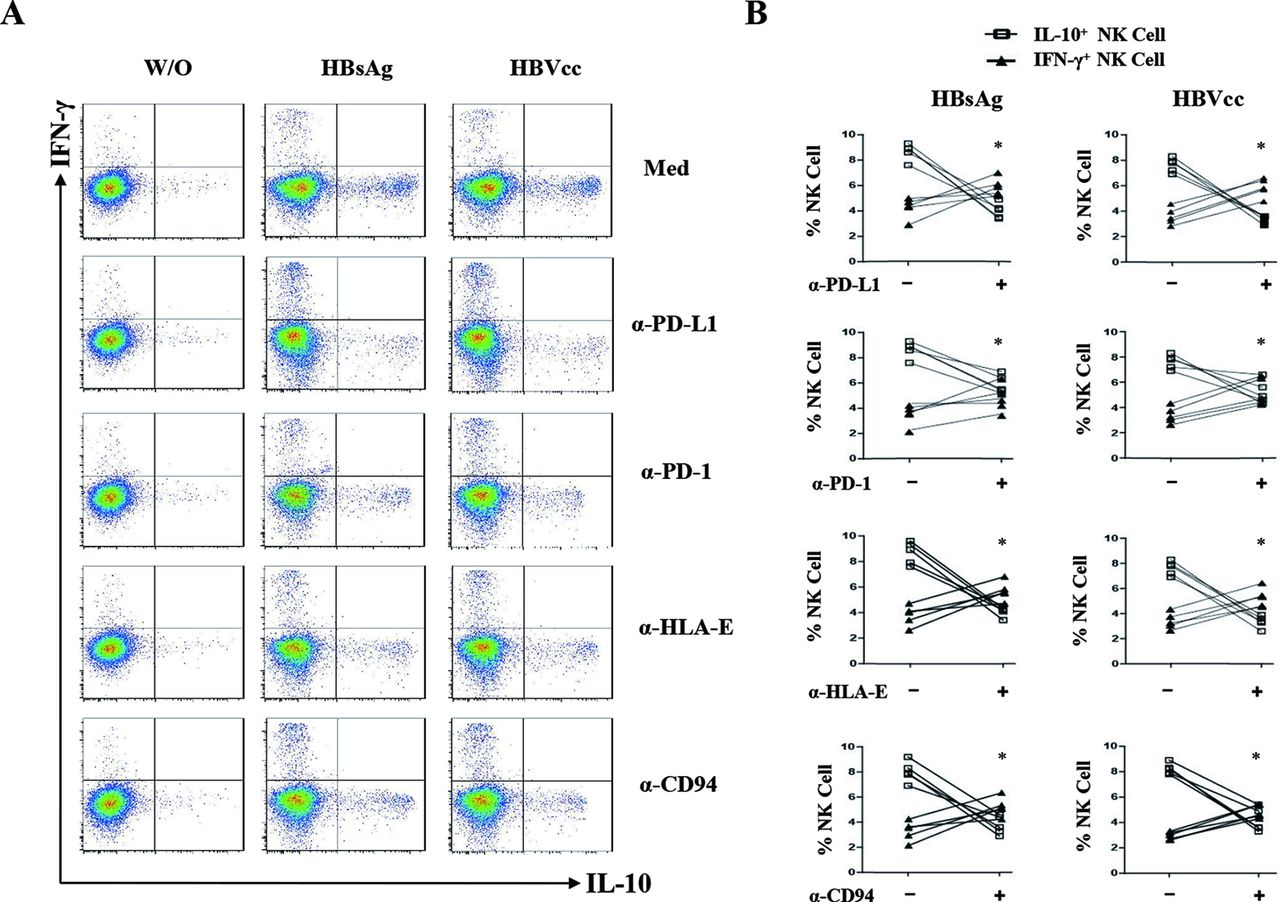
HBsAg-induced and HBVcc-induced NK-reg differentiation through monocytes is mediated by PD-L1/PD-1, HLA-E/CD94. Purified monocytes and NK cells were cocultured for 24 hours with HBsAg (10 μg/mL) or HBVcc (107 copies/mL) in presence of PD-L1, PD-1, HLA-E, CD94, IL-10, TNF-α and TGF-β antibodies. NK cells were identified by CD3 and CD56 staining, and the expression of IL-10 and IFN-γ were examined by intracellular cytokine staining. (A) A representative experiment from five independent experiments is shown. (B) Statistical analysis for the expression of IL-10 and IFN-γ on NK cells. The error bars represent SEM. *p<0.05. HBsAg, hepatitis B surface antigen; HBVcc, cell culture of HBV; IL, interleukin; NK, natural killer; NK-reg, regulatory NK cell; TGF-β, transforming growth factor-β.
HBV-induced NK-reg cells inhibit autologous CD4+ and CD8+ T cell activation via IL-10
We next investigated whether HBV-induced NK-reg cells inhibited T cell activation. The CFSE-labelled autologous T cells were cocultured with NK cells, which were repurified from monocyte/NK cell coculture with or without HBsAg/HBVcc, and stimulated with PHA in presence or absence of anti-IL-10 antibody. CD4+ and CD8+ T cell proliferation was examined by flow cytometry. The supernatants of NK/T cell cocultures were collected to measure IFN-γ by ELISA. We observed that both HBsAg-induced and HBVcc-induced NK cells via monocytes significantly suppressed the proliferation of CD4+ T cells and CD8+ T cells (figure 7A–C, p<0.05), and IFN-γ production (figure 7D, p<0.05), compared with control. When IL-10 antibody was added to NK/T cells coculture, we observed that neutralisation of IL-10 restored the proliferation of CD4+ T cells and CD8+ T cell (p<0.05), as well as IFN-γ secretion (p<0.05). These results indicate HBV-induced monocytes programme NK cells to inhibit autologous CD4+ and CD8+ T cell activation in an IL-10 dependent manner.
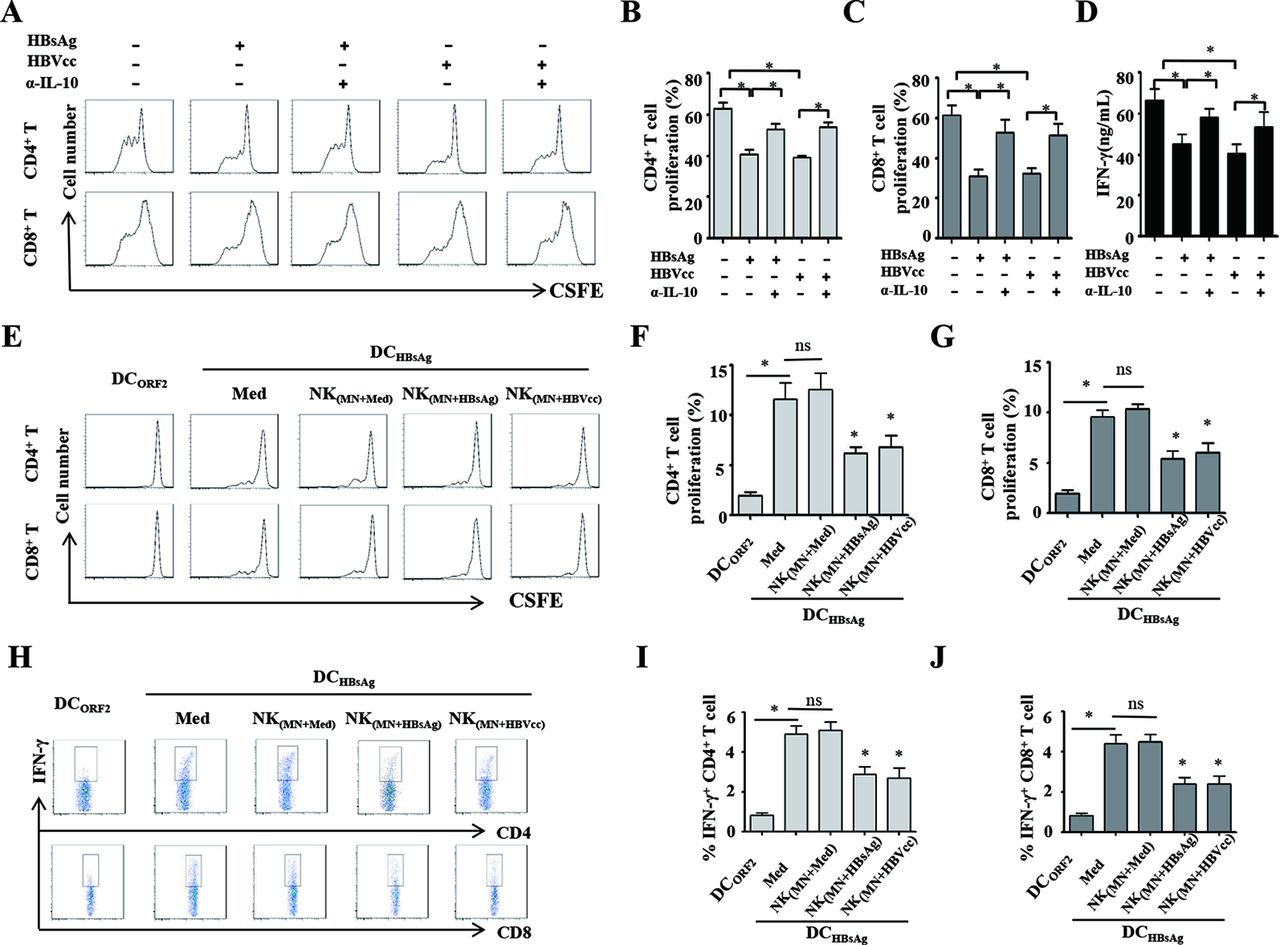
HBV-induced NK cells via monocytes inhibit autologous CD4+ and CD8+ T cells activation in IL-10 dependent manner. NK cells repurified from monocytes/NK cells coculture with or without HBsAg/HBVcc were cocultured with CFSE-labelled autologous T cells and stimulated with PHA in presence of anti IL-10 antibody. (A–C) CD4+ and CD8+ T cell proliferation was examined by flow cytometry. (D) The supernatants of NK/T cell coculture were collected for IFN-γ detection by ELISA. To investigate the regulatory effect of NK cells on HBV-specific T cell response, monocytes, NK cells and T cells were purified from patients with CHB. T cells were cocultured with autologous DCs loaded with either HBsAg or HEV-ORF2 protein in the presence of NK cells, which were repurified from monocyte/NK cell coculture with or without HBsAg/HBVcc. (E–G) CD4+ and CD8+ T cell proliferation was examined by flow cytometry. (H–J) IFN-γ expression of CD4+ and CD8+ T cells were examined by intracellular cytokine staining. Shown is one of five representative experiments. The bars represent the SEM. *p<0.05. CHB, chronic HBV infection; DCs, dendritic cells; HBsAg, hepatitis B surface antigen; HBVcc, cell culture of HBV; HEV-ORF2, hepatitis E virus open reading frame 2; IL, interleukin; NK, natural killer.
To further investigate the regulatory effect of NK cells on HBV-specific T cell response, we purified monocytes, NK cells and T cells from patients with CHB patients and generated DCs from monocytes. T cells were cocultured with autologous DCs loaded with either HBsAg or HEV-ORF2 protein (negative control), and T cell proliferation and IFN-γ production were examined by flow cytometry and intracellular staining. We observed that HBsAg-loaded DCs obviously induce HBsAg-specific proliferation and IFN-γ production by CD4+ and CD8+ T cells. While we added the autologous NK cells, which were repurified from monocyte/NK cell coculture with or without HBsAg/HBVcc, into T cell/DCs coculture, the results showed that both HBsAg-induced and HBVcc-induced NK cells via monocytes significantly suppress HBsAg-specific proliferation (figure 7E–G, p<0.05) and IFN-γ production (figure 7H–J, p<0.05) by CD4+ and CD8+ T cells.
Discussion
The course of HBV infection is complicated. Three phases of CHB are now widely accepted: an immune tolerant phase, an immune active phase and an inactive phase.29 Immune tolerance is a serious problem in CHB carriers, who are at high risk of developing cirrhosis and HCC later in life.30 NK cells and monocytes are two major components of innate immunity. HBV persistent infection is thought to result from inefficiencies of innate and adaptive immune responses. However, the roles of monocytes and NK cells in HBV persistent infection have not been well understood. In the present study, we found that monocytes from patients with CHB expressed higher levels of suppressive cell surface molecules (PD-L1 and HLA-E) and anti-inflammatory cytokines (IL-10 and TGF-β), compared with healthy individuals. On LPS stimulation, monocytes from patients with CHB expressed and secreted much more TNF-α, IL-10 and TGF-β compared with healthy controls (figure 1). These findings suggest that in patients with CHB, monocytes display markers of chronic activation (PD-L1, HLA-E, TNF-α, IL-10 and TGF-β), consistent with a role in perpetuating inflammation and/or inhibiting immunity. Recently, myeloid-derived suppressor cells (MDSCs) were initially defined as CD14+ monocytes to suppress lymphocyte activation. A higher percentage of monocytic MDSCs (mMDSCs) has been detected in the peripheral blood of patients with CHB.31 A correlation between the frequencies of mMDSCs and the level of HBsAg in the sera of patients with CHB was observed, and this correlation was confirmed in HBsAg-stimulated human PBMCs and in HBV mouse models.32 33 Granulocytic MDSCs have also been reported to expand in CHB and inhibit T cell activation in a partially arginase-dependent manner.34
In CHB, NK cells exhibited selective defects of antiviral function, including a conserved or enhanced cytolytic activity and a diminished cytokine production that may contribute to both immunopathology and viral persistence.35 36 In this study, we observed that the expression levels of CD94 and PD-1 on NK cells from patients with CHB were significantly higher than in healthy controls. CD94 is an inhibitory receptor that suppresses NK cell activation by engagement with HLA-E,37 and PD-1 binding PD-L1 to induces NK cell exhaustion.38 We also found higher mRNA expression levels of TGF-β and IL-10, lower expression of IL-12, IL-18 and T-bet in NK cells from the patients with CHB. Furthermore, purified NK cells from patients with CHB express and produce more IL-10 on PHA stimulation, compared with healthy control (figure 2). Our results indicate that NK cells reduced hallmarks of the type 1 response (less T-bet, IL-12 and IL-18 message) but increased RNA and protein level of IL-10. Consistent with our findings, it has been reported that the expression of inhibitory receptors such as NKG2A is elevated, while activating receptors, CD16 and NKp30 are downregulated on NK cells in CHB, and this correlates with serum HBV DNA load.15 39 Active patients with CHB exhibited increased NKG2A expression on NK cells, and the NKG2A upregulation functionally inhibited NK cells, as NKG2A blockade markedly enhanced cytotoxicity in NK cells from active patients with CHB.40
The role of HBsAg in monocytes remains controversial. HBsAg can selectively inhibit TLR2 ligand-induced IL-12 production in monocytes/macrophages by blocking the JNK-MAPK pathway.6 Others reported that HBsAg has an immunostimulatory effect by inducing TNF-α and IL-10 production.41 In this study, using monocytes from healthy donors stimulated with HBsAg or HBVcc in vitro, we discovered that HBV employed HBsAg to induce monocyte PD-L1 and HLA-E expression, and anti-inflammatory cytokine (IL-10 and TGF-β) secretion (figure 3). This result shows that the application of either HBV or HBsAg reproduces the effects of in vivo HBV infection, in terms of TNF-α, IL-10, TGF-β, HLA-E and PD-L1. We further observed that HBsAg increases message of NFκB and MyD88 and that the effect of HBsAg on monocytes is blocked by either NFκB or MyD88 inhibitors (figure 4). It indicates that HBsAg induces immunosuppressive monocytes via the MyD88/NFκB signalling pathway. It has been reported that HBsAg induces monocytes towards mMDSCs in an ERK/IL-6/STAT3 signalling-dependent manner.32 These findings suggest multiple signalling pathways are involved in the response of monocytes to HBsAg.
Activated monocytes interact with lymphocytes in part through cell surface molecules.42 Our previous study has addressed the impact of TLR signalling on the crosstalk between NK cells and Kupffer cells and revealed how the liver displayed a downregulated inflammatory response to constitutive bacterial elements through the secretion of IL-10 yet retains a vigorous response to viral challenge.43 IL-12 and TNF-α released by activated monocytes and macrophages were identified as powerful inducers of IFN-γ by NK cells.44 The immune regulation functions of NK cells have been a focus of recent attention. With appropriate stimulation, human and mouse NK cells have been observed to produce the immune regulatory cytokine IL-10.45 In this study, we used a monocytes/NK cells coculture system and found that HBV-induced IL-10-producing NK cells are mediated by monocytes in a cell–cell contact dependent manner (figure 5). We further identified that the critical signalling molecules of HBV-induced IL-10-producing NK cells are PD-L1/PD-1 and HLA-E/CD94 (figure 6), which are expressed on monocytes and NK cells, respectively. PD-L1 and PD-1 are coinhibitory molecules that mediate T cell dysfunction in HBV and other chronic viral infections, and inhibition of this interaction restores T cell function.46 47 PD-L1/PD1 and HLA-E/CD94 interaction were also reported in the context of tumour immune escape.36 HBV-induced pDCs and KCs alter NK cell function in CHB.48–50
NK cells act as killers towards tumour or virus-infected cells and as regulatory cells to affect the adaptive immune response.51 52 NK-reg expressing intracellular TGF-β and IL-10 but no IFN-γ expression were first defined in a tumour escape study. Tumour-infiltrating NK-reg cells can mediate tumour immune escape or direct killing.13 Recent studies showed that decidual NK-reg cells can mediate maternal-fetal immune regulation or vascular remodelling, while liver NK-reg cells can mediate immune tolerance or immune injury.53 NK-reg cells inhibited antigen-induced proliferation and IFN-γ production by T cells.54 In this study, we observed that HBV-treated monocytes induced IL-10 and IFN-γ expression in NK cells, and in monocytes in direct coculture, blockade of PD-L1/PD-1 and HLA-E/CD94 significantly inhibited the IL-10 expression and increased IFN-γ in NK cells (figures 5 and 6). The inhibition of IFN-γ production by IL-10+ NK-reg cells has been clarified as previous studies.55 NK cells have been assumed to participate in adaptive immune responses by an indirect mechanism that involves their secretion of cytokines and chemokines.56 NK cells enhance the function of CD4+ T cells and promote the differentiation of Th1 cells by secreting IFN-γ.57 In the present study, we found that the HBV-induced NK cells via monocytes inhibit autologous T cell activation in IL-10 dependent manner as well as HBV-specific T cell response (figure 7), suggesting that HBV-induced NK-reg cells via monocytes directly inhibit the antiviral effect of NK cells and downregulate T cell immune response. It has been reported that in vitro removal of NK cells augments circulating CD8+ T cell responses directed against HBV but not against well-controlled viruses in patients with CHB.58
To the best of our knowledge, this is the first description of an immunosuppressive cascade in which NK-reg cells mediate between HBV-exposed monocytes and T cells as shown in figure 8. Future investigation needs to address how to manipulate the function of these NK-reg cells in the clinical setting. Possible manipulation strategies to enhance immune responses may include altering NK-reg cell activity by means of monocytes depletion, PD-L1/PD-1 and HLA-E/CD94 blockade as well as blocking immunosuppressive cytokine function.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic outline of HBV initiates an immunosuppressive cascade. Consistent with the experimental data, HBV employ HBsAg to induce suppressive monocytes, which express inhibitory molecules PD-L1 and HLA-E, secreting IL-10, TNF-α and TGF-β through MyD88/NFκB signalling. Engagement of PD-L1/PD-1 and HLA-E/CD94 educates IL-10-producing regulatory NK cells, which inhibit autologous NK cell activation, as well as CD4+ and CD8+ T cells activation in IL-10 dependent manner. HBsAg, hepatitis B surface antigen; IL, interleukin; NK, natural killer; TGF-β, transforming growth factor-β.
Acknowledgments
none
References
Footnotes
Original reference: none
no
Contributors HL planned and performed the experiments, analysed the data, wrote the paper. NZ, HS, YY, AC and TL performed the experiments. ZW, GW and JN contributed reagents and materials. INC and LS interpreted and edited the manuscript. ZT designed, interpreted and funded the study and wrote the manuscript.
Funding This work was supported in part by National Natural Science Foundation of China (81373143 and 81571535 to ZT) and by grants from the Jilin Provincial Natural Science Foundation of China (No. 20140520014JH to HL).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The IRB of Jilin University, The First Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.