Article Text

Abstract
Objective There are few studies which characterised the molecular alterations in premalignant colorectal adenomas. Our major goal was to establish colorectal adenoma genome atlas and identify molecular markers of progression from colorectal adenoma to adenocarcinoma.
Design Whole-exome sequencing and targeted sequencing were carried out in 149 adenoma samples and paired blood from patients with conventional adenoma or sessile serrated adenoma to characterise the somatic mutation landscape for premalignant colorectal lesions. The identified somatic mutations were compared with those in colorectal cancer (CRC) samples from The Cancer Genome Atlas. A supervised random forest model was employed to identify gene panels differentiating adenoma from CRC.
Results Similar somatic mutation frequencies, but distinctive driver mutations, were observed in sessile serrated adenomas and conventional adenomas. The final model included 20 genes and was able to separate the somatic mutation profile of colorectal adenoma and adenocarcinoma with an area under the curve of 0.941.
Conclusion The findings of this project hold potential to better identify patients with adenoma who may be candidates for targeted surveillance programmes and preventive interventions to reduce the incidence of CRC.
- colorectal adenoma
- somatic mutation
- sessile serrated adenoma
- conventional adenoma
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Large-scale sequencing of colorectal cancer has revealed potential driver genes.
Conventional adenoma may progress to adenocarcinoma.
Sessile serrated adenoma also contributes to adenocarcinoma development via different molecular mechanisms.
What are the new findings?
Conventional adenoma and sessile serrated adenoma had similar mutation frequencies, but the genes involved substantially differed.
Both novel and known colorectal cancer-related mutations with driver patterns were observed in adenomas.
A 20-gene panel can distinguish colorectal adenoma from adenocarcinoma.
How might it impact on clinical practice in the foreseeable future?
The identified novel driver mutations for conventional adenoma and sessile serrated adenoma could be targets to guide early diagnosis and prevention of colorectal cancer.
Introduction
Cancer is a progressive disease that results from the accumulation of genetic and molecular changes over many years. Many cancers were detected and treated at an advanced stage using chemotherapy and radiation with disappointing results. In order to avoid such outcome, the better strategy is to detect cancer as it develops, at its earliest stages, because it allows for a preventive intervention to stop or even reverse the process of tumourigenesis.
Most epithelial cancers are preceded by premalignant lesions. Therefore, detection and removal of premalignant lesions has become one of the most commonly used preventive measures against tumour progression. This paradigm is especially true for colorectal cancer (CRC). Removal of premalignant lesions via colonoscopy has long been the gold standard of CRC prevention. However, colonoscopy is a financial burden and a source of complications and discomfort to patients. Previous literature has suggested that about 25% of asymptotic and average-risk patients develop colon adenomas,1 a precursor to CRC. But the annual rate of transition to CRC was between 2.5% and 5.6% even for advanced adenomas.2 Despite these relatively low rates of transition to CRC, each colonoscopy with polypectomy could cost as high as $846, and treatment for complications associated with colonoscopy could cost from $320 to $12 446 in 2009.3 One analysis showed that routine use of colonoscopy screening generates its largest cost to the healthcare system when adenomas are found at baseline colonoscopy because these patients are subject to future surveillance colonoscopies. Moreover, current guidelines for surveillance colonoscopy are based on empirically generated descriptors that are imprecise.4 Thus, methods of improving risk stratification based on the molecular signature of the adenoma are highly desirable. For individuals with low risk of adenoma progression to CRC, a less frequent screening strategy could be applied to reduce the economic and physical burden.
Previously, large-scale sequencing projects in colorectal tumour tissues have advanced the understanding of CRC pathology.5 An increasing body of evidence has indicated that certain driver mutations can also be identified in benign and premalignant conditions.6 To establish colorectal adenoma genome atlas, identify molecular signatures, and create prediction model to predict malignant progression of colorectal adenomas, we carried out whole-exome sequencing (WES) and targeted sequencing (TS) and compared these sequencing data with those available from The Cancer Genome Atlas (TCGA).5
We hypothesised that mutation profiles of a limited number of genes could clearly classify conventional adenoma (CNAD) and CRC. In order to increase the statistical power of our analysis, we pooled the WES and our own TS data for CNAD and compared it with the CRC WES data from TCGA. Because we selected genes of interest using TS, popular methods for determining mutation significance that depend on estimates of the background mutation rates for neighbouring genes could not be applied. Therefore, we chose to determine the significance of mutations based on clustering patterns as suggested previously by Vogelstein et al 7 and implemented by Van den Eynden et al.8 This method was based on the observation that tumour suppressor genes often have loss-of-function truncation mutations (non-sense, splice site, and non-stop SNVs and frameshift indels) without particular hot spots, whereas oncogenes often have gain-of-function mutations that modify crucial protein domains through substitution of single amino acids (missense SNVs and in-frame indels).
In addition to the hypothesis about differentiating colorectal adenoma and adenocarcinoma merely based on somatic mutations, we also hypothesise that certain mutations might be more prevalent in adenomas than in cancers due to differences in microenvironments among different stages of tumourigenesis. Our investigation of somatic mutations in premalignant lesions will provide a deeper understanding of the tumourigenic process in CRC and reveal potential targets for surveillance and prevention.
Materials and methods
Study population
Study participants were recruited from The University of Texas MD Anderson Cancer Center between 2010 and 2014 during their routine colonoscopy as part of the Premalignant Genome Atlas project. A written informed consent was obtained prior to participation for each participant. There were no age, gender and ethnicity restrictions; however, patients with a diagnosis of Lynch syndrome were excluded. After consenting, each participant provided samples of blood, polyp and adjacent normal tissue samples if polyps were found during colonoscopy. When a lesion or polyp was removed, a portion of it was flash-frozen and hand-delivered to our laboratory for storage. Each sample was labelled with a unique study identification number.
Epidemiological data were collected from a standard questionnaire through in-person interview by trained MD Anderson staff interviewers, and clinical and pathological data were abstracted via medical chart review to confirm the diagnosis. Adenoma was considered advanced if (1) its diameter was larger than 1 cm; (2) high-grade dysplasia was reported; or (3) it had a significant villous component.2
WES and TS
This article presents the results from two independent projects. One was conducted to characterise previously reported cancer driver genes in 100 pairs of CNAD samples and corresponding blood; the other was an exploratory WES project performed to identify novel driver mutations in 35 CNAD and 14 sessile serrated adenoma (SSA) samples and their corresponding blood samples. Probability to detect mutations with various population prevalence has been shown in online supplementary figure 1. DNA was isolated from both blood and tissue samples and sent to the Baylor College of Medicine Human Genome Sequencing Center for sequencing using a HiSeq2000 system (Illumina, San Diego, California). The median depth for WES was 84× to 160×; for TS, the median depth was from 83× to 199×. Details of the experimental protocols and analytical methods can be found in the online supplementary materials and methods and supplementary table 1. This study also included TCGA WES data on paired tumour tissues and blood from patients with colorectal adenocarcinoma for comparison. We downloaded WES data from the Cancer Genomics Hub for 460 patients, including 330 colon and 130 rectal adenocarcinomas.9 Of the 460 pairs included in the WES data, 378 passed quality control (274 colon cancers and 104 rectal cancers).
Identification of mutation signature for adenoma and CRC
The Student’s t-test was used to compare the mutation rates in adenomas and CRC. The Fisher’s exact test was employed to determine differences in mutation prevalence among patient samples with different pathological and clinical features. Classification and regression tree analysis was performed to discover differentially mutated genes in SSA and CNAD samples.10 Supervised learning with random forest and permutation tests for variable importance was performed using the randomforest11 and rfPermute12 packages on a pooled data set containing both our adenoma and the TCGA CRC data sets. Permutation of the random forest class labels was performed for 1000 iterations to provide a better estimate of variable importance than classic random forest model. A reduced model was constructed using important variables identified in the random forest analysis with a permuted p value <0.05. For the pooled analysis of CRC and adenoma, we further filtered the mutations using VCRome (Roche, Pleasanton, California) and custom panel probes to ensure similar coverage of variants.
Results
Patient characteristics and sequencing metrics
Among the 135 CNADs included in the present study, 30 were advanced lesions, and 104 were non-advanced lesions. We were not able to determine the classification of one CNAD owing to missing information. The stage distribution of CRC was as follows: 20% stage I, 39% stage II, 28% stage III and 13% stage IV. The mean age of the patients with CNAD was 59.3 years, and 43.6% of the patients overall were women. The patient characteristics of the two cohorts are detailed in online supplementary tables 2 and 3.
For WES in CNAD samples, 72.27%–78.13% of reads were mapped to VCRome targets, and the median coverage was between 84x and 160×. Whereas the adenoma samples had an average non-silent somatic mutation rate of 1.6 per million bases (Mb), the average rate in the TCGA CRC data set was significantly higher (10.6/Mb, p=7.47×10−15). Based on mutation rate, MLH1 and microsatellite instability status, we considered the 66 (17.5%) cases with the highest non-silent mutation rates as hypermutaters (patients with unusual frequency of mutations due to defect in DNA repair mechanisms), resulting in a cut-off non-silent mutation rate of 11.6/Mb (figure 1). Limiting the samples to 312 (82.5%) non-hypermutaters brought the non-silent mutation rate down to 4.6/Mb and produced an even more significant distinction between CRC and adenoma samples than the rate with hypermutater (p<2.2×10−16).
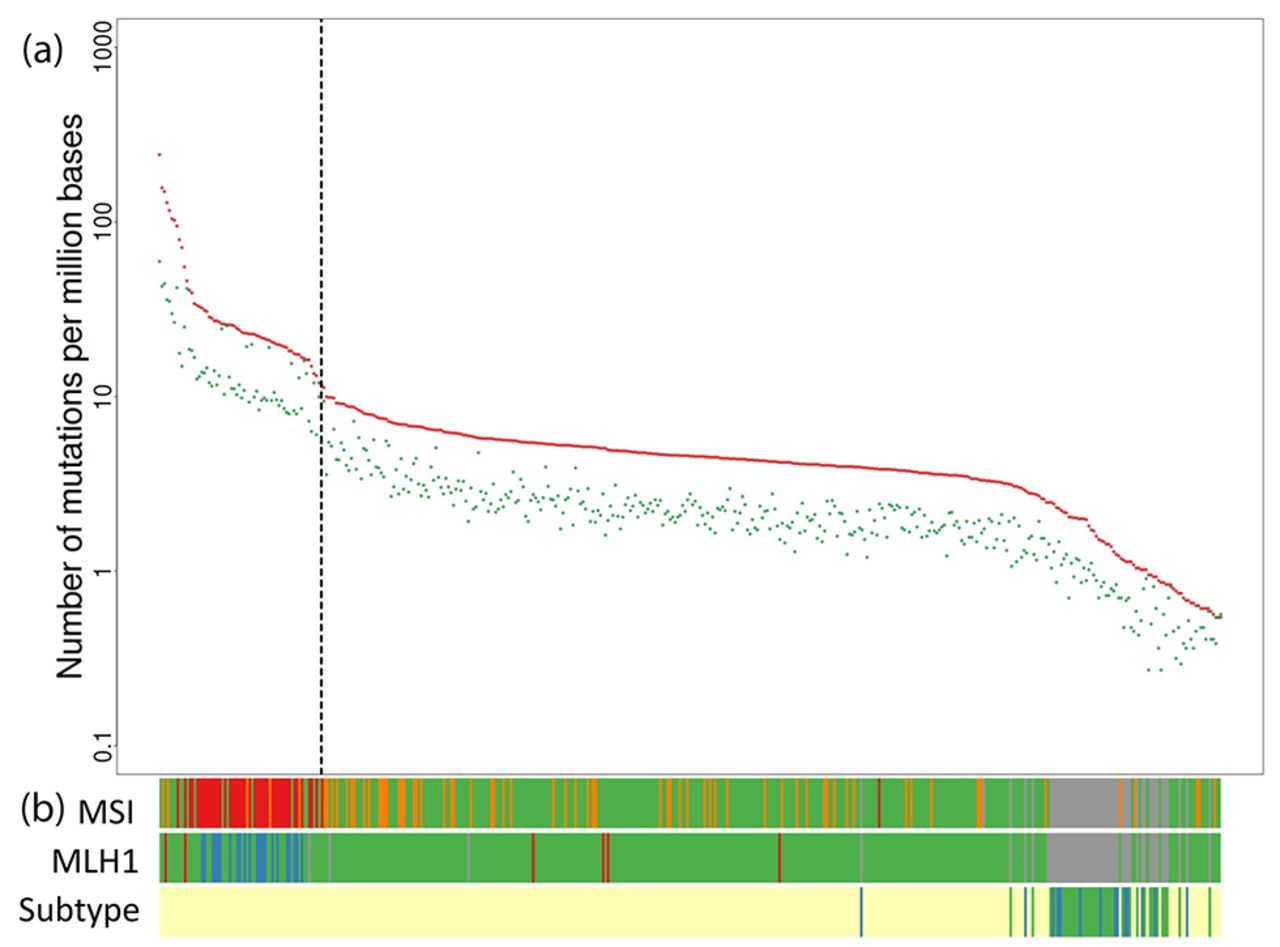
Frequency of somatic mutations in patients with colorectal cancer or adenoma. (A) Number of mutations per million base pairs: red, non-silent mutations; green, silent mutations. (B) Microsatellite instability status (MSI): red, MSI high; orange, MSI low; green, microsatellite stable; grey, data not available. MLH1 expression (MLH1): red, upregulation; blue, downregulation; green, normal; grey, data not available. Pathological subtypes (Subtype): yellow, colorectal cancer; blue, sessile serrated adenoma; green, conventional adenoma.
No significant differences in the somatic mutation rate were observed for CNADs and SSAs (1.5 and 1.7/Mb, respectively; p=0.470). We also found no difference in the non-silent mutation rates in non-advanced and advanced adenomas (1.6 and 2.0/Mb, respectively; p=0.304). In the following sections, we focus on non-silent somatic mutations with mutation patterns fitting those for oncogenes and tumour suppressor genes as determined using the R package SomInaClust.8
Discovery of potential somatic driver mutations in CNAD and SSA using WES data
For CNAD, four genes (catenin beta 1 (CTNNB1); keratin-associated protein 4–5 (KRTAP4-5); golgin A8 family member B (GOLGA8B); and transmembrane protease, serine 13 (TMPRSS13)) had the oncogene pattern. The gene APC, WNT signalling pathway regulator (APC) was the only gene fitting the tumour suppressor gene pattern, with a q value <0.05 (figure 2A). The most frequently non-silently mutated gene was APC, which was mutated in 16 (45.7%) patients, all 16 of these patients carried at least one truncating mutation (non-sense, frameshift, splice site or non-stop mutations). KRTAP4-5 was mutated in six CNAD samples with all mutations being missense and located at a known single-nucleotide variant (SNV) site (rs411367). CTNNB1 mutations were also exclusively missense mutations, and two of these mutations were at SNV rs121913409, which has been reported to be pathogenic in liver cancer.13 Two out of 3 GOLGA8B-mutated CNADs carried a missense SNV (rs200544945), and two out of five missense mutations in TMPRSS13 were found at a rare SNV, rs61900347. A closer examination of 96 CRCs with mutation frequencies less than or equal to that of adenomas demonstrated that three of the five CNAD driver genes (KRTAP4-5 and TMPRSS13 and APC) showed driver patterns as well while two genes (CTNNB1 and GOLGA8B) did not reach statistical significance as driver genes possibly due to smaller sample size (online supplementary figure 2). Among the 61 genes with statistically significant (q<0.05) driver pattern in non-hypermutater CRCs, 13 were significantly more prevalent in non-hypermutater CRCs compared with CNAD as shown in online supplementary table 4. However, only 4 of the 61 driver genes (TP53, NHEDC1, PIK3CA and KRAS) showed significant differences in prevalence between CNAD and the low mutation frequency CRC indicating that this subgroup had mutation profile more similar to that of CNAD. The result seemed to suggest that the four aforementioned genes could drive CNAD progression to malignancy despite the similarity in mutation profiles.

Frequently mutated genes with driver patterns demonstrated by WES and TS in adenomas. (A) CNAD WES. (B) SSA WES. (C) CNAD TS. (D) CNAD WES and TS. Driver gene q value: false-discovery rate of driver gene probability. Genes in panels (A) and (B), p<0.05; genes in panels (C) and (D), q<0.1. CNAD, conventional adenoma; OG, oncogene; SSA, sessile serrated adenoma; TS, targeted sequencing; TSG, tumour suppressor gene; UTR, untranslated region; WES, whole-exome sequencing.
For SSA, one potential driver gene was found with the oncogene pattern: B-Raf proto-oncogene, serine/threonine kinase (BRAF) (figure 2B). Seven patients had somatic mutations at the known pathogenic locus V600E, which was enriched in both SSAs and hypermutaters. Although KRTAP4-5 did not have a statistically significant mutation pattern of driver genes after adjusting for multiple comparison, possibly because of the small sample size, all three KRTAP4-5 mutations in SSAs were located at rs411367, as in CNADs, suggesting that the driver mutation in CNAD could also be a driver in SSA. Because of the design of the targeted panel, we were unable to validate the prevalence of KRTAP4-5 mutations. However, our WES suggested KRTAP4-5 is functional in CRC tumourigenesis in both CNAD and SSA, which warrants further investigation.
Classifiers between CNAD and CRC identify potential driver genes
Profiles of the mutations identified by TS alone and combining TS and WES in CNADs were shown in figure 2C,D. With the larger sample size available for this step, we discovered three genes with driver mutation patterns—KRAS, F-box and WD repeat domain containing 7 (FBXW7), and SRY-box 9 (SOX9)—in addition to those implicated in WES. Among the three additional genes, KRAS followed the oncogene pattern, with missense mutations at the two known pathological SNVs, rs12191352914 and rs121913530.15 16 Both FBXW7 and SOX9 displayed the mutation patterns of tumour suppressor genes. With the larger sample size, we also discovered that two recurrent mutations of CTNNB1, at rs121913412 and rs121913409, were enriched in CNAD. These SNVs have been suggested to be pathogenic in other types of cancer.13 17 18
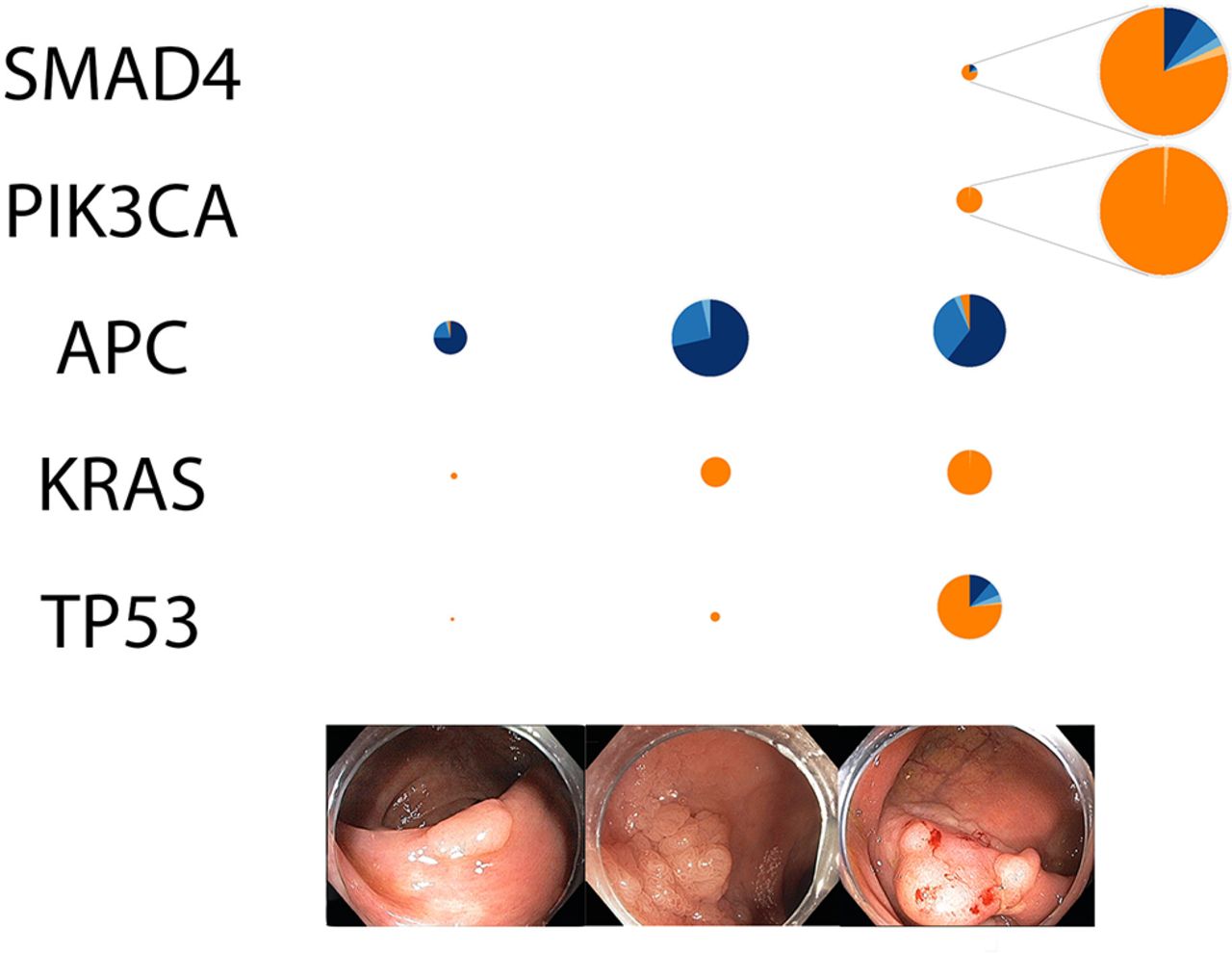
We used a trend test to identify genes with a consistent trend in mutation prevalence among non-advanced CNAD, advanced CNAD and CRC. We found five genes—TP53, PIK3CA, KRAS, APC and SMAD family member 4 (SMAD4)—had a statistically significant trend of mutation prevalence towards CRC after adjusting for multiple comparison (figure 3).19 We observed differences in the composition of non-silent mutations. For instance, TP53 mutations in CNADs were exclusively missense, whereas one quarter of the TP53 mutations in CRCs were truncating mutations. Upon taking a closer look into the missense mutations in CNADs, we found two sites, rs28934578 (R175H) and rs28934576 (R273H), that were recurrently mutated in non-hypermutater (26 at R175H and 9 at R273H) and were known to modify the conformation or the DNA-binding domain of TP53. 20

Prevalence and composition of non-silent mutations in genes displaying a significant trend with disease progression. Left: non-advanced adenoma. Middle: advanced adenoma. Right: colorectal cancer. The diameters of the pie charts indicate relative prevalence of non-silent mutations. Colour: mutation type. All genes had trend test q<0.1.
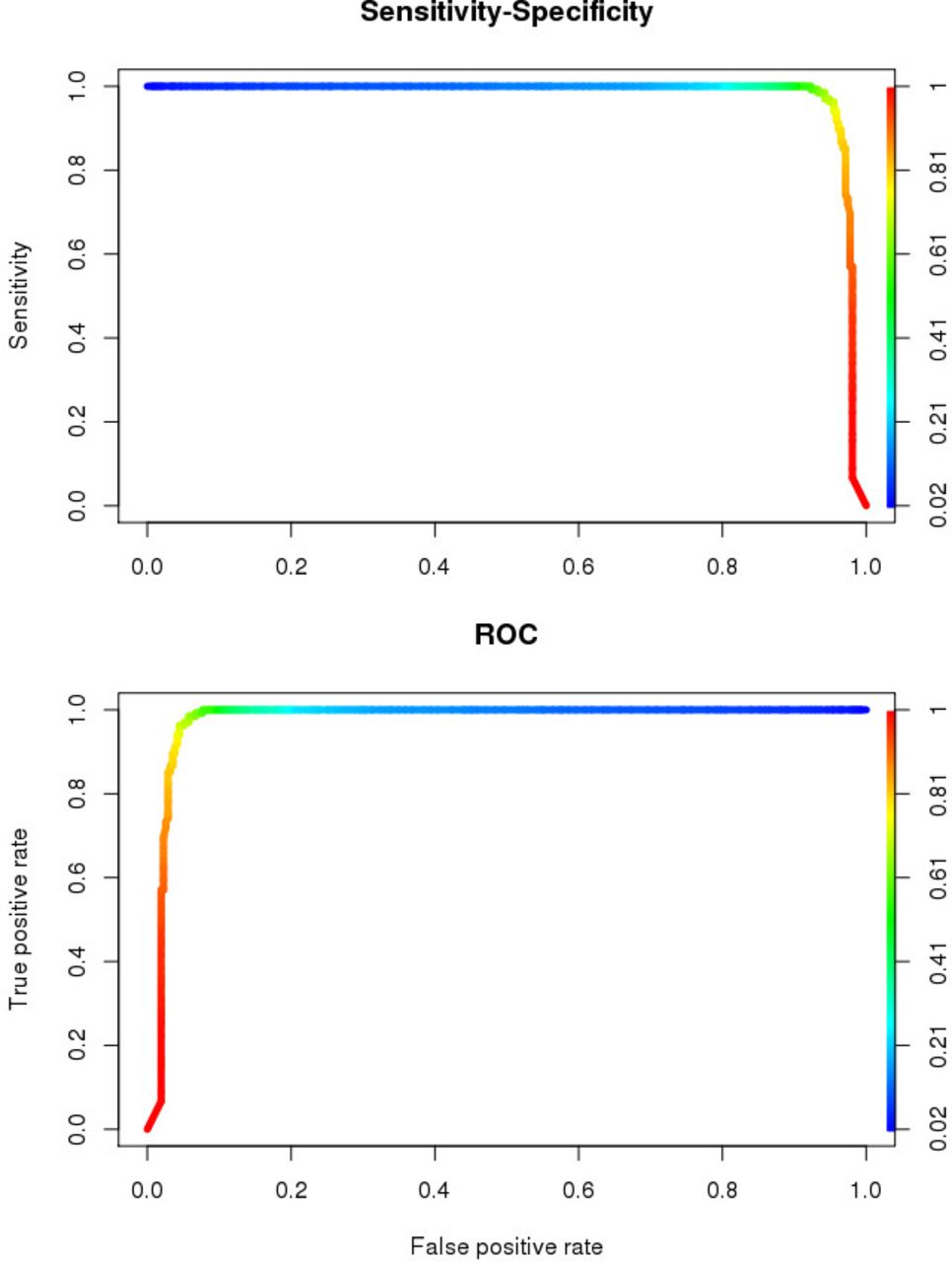
To identify the most important genes that significantly differentiate between 135 CNADs and 312 non-hypermutater CRCs, we performed permuted random forest and identified 20 significantly important genes: APC, ATM serine/threonine kinase (ATM), cell division cycle 27 (CDC27), CUB and Sushi multiple domains 1 (CSMD1), CUB and Sushi multiple domains 3 (CSMD3), CTNNB1, FAT atypical cadherin 4 (FAT4), FBXW7, KRAS, LDL receptor-related protein 1B (LRP1B), mediator complex subunit 12 (MED12), sodium leak channel, non-selective (NALCN), neuroblastoma RAS viral oncogene homologue (NRAS), PIK3CA, ryanodine receptor 3 (RYR3), SMAD4, SOX9, spectrin repeat containing nuclear envelope protein 1 (SYNE1), TMPRSS13 and TP53. We then used these genes to create a classifier which differentiates samples into adenomas or CRC. The area under the receiver operating characteristic curve of our classifier was 0.941, and the error rate was 14.54% (figure 4). The five genes with significant consistent trend mentioned above were in this gene panel (online supplementary table 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prediction model for malignant progression from conventional adenoma to non-hypermutated colorectal cancer. ROC, receiver operating characteristic.
Discussion
In this study, we identified potential driver mutations of adenomas via WES and profiled the identified driver genes using TS. To the best of our knowledge, the current study has an unparalleled scale of both WES and TS of colorectal adenomas. We also reanalysed publicly available CRC data from TCGA using the same pipeline to ensure valid comparisons and the most up-to-date variant discovery. Our study had six major findings. First, WES revealed similar somatic mutation frequencies in CNAD and SSA. Second, all adenomas included in WES were non-hypermutaters, with an average non-silent somatic mutation frequency significantly lower than that of CRC non-hypermutaters. Third, CNAD and SSA had both shared and unique driver genes, potentially reflecting differences in underlying biology of these lesions. Fourth, TS confirmed the WES findings and gave a better estimate of population prevalence of mutations in genes of interest. Fifth, a subset of mutations exhibited excellent accuracy for distinguishing between adenoma and CRC. Finally, genes displaying a consistent trend in mutation prevalence among non-advanced CNAD, advanced CNAD and CRC could reflect the progress towards malignancy. Collectively, out results established the understudied mutation atlas for adenomas.
We also discovered for the first time that GOLGA8B, TMPRSS13 and KRTAP4-5 have a driver pattern in CNAD. The biological function of both GOLGA8B and TMPRSS13 in the development of premalignant lesion in the large intestine is largely unknown. GOLGA8B resides in a region that is frequently found to be deleted in newborns with epilepsy and intellectual disability.21 TMPRSS13 is also referred to as mosaic serine protease large form (MSPL), and it has been shown to activate prohepatocyte growth factor (pro-HGF).22 Furthermore, the activation of HGF receptor (encoded by MET proto-oncogene, which is a receptor tyrosine kinase for HGF) by HGF has been shown to rescue CRC cells from the epidermal growth factor receptor inhibitor cetuximab.23 Whether TMPRSS13 promotes adenoma development and progression via HGF/MET axis remains to be investigated. KRTAP4-5 belongs to the keratin-associated protein family, which contributes to hair structures. A recent publication reported that depletion of the related gene KRTAP5-5 in mammary cancer cells reduced their invasion potential.24
A widely accepted model of progression from normal epithelium to adenoma to adenocarcinoma was first proposed in 1988. In that model, several genetic alterations, for instance, 5q, 17p, 18q loss and RAS mutations, became more prevalent as lesions progressed from class II and class III adenomas to adenocarcinoma.25 Subsequent studies refined this model and identified additional genetic mutations involved in this process including APC, CTNNB1, CDC4 (FBXW7), PIK3CA, TP53 and SMAD4.26 In our study, we also confirmed the presence of truncating mutations in APC and activating mutations of CTNNB1 in CNAD. CTNNB1 encodes for β-catenin, and CTNNB1-mutated CRCs tend to be highly invasive in patients with Lynch syndrome.27 Consistent with previous findings,28 the prevalence of CTNNB1 mutation in the current study decreased from 12% in non-advanced CNAD to 7% in advanced CNAD to 3% in CRC. The higher prevalence of driver mutations in premalignant lesions than in malignant lesions was also reported and discussed by a recent review.6 One of the examples of this counterintuitive phenomenon, referred to as oncogene-induced senescence, was the 70%–88% prevalence of a mutation of BRAF at V600E in melanocytic nevi, a much higher rate than the 40%–45% found in melanoma samples.6 The potential mechanisms underlying oncogene-induced senescence included DNA damage, p38 activation and formation of heterochromatic foci.29 Whether the presence of recurrent CTNNB1 mutations in adenoma reflects an early event in polyposis, a bystander mutation or a trigger for oncogene-induced senescence remains to be elucidated. In animal studies, heterozygous activating CTNNB1 mutations were unable to deregulate Wnt pathway or confer crypt progenitor cell phenotype unless E-cadherin expression was inhibited, suggesting that -CTNNB1 mutations alone might not be sufficient to drive tumourigenesis.30
Previous efforts to identify novel mutations in adenomas using next-generation sequencing31–34 are summarised in online supplementary table 6. Several of the genes in our classifier have previously been found in prior studies on mutations of adenoma (APC, CSMD1, CSMD3, CTNNB1, FAT4, FBXW7, KRAS, LRP1B, NRAS, RYR3, SOX9, SYNE1 and TP53). We reported several genes that were found to be mutated in CNAD for the first time (ATM, CDC27, MED12, NALCN and TMPRSS13). Moreover, the absence of mutations in PIK3CA and SMAD4 in CNAD but relatively high frequency in CRC may provide useful translational application as molecular classifier. Among the previously unreported genes, TMPRSS13 exhibited a driver-gene pattern in CNADs; most of the other genes were predominantly found in CRC. Factors that could contribute to these discrepancies include but are not limited to sample size, sequencing technology and analysis software. In addition to the aforementioned reports, a group of our colleagues recently published a study of 25 familial adenomatous polyposis (FAP) samples that displayed a mutation rate similar to that in the CNAD samples in the current study.35 Both FAP and CNAD had highly prevalent truncating mutations of APC; FBXW7 mutations were missense in FAP but truncating in CNAD. Transcription factor 7-like 2 (TCF7L2) was frequently mutated only in FAP, and no non-silent CCR-4-NOT transcription complex subunit 3 (CNOT3) or protein-L-isoaspartate (D-aspartate) O-methyltransferase domain containing 1 (PCMTD1) mutations occurred in CNAD. These differences in mutated genes may contribute to the distinct disease patterns observed in hereditary and sporadic adenomas.
In addition to developing a more comprehensive model of tumourigenesis which takes into account the heterogeneity of adenoma histology, the current study also investigated the mutations in a pathologically distinct subgroup of adenomas with tumorigenic potentials, SSAs.36 SSA is thought to progress into hypermutated CRC via the serrated pathway.37 In our study, somatic BRAF mutations at rs113488022 (V600E) were found in 50% of SSAs. This somatic mutation was also found in 7 of 312 non-hypermutated and 25 of 66 hypermutater CRCs, suggesting enrichment of mutations at this locus in SSA and hypermutaters. Although the prevalence of KRTAP4-5 mutations did not reach significance, possibly because of our small sample size, KRTAP4-5 was the only gene found to be frequently mutated in both CNADs and SSAs. Aggregation of somatic missense mutations at rs411367, which is found in 1% of the general population, is consistent with the pattern found in driver mutations. Future validation of KRTAP4-5 missense mutations is required to determine whether this gene serves as a driver in both SSA and CNAD.
In a previous report, analysis of 33 SSA-associated carcinomas, 79% were MLH1 deficient.38 In addition, epigenetic MLH1 downregulation has been implicated in hypermutated CRC.5 These results seem to support the hypothesis that SSA is the precursor for hypermutated CRC. However, our WES demonstrated no significant difference in the mutation rates in SSAs and CNADs. In addition, we found no mutations of APC in SSAs. This finding appears to contradict the hypothesis that SSA is the origin of hypermutated CRC because APC is frequently mutated in hypermutated CRCs.5 Whether an SSA obtains APC mutation after it progresses to a more advanced lesion or CRC carrying APC mutations develops exclusive from CNAD warrants further investigation
In addition to performing WES, we determined the prevalence of somatic mutations in known driver genes in an additional 100 non-advanced and advanced CNAD samples using TS. The combination of mutations found in genes covered by both WES and TS provided further insight into when these mutations occur. Among genes with a consistent trend, we observed three mutation patterns: (1) monotonic increasing or decreasing (TP53, CTNNB1 and KRAS); (2) increasing or decreasing from non-advanced to advanced CNAD but remaining similar in advanced CNAD and CRC (FBXW7); and (3) remaining similar in non-advanced and advanced CNADs but increasing or decreasing from advanced CNAD to CRC (PIK3CA and SMAD4). Whether the third group of genes could predict progression from CNAD to CRC requires further study.
To identify the mutational signature of CNAD and compare it with that of CRC, we aggregated the WES data on adenocarcinomas from TCGA and our sequencing of adenomas. We expected batch effects might lead to false discovery of differently mutated genes, so we applied several steps to minimise these effects, such as using capture probes synthesised by the same supplier (NimbleGen, Roche) and sequencing using an Illumina Hiseq 2000 at a similar depth (~100×) and in the same sequencing centre following the same protocol. We also processed the raw data of TCGA and our own project through the same pipeline. When we combined the TS and WES results, we only selected common exon regions for both capture probes. Further validation studies in independent cohorts are needed to evaluate the performance of this random forest classifier and establish whether our findings are generalisable.
We believe that our findings illuminated genetic alterations that mark fundamental differences among different types of adenomas and CRC. Our results are based on cross-sectional study; we will validate the findings in prospective cohort. We will continue to follow the patients in our cohort and determine whether adenomas with genetic alterations similar to those found in CRC are associated with increased cancer risk. If carrying adenomas with unfavourable genetic alterations does increase cancer risk, closer surveillance would be recommended even if the adenomas do not fit the pathological criteria for advanced adenoma. In addition, the novel driver genes we reported in this article could be crucial players in the tumourigenesis process, and we plan to collaborate with our colleagues and study the functions of these genes in both cell line and animal models to better characterise their roles.
In conclusion, the data from the current project provide novel insights into potential driver genes involved in progression from colorectal adenoma to adenocarcinoma. The genes that differed in the mutational profiles of CNAD and CRC could serve as gene panels for early surveillance. Furthermore, genes with a clear trend towards malignancy could serve as molecular ‘clocks’ to indicate how far an adenoma has progressed towards tumourigenesis. These findings will improve our understanding of the underlying biology of CRC, risk stratification, and design of prevention and surveillance programmes for CRC.
Acknowledgments
We thank Dr Stanley R Hamilton for his advisory role on this study; the Human Genome Sequencing Center at Baylor College of Medicine for generating the sequencing data; and the staff of the Premalignant Genome Atlas and Center for Translational and Public Health Genomics, Duncan Family Institute for Cancer Prevention, and MD Anderson Cancer Center for recruitment, data collection, management, and biospecimen collection and processing.
References
Footnotes
SK, LM and XW contributed equally.
S-HL, GSR and CH contributed equally.
Contributors Study design: GSR, XW, SK, LM, EH. Study recruitment, data and biospecimen collection and management: GSR, JRS, LM, XW. Bioinformatic analysis: SHL, CH, YY, JSC, HL, AF. Statistical analysis: SHL, YY. Biological annotation: SHL, JG, MATH, XW. Primary results interpretation: SHL, DGM, JM, AF, SK, XW. Manuscript drafting: SHL, YY, XW. All authors contributed to and critically reviewed the manuscript.
Funding This work was supported in part by the Colorectal Cancer Moon Shot Program, the Premalignant Genome Atlas Program, and the Center for Translational and Public Health Genomics, Duncan Family Institute for Cancer Prevention and Risk Assessment, The University of Texas MD Anderson Cancer Center.
Competing interests None declared.
Patient consent This manuscript only contains deidentified information which does not track back to patients. All patients have signed an MD Anderson consent form when they enrolled in our study.
Ethics approval Institutional Review Board of MD Anderson Cancer Center.
Provenance and peer review Not commissioned; externally peer reviewed.