Article Text

Abstract
Functional gastrointestinal disorders (FGIDs) and IBDs are two of the most prevalent disorders of the GI tract and consume a significant proportion of healthcare resources. Recent studies have shown that membrane-bound guanylate cyclase-C (GC-C) receptors lining the GI tract may serve as novel therapeutic targets in the treatment of FGIDs and IBDs. GC-C receptor activation by its endogenous paracrine hormones uroguanylin and guanylin, and the resulting intracellular production of its downstream effector cyclic GMP, occurs in a pH-dependent manner and modulates key physiological functions. These include fluid and electrolyte homeostasis, maintenance of the intestinal barrier, anti-inflammatory activity and regulation of epithelial regeneration. Studies of the GC-C paracrine signalling axis have revealed the therapeutic potential of these receptors in treating GI disorders, including chronic idiopathic constipation and irritable bowel syndrome–constipation. This review focuses on the evolving understanding of GC-C function in health and disease, and strategies for translating these principles into new treatments for FGIDs and IBDs.
- uroguanylin
- plecanatide
- linaclotide
- guanylate cyclase C
- chronic idiopathic constipation
- irritable bowel syndrome-constipation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- uroguanylin
- plecanatide
- linaclotide
- guanylate cyclase C
- chronic idiopathic constipation
- irritable bowel syndrome-constipation
Introduction
Initially discovered in the 1970s as the intestinal receptor for exogenous diarrhoeagenic bacterial heat-stable enterotoxins (STs),1 2 the guanylate cyclase-C (GC-C) receptor and its associated signalling pathways contribute to the maintenance of normal physiological functioning of the GI tract. Indeed, activation of the GC-C receptor by its endogenous intestinal paracrine hormones uroguanylin and guanylin plays a key role in fluid and ion homeostasis, maintenance of the intestinal barrier, inhibition of inflammation, visceral pain signalling and tumourigenesis.3–13 In that context, disruptions in GC-C signalling pathways have been linked to numerous GI disorders including IBDs and colorectal cancers (CRCs), and genetic mutations that result in loss or gain of function in this receptor have been associated with constipation or diarrhoea, respectively.10 13–20
The multiple functions of the GC-C receptor and its ligands have advanced these receptors as attractive targets for therapy in diverse gastrointestinal diseases, including functional gastrointestinal disorders (FGIDs) (IBS-C and CIC) and IBDs (figure 1).3 FGIDs and IBDs are two of the most prevalent disorders of the GI tract. Irritable bowel syndrome (IBS) affects 10% to 20% of the population in developed countries.21 The diagnosis of IBS is traditionally based on symptoms of recurrent abdominal pain or discomfort, occurring with two or more of the following: associated with defecation, onset associated with a change in the frequency or form (consistency) of bowel movements. It is estimated that 5%–10% of adults have IBS with constipation (IBS-C), that at least 15% of adults have functional or chronic idiopathic constipation (CIC)22 and that there is overlap of these disorders. Commonly used diagnostic criteria for functional constipation are shown in box 1.23 CIC is characterised by a variety of bowel symptoms including reduced bowel movement (BM) frequency, hard stools, straining during defecation and a sense of incomplete evacuation after defecation, as well as abdominal symptoms of bloating and discomfort.24 25 Therefore, the primary symptoms of the two conditions overlap significantly. The definition of CIC was operationalised in trials of the two GC-C agonists, linaclotide (five trials) and plecanatide (four trials), as summarised in a recent publication on their efficacy and tolerability.26
Functional constipation, Rome IV criteria that need to be fulfilled for the last 3 months with symptom onset at least 6 months prior to diagnosis
Must include two or more of the following:
Straining during more than one-fourth (25%) of defecations.
Lumpy or hard stools (Bristol Stool Form Scale (BSFS) 12) more than one-fourth (25%) of defecations.
Sensation of incomplete evacuation more than one-fourth (25%) of defecations.
Sensation of anorectal obstruction/blockage more than one-fourth (25%) of defecations.
Manual manoeuvres to facilitate more than one-fourth (25%) of defecations (eg, digital evacuation, support of the pelvic floor).
Fewer than three spontaneous bowel movements per week.
Loose stools are rarely present without the use of laxatives.
Insufficient criteria for irritable bowel syndrome.

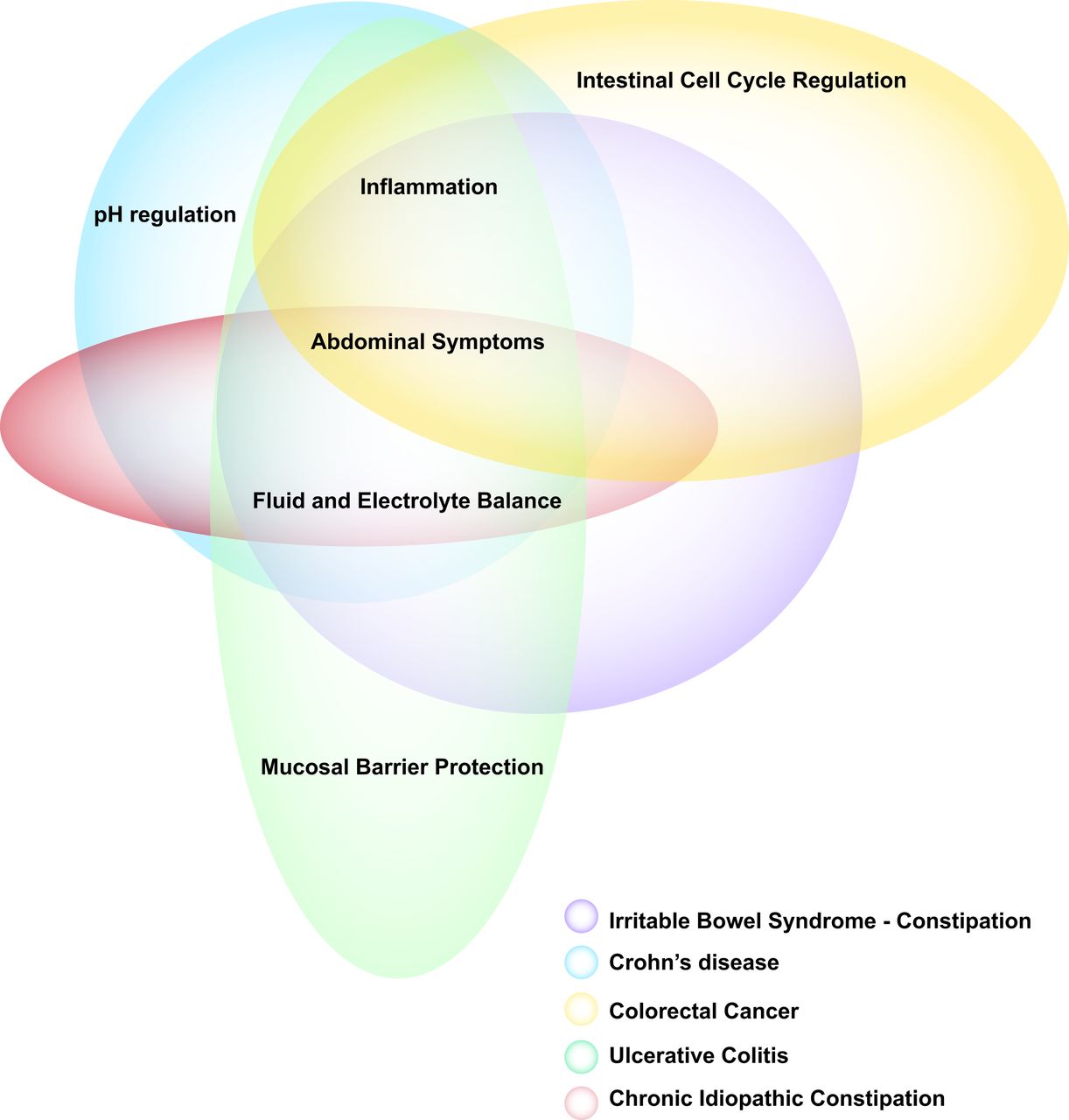
Guanylate cyclase-C (GC-C) functional overlap with numerous symptoms found in GI disorders. Research on various GI disorders has elucidated the importance of the GC-C receptor and the physiological pathways in which it is involved. Dysregulation of the GC-C–cGMP signalling axis has been associated with dysfunction in the mucosal barrier (eg, UC); fluid and electrolyte balance and pH regulation (eg, chronic idiopathic constipation, IBS with constipation); and epithelial cell proliferation, migration, differentiation and apoptosis (eg, colorectal cancer). These pathways contribute to the abdominal symptoms and/or inflammation involved in GI diseases.
FGIDs significantly reduce patients’ quality of life and pose a significant burden on healthcare resources.3 27–29 Both IBS and IBD are chronic GI disorders characterised by changes in bowel function, and alterations in mucosal and immune functions, microbiota and visceral sensitivity.3 A ligand-based approach to therapy using synthetic analogues of GC-C agonists has been enhanced by an expanded understanding of the GC-C signalling pathways as well as considerations of the structural, conformational and dynamic features of ligand–receptor interactions in this pathway. Indeed, synthetic analogues of GC-C ligands are effective in providing relief, and are well tolerated, when used in the clinical management of CIC and IBS-C.29 30 This review summarises the role of the GC-C system in correcting pathobiological mechanisms contributing to these diseases.
Intestinal pH shapes GC-C function
Distinct and tightly regulated pH environments have well-established roles in physiological processes in different regions of the GI tract. For example, in the stomach, gastric acidity is important for initiating food digestion and providing a chemical barrier against ingested micro-organisms.31 Acidic gastric contents empty into the duodenum where they mix with alkaline secretions from the pancreas and biliary tract, producing a moderate rise in pH that protects the intestinal columnar epithelium from the deleterious effects of acid, while creating the pH optimum for pancreatic enzymes to enhance nutrient absorption,32 particularly the absorption of fat since pancreatic lipase is irreversibly inactivated below pH 4.0. There is a steady rise in pH across the rostral–caudal axis of the GI tract, from approximately pH 5–6 in the duodenum and proximal jejunum to an alkaline pH of ~7.8 in the distal ileum, the latter pH increase reflecting chloride–bicarbonate ion exchange in this region.32 With the exception of a slight drop in the caecum, pH values remain close to neutral throughout the colon, providing a supporting environment for commensal bacteria (figure 2A).31 Intraluminal pH values are tightly regulated by a number of ion channels, including the cystic fibrosis transmembrane conductance regulator (CFTR) and the Na+/H+ exchanger, which in turn are regulated by GC-C activation.33–36

Variations in pH, guanylate cyclase-C (GC-C) signalling axis components and fluid homeostasis along the rostral–caudal axis of the intestine. (A) Intestinal pH values gradually rise in small intestine, with a drop in the caecum reflecting micro-organism populations. (B) Uroguanylin expression peaks in distal jejunum and is minimal in colon. (C) Guanylin levels rise along the distal small intestine and peak in the caecum before sharply falling in the distal colon. (D) GC-C is ubiquitously expressed in both the small and large intestine. (E) In a healthy individual, fluid volumes are balanced by secretory and absorption mechanisms, with highest net secretion in proximal small intestine and highest net absorption in colon.
In that context, the GC-C–cGMP signalling axis is selectively regulated by the changing pH environments across the rostral–caudal axis of the intestine, in part, mediated by compartmentalised expression of its endogenous paracrine hormones. Thus, while GC-C is expressed along the length of the GI tract, it is differentially activated by uroguanylin in small intestine and by guanylin in colorectum in a pH-dependent manner (figure 2B–D). Uroguanylin is a 16 amino acid peptide that activates GC-C with maximum potency in the slightly acidic (pH 5–6) environments of the duodenum and proximal jejunum (figure 3).37 It is expressed by tuft-like epithelial cells primarily in the proximal small intestine, with lower levels in stomach, distal small intestine and colorectum.38 39 In contrast, guanylin is a 15 amino acid peptide that activates GC-C in neutral to slightly basic pH environments and is principally expressed in the colorectum by goblet cells (figure 3).37–39 Both peptides are secreted as unprocessed pro-hormones that are cleaved to mature forms that are stabilised by two disulfide bonds compatible with structural flexibility and isomerisation.40–43

Structures of GC-C agonists. Both uroguanylin and guanylin are active endogenous ligands of GC-C. These peptides have many structural conformations defined by two disulfide bonds, in striking contrast to the three disulfide bonds and rigid structure of STs. Further, uroguanylin has pH-sensing aspartate residues in its N terminus (*), which allow the ligand to preferentially bind GC-C in acidic (pH 5–6) environments of the proximal small intestine, as well as a COOH-terminal leucine (**) compared with guanylin. Plecanatide is the synthetic analogue of uroguanylin, retaining two intrachain disulfide bridges and two acid-sensing amino acids in the N terminus, which together confer preferential biological activity in acid pH environments. In contrast, linaclotide is the synthetic analogue of STs, retaining three intrachain disulfide bridges in the absence of acid-sensing residues in the N terminus, conferring a rigid structure whose activity is pH independent.
pH sensitivity of GC-C hormones
Uroguanylin contains acid-sensing residues glutamate and aspartate at the N terminus. Glutamate and aspartate exhibit ionisation and hydrogen bonding properties with steep pH dependencies.44 In acidic pH environments, these residues produce a greater percentage of uroguanylin in the conformation that binds GC-C with high affinity.37 40 42 43 When these residues are removed, uroguanylin adopts characteristics similar to those of guanylin, with greater affinity for GC-C in neutral-alkaline pH environments.37 These physicochemical characteristics impart uroguanylin with 100-fold greater potency than guanylin in stimulating GC-C-mediated cGMP accumulation at pH 5, while guanylin is 3-fold more potent than uroguanylin in stimulating cGMP production at pH 8 in stimulating intracellular cGMP accumulation and transepithelial chloride secretion.37 Together, these pH-sensitive structures coupled with selective expression of peptides across the rostral–caudal axis of the bowel ensure optimal ligand-dependent GC-C–cGMP signalling to regulate intestinal fluid and electrolyte homeostasis.
pH insensitivity of STs
Originally, GC-C was discovered as an intestinal receptor for the family of diarrhoeagenic bacterial heat-stable enterotoxins, collectively referred to here as STs.1 2 41 STs are a major cause of diarrhoeal disease and are associated with morbidity and mortality, especially in children less than 5 years of age.45–47 The toxin is secreted as a 72 amino acid precursor protein and is later cleaved into a mature peptide.48 Enterotoxigenic bacteria colonise the gut and release STs that drive intestinal hypersecretion and simultaneously inhibit fluid absorption.1 2 34–36 41 49 Unlike uroguanylin, STs lack pH-sensitive amino acids in the N terminus, abrogating pH checkpoints that normally regulate endogenous GC-C ligand binding along the GI tract.37 Additionally, STs contain an extra intramolecular disulfide bridge locking the core 13 amino acid sequence into a stable, high-affinity receptor-binding isomer.50 51 This structural rigidity, and its pH independence, result in maximum biological activity and directly correlate with the ability of ST to cause unchecked fluid secretion leading to diarrhoea.50 Infections by ST-producing bacteria are especially dangerous in infants where increased expression of GC-C and elevated susceptibility to dehydration lead to higher mortality rates.45–47 52
Biology of the GC-C signalling axis
Fluid and electrolyte homeostasis
Agonist binding to the extracellular domain of GC-C activates its catalytic domain and leads to the conversion of guanosine triphosphate to cyclic GMP (cGMP) (figure 4A).41 The resulting cGMP targets a variety of effector proteins including phosphodiesterases, protein kinases and ion channels.41 After cGMP directly activates protein kinase G II and cross-activates protein kinase A, these mediators stimulate CFTR channels, leading to efflux of Cl− and HCO3 − ions into the intestinal lumen.34–36 This efflux of anions results in a net osmotic increase, driving water into the GI tract.34–36 Additionally, cGMP accumulation inhibits fluid absorption by directly inhibiting the apically located Na+/H+ exchanger-3 (NHE3) allowing Na+ (and water) to remain in the intestinal lumen.33 34 36 53 This overall net secretion is driven primarily in the duodenum and proximal jejunum. In the colon, GC-C signalling inhibits absorption via a cGMP-mediated protein kinase-independent mechanism.39 41 54 Appropriate secretion of fluid into the proximal regions of the small intestine is crucial for process of digestion and contributes to the eventual formation and consistency of stool; in addition, fluid secreted into the intestine indirectly stimulates intestinal propulsion.55
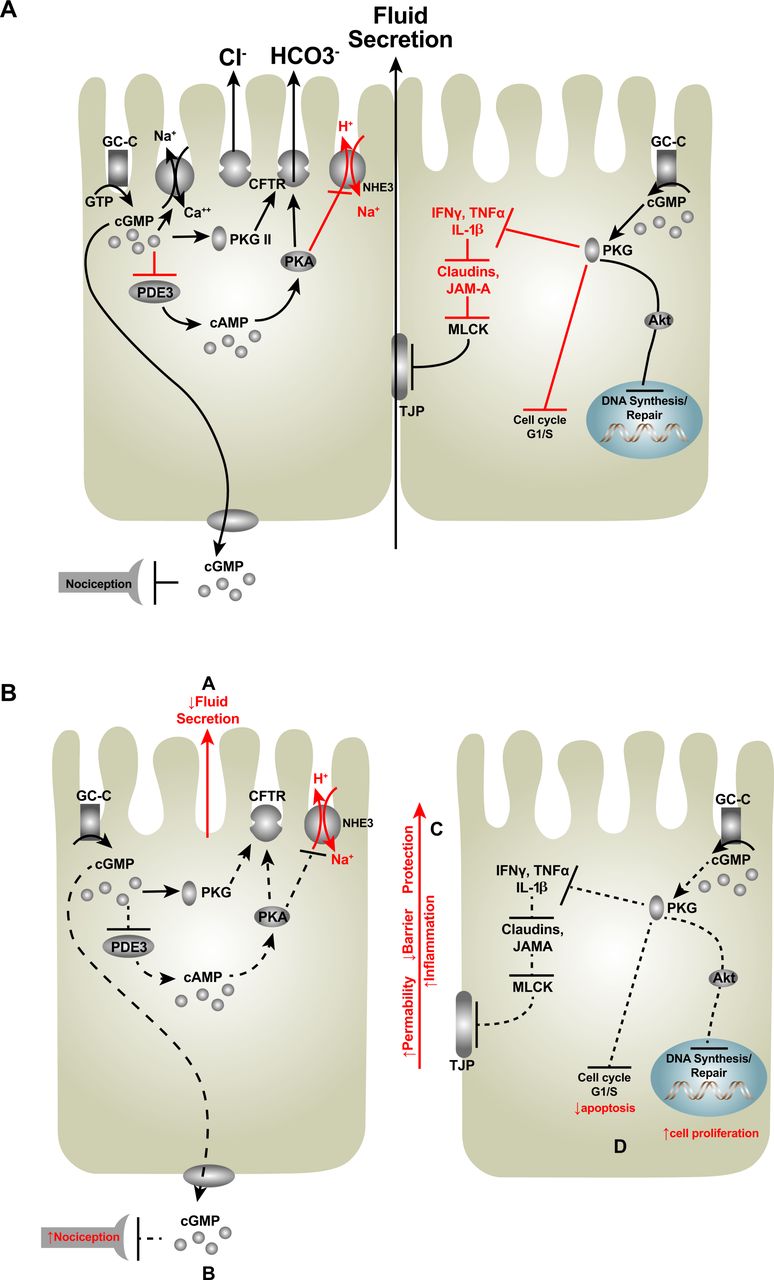
{kind=link}
{kind=link}
{kind=link}
{kind=link}
GC-C activation, intracellular effects. (A) The GC-C receptor is activated by both endogenous and exogenous ligands, which increase intracellular cGMP levels. The subsequent regulation of protein kinases, phosphodiesterases and ion channels modulates a number of downstream physiological mechanisms including fluid and electrolyte secretion, barrier function, inflammation and proliferation. (B) Dysregulation of this signalling pathway in turn produces epithelial dysfunction, which may contribute to chronic constipation, IBDs and intestinal tumourigenesis. CFTR, cystic fibrosis transmembrane conductance regulator; GC-C, guanylate cyclase-C; GTP, guanosine triphosphate; MLCK, myosin light chain kinase; PDE3, phosphodiesterase 3; NHE3, Na+/H+ exchanger-3; PKA, protein kinase A; PKG II, protein kinase G II; TJP, tight junction protein; TNFα, tumour necrosis factor alpha.
Effects of mutations in the GC-C gene, GUCY2C
In the context of the role of GC-C in epithelial secretion, mutations in the GUCY2C gene encoding GC-C are associated with imbalances in intestinal fluid and electrolyte homeostasis. These mutations have provided insight into the structure–function relationship of the receptor.56 One example of this is a gain-of-function mutation in GC-C in a Norwegian family with chronic mild diarrhoea.15 This autosomal dominant mutation in the extracellular domain increases the binding affinity of GC-C for all ligands. Moreover, affected family members were more susceptible to IBS, IBD, small bowel obstruction and oesophagitis. Another gain-of-function group of mutations was linked to congenital sodium diarrhoea (CSD), in which GC-C is constitutively active, elevating cGMP levels in the absence of ligand binding.17 Of note, the other major cause of CSD is loss-of-function autosomal recessive mutations in the Na+/H+ exchanger, which is a key downstream target of GC-C–cGMP signalling.57
Conversely, inactivating mutations of GC-C associated with decreased cGMP production were discovered in newborns born to families with a history of meconium ileus (figure 4B).18 Though commonly associated with cystic fibrosis, meconium ileus is a distinct diagnosis characterised by neonatal intestinal obstruction. A recent study reported two separate autosomal recessive GUCY2C loss-of-function mutations that cause meconium ileus in unrelated Bedouin families.18 In one family, a missense mutation produced a change in the extracellular ligand binding domain associated with a 40% decrease in ligand-induced cGMP production. In the second family, a nonsense mutation inactivated the catalytic domain of GC-C preventing cGMP production. More recently, loss-of-function mutations in GUCY2C were identified in a Lebanese family demonstrating altered affinity in receptor ligand binding.19 Ultimately, these mutations were associated with decreased fluid and electrolyte secretion, resulting in meconium ileus.18 19 Interestingly, this loss-of-function mutation in GC-C provides protection against enterotoxigenic diarrhoea demonstrating the structural specificity required for the receptor to respond to the relatively rigid molecular structure of ST, with the extra intramolecular disulfide bridge. Indeed, mice homozygous or heterozygous for inactivating mutations in GC-C exhibited lower mortality rates in response to ST-induced secretory diarrhoea compared with wild-type animals.58 59
Emerging roles for the GC-C axis
Recent insights have revealed a role for the GC-C signalling axis in GI functions beyond fluid and electrolyte homeostasis, including the pathophysiology of visceral pain, mucosal barrier maintenance, inflammation and tumourigenesis (table 1).
Expanding pathophysiological roles of the GC-C signalling axis in intestine based on animal models of human diseases
Visceral pain
The GC-C signalling axis modulates afferent pathways involved in GI pain sensation.4 12 60 Abdominal pain is a main symptom of FGIDs and IBDs, and GC-C-based therapies promote visceral analgesia in animal models and in clinical trials for patients with these conditions.4 30 The intracellular cGMP that is produced on ligand binding to GC-C on epithelial cells can be transported into the extracellular space through MRP4 cyclic nucleotide efflux pumps located on the basolateral membrane, decreasing conduction of submucosal afferent nociceptive neurons and attenuating the sensation of visceral pain.4 61 Also, GC-C signalling may contribute to analgesia indirectly by reducing inflammation through maintenance of the intestinal barrier, restricting luminal factors from gaining access to nociceptive or immune mechanisms in the lamina propria and beyond.10 13 62 63 Moreover, given its key role in regulating ion flux, pH and water secretion, GC-C and its ligands may establish luminal microenvironments that maintain the intestinal microbiome. This is supported by a recent report that genetic GC-C activation influences the intestinal microbiota in patients with familial diarrhoea and increased susceptibility for Crohn’s disease, including increases in Enterobacteriaceae and loss of Bifidobacterium and Faecalibacterium prausnitzii; the latter change is frequently associated with Crohn’s disease.64
Mucosal barrier
The intestinal barrier maintains mucosal immune homeostasis and prevents inflammation. Loss of GC-C or uroguanylin in mice increases intestinal epithelial permeability and inflammation, in part by weakening tight junctions through activation of myosin light chain kinase, reducing claudin-2 and junctional adhesion molecule-A levels, and activating serine/threonine kinase 1, which reduces occludin and claudin-4.10 65 This dual role of barrier maintenance with selective permeability is supported, in part, by the GC-C signalling axis.66
Inflammation
Dysregulation of the GC-C signalling axis in early stages of inflammation may accelerate disease progression in IBD.10 13 62 67 Thus, reducing the expression of GC-C, uroguanylin or guanylin increased intestinal inflammation, reflected by higher UC disease activity indices.68 Similarly, guanylin expression was reduced in mucosal biopsies from patients with UC.14 67 The inflammatory cytokine tumour necrosis factor alpha suppresses the expression of guanylin mRNA and protein.62 Decreased guanylin expression was detected prior to colitis-induced goblet cell depletion, which occurs at a very early stage of inflammation.62 Indeed, reduced guanylin expression precedes epithelial damage that results in mucin depletion and inflammation and predisposition to the phenotype of UC.62
Tumourigenesis
Under normal conditions, the intestinal epithelium rapidly regenerates, undergoing cycles of proliferation, migration, differentiation and apoptosis that leads to complete epithelial renewal every 3 to 5 days in the small intestine and every 5 to 7 days in the colon.69 GC-C–cGMP signalling is an essential regulator of those key processes contributing to the homeostasis of the crypt–surface axis in small and large intestines. Indeed, silencing of that axis results in hyperproliferation, with hyperplasia of proliferating crypts, accelerated migration, reduced differentiation along the secretory lineage and reduced apoptosis.8 9 11 70–73 Further, loss of GC-C–cGMP signalling reduces DNA damage sensing and repair, resulting in accumulation of driver mutations associated with transformation.8 9 In that context, guanylin is the most commonly lost gene product in intestinal tumourigenesis, it is lost at the earliest stages of transformation and the loss of guanylin is conserved across species affected by CRC.11 20 74 Indeed, the paracrine hormone hypothesis of CRC suggests that transformation initiates as a disease of guanylin insufficiency that silences the GUCY2C tumour suppressor.69 Counteracting this loss by oral or genetic hormone replacement opposes the development of tumourigenesis in mice.11 16 69 These observations form the basis for clinical trials exploring the utility of oral GC-C agonists as a chemopreventive strategy for CRC in humans.75 76
Targeting GC-C for FGIDs
First-line treatments for FGIDs are diet and lifestyle changes, followed by treatment with osmotic and stimulant laxatives, which are effective for some patients, primarily those with reduced frequency of bowel movements.3 27–29 However, no single or combined treatment works for all patients, and there remains a significant unmet clinical need for new and differentiated treatment options.77
The role of the GC-C–cGMP signalling axis in regulating fluid and electrolyte homeostasis across the rostral–caudal axis of the intestine, and the associated pathophysiological secretory consequences of GC-C dysfunction contributing to diarrhoeal diseases, provide the biological foundation for the utility of GC-C ligand analogues to treat CIC and IBS-C. GC-C has emerged as a therapeutic target for the treatment of CIC and IBS-C, reflecting its impact on fluid and electrolyte secretion, visceral analgesia and inflammation.3 21 29 50 51 Both linaclotide and plecanatide are approved by the Food and Drug Administration (FDA) for the treatment of CIC and IBS-C.
Linaclotide (Linzess)
Linaclotide (Ironwood Pharmaceuticals, Cambridge, Massachusetts, USA) is a 14 amino acid synthetic analogue of ST differing in structure by a single tyrosine/leucine substitution (relative to ST) in its pharmacophore (figure 3).50 51 Like ST, linaclotide is stabilised by three disulfide bonds that lock the ligand into a constitutively active conformation.50 As a GC-C agonist, linaclotide activates the cGMP signalling cascade, increasing fluid and electrolyte secretion in the intestinal lumen.50 51 Increased luminal fluid hydrates stool, relieves symptoms of constipation and may contribute to the relief of visceral analgesia.4 51 In two placebo-controlled CIC clinical trials, treatment with linaclotide increased complete spontaneous bowel movements from baseline.30 In addition, linaclotide improved stool consistency, reduced straining and decreased bloating and abdominal discomfort. In both studies, the primary endpoint was the percentage of patients who were overall responders, defined as three or more complete spontaneous bowel movement (CSBMs) per week and an increase of at least one CSBM per week from baseline for nine or more weeks during the 12-week treatment period.78 Among patients treated with 145 µg linaclotide, 21.2% and 16.0%, in respective studies, were considered overall CSBM responders, a significant improvement over 3.3% and 6.0%, respectively, in placebo groups. Similarly, among patients receiving 290 µg of linaclotide, 19.4% and 21.3% were overall CSBM responders, respectively. Further, both studies demonstrated the absence of rebound deterioration of bowel and abdominal symptoms below baseline (untreated) after discontinuation of linaclotide. Diarrhoea was the most common adverse event reported, with combined rates of 16.0% in the 145 µg groups, 14.2% in the 290 µg groups and 4.7% in the placebo groups. The FDA recently approved a 72 µg dose of linaclotide for chronic constipation.79
Linaclotide also has demonstrated efficacy in the treatment of IBS-C. In the two phase III clinical trials conducted in the USA, the dose tested was 290 µg/day.80 81 An earlier phase IIB trial had documented efficacy of linaclotide daily doses of 75, 150, 300 and 600 µg relative to placebo.82 It is therefore interesting to note recent evidence that the optimal dose to achieve efficacy in Japanese patients with IBS-C appears to be 500 µg/day based on the global assessment of relief of IBS symptoms, although the responses based on improvement in bowel function endpoints over 12 weeks were observed with the 125, 250 and 500 µg/day doses.83
Plecanatide (Trulance)
Plecanatide (Synergy Pharmaceuticals, New York, New York, USA) is a 16 amino acid peptide analogue of uroguanylin, with aspartate in the third position of the N terminus replaced by glutamate (figure 3).29 Plecanatide preferentially binds GC-C with high affinity in the slightly acidic environments (pH 5–6) of the duodenum.29 Like linaclotide, plecanatide has no oral bioavailability and is not absorbed systemically, eliminating the potential for off-target effects associated with other treatments for FGIDs.84 Plecanatide has the same mechanism of action as uroguanylin, stimulating the cGMP signalling axis in a pH-sensitive manner to induce fluid and electrolyte secretion and relieve constipation.29 Like linaclotide, plecanatide also produces visceral analgesia,63 suggesting a drug class effect reflecting the mechanism of action (increased intracellular cGMP). The efficacy, safety and tolerability of plecanatide in CIC have been assessed in two double-blind, placebo-controlled, 12-week clinical trials and have been published.85 86 In both, the primary endpoint was the percentage of durable overall CSBM responders defined as patients who experienced three or more CSBMs per week and an increase of at least one CSBM per week over baseline for 9 of the 12 treatment weeks of the study, with responses in at least three of the last 4 weeks. Among patients treated with 3 mg plecanatide, 21.0% and 20.1% were considered durable overall CSBM responders, a significantly greater percentage compared with the 10.2% and 12.8% in the placebo groups, respectively. Similarly, among patients who received 6 mg plecanatide, 19.5% and 20.0% were durable overall CSBM responders. Again, diarrhoea was the most common adverse event reported, with rates of 4.6% in the 3 mg group, 5.1% in the 6 mg group and 1.3% in the placebo group. Further, both studies revealed that abdominal bloating and discomfort were improved, without rebound deterioration of symptoms below baseline (untreated) after discontinuation of plecanatide. It has been suggested that lower rates of diarrhoea achieved with plecanatide may reflect the pH-sensitive structure of that peptide, with activity limited to the acid compartment of the small intestine like uroguanylin; this may contrast with the effects of linaclotide, which are45 pH-independent and may result in activity across the rostral–caudal axis of the intestine, like STs.85 86 The efficacy of plecanatide 3 or 6 mg/day for IBS-C has been based on three trials presented in abstract format26 or, in the case of unpublished studies (NCT02387359; NCT02493452), assessed in a recent systematic review and meta-analysis based on United States Securities and Exchange Commission filings of drug sponsors.26 87
Conclusion
Progress in the development of treatment options for CIC and IBS-C are being informed by the evolving understanding of the normal physiology of the gut and the complex pathophysiology of these disease states, coupled with improved definitions of clinically meaningful treatment outcomes.3 21 77 78 The GC-C–cGMP signalling axis has emerged as a key mechanism regulating normal fluid and electrolyte homeostasis across the rostral–caudal axis of the intestine, making it a logical, mechanism-based target to treat chronic constipation syndromes. Although the precise role of dysfunction in the GC-C signalling pathway in the pathophysiology of CIC or IBS-C has yet to be completely defined, recent preliminary studies revealed reduced GC-C mRNA and protein expression in rectal mucosa of patients with CIC and IBS-C consistent with a role for loss of cGMP signalling contributing to these diseases.88 89 In turn, these molecular insights provide the mechanistic framework for the clinical efficacy of GC-C ligand analogues in improving bowel function in IBS-C and CIC.30 85 86 Moreover, those clinical studies provided a remarkable opportunity for reverse translation from bedside to bench, revealing for the first time the role of the GC-C signalling axis in modulating visceral pain.4 12 60 Taken together, these observations suggest that linaclotide and plecanatide will expand treatment options available to patients with CIC and IBS-C.3 Beyond constipation, the emerging role of the GC-C–cGMP signalling axis in regulating inflammation, barrier function and tumourigenesis underscores active development programmes exploring the future potential of GC-C ligands for the prevention and treatment of IBD and CRC.63 69 75
References
Footnotes
Contributors SAW and MC jointly conceived the elements of this review, collaborated in its writing and revising, and approved the final version of the manuscript.
Funding SAW is supported by grants from the National Institutes of Health (R01 CA206026, R01 CA204881, P30 CA56036), Synergy Pharmaceuticals, Inc. and Targeted Diagnostic and Therapeutics, Inc. MC is supported by a grant from the National Institutes of Health (R01DK92179).
Competing interests SAW is the Chair of the Scientific Advisory Board and a member of the Board of Directors, both uncompensated, of Targeted Diagnostics & Therapeutics, Inc., which provides funding to his laboratory. Also, he is a consultant to Synergy Pharmaceuticals, Inc., which provides funding to his laboratory. He is participating in NCI-funded studies of the use of linaclotide to prevent colorectal cancer in which the drug is generously provided by Ironwood Pharmaceuticals, Inc. He is the Samuel MV Hamilton Professor of Thomas Jefferson University. MC has served as an adviser to Ironwood Pharmaceuticals, Inc. and Synergy Pharmaceuticals, Inc., with compensation to his employer, Mayo Clinic.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.