Article Text

Abstract
Objective The initial colonisation of the human microbiota and the impact of maternal health on neonatal microbiota at birth remain largely unknown. The aim of our study is to investigate the possible dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus (GDM) and to estimate the potential risks of the microbial shift to neonates.
Design Pregnant women and neonates suffering from GDM were enrolled and 581 maternal (oral, intestinal and vaginal) and 248 neonatal (oral, pharyngeal, meconium and amniotic fluid) samples were collected. To avoid vaginal bacteria contaminations, the included neonates were predominantly delivered by C-section, with their samples collected within seconds of delivery.
Results Numerous and diverse bacterial taxa were identified from the neonatal samples, and the samples from different neonatal body sites were grouped into distinct clusters. The microbiota of pregnant women and neonates was remarkably altered in GDM, with a strong correlation between certain discriminatory bacteria and the oral glucose tolerance test. Microbes varying by the same trend across the maternal and neonatal microbiota were observed, revealing the intergenerational concordance of microbial variation associated with GDM. Furthermore, lower evenness but more depletion of KEGG orthologues and higher abundance of some viruses (eg, herpesvirus and mastadenovirus) were observed in the meconium microbiota of neonates associated with GDM.
Conclusion GDM can alter the microbiota of both pregnant women and neonates at birth, which sheds light on another form of inheritance and highlights the importance of understanding the formation of early-life microbiome.
- GDM
- microbiota
- neonate
- pregnancy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The maternal microbiota changes dramatically during and after pregnancy.
The microbiota of the pregnant woman can vertically transmit to the offspring.
The neonatal microbiota will affect the newborn’s physiology and future health.
What are the new findings?
Initial colonisation and body site-specific differentiation of the neonatal microbiota may have taken place before birth.
Community homogeneity across multiple sample types associated with gestational diabetes mellitus (GDM), and concordant variations occurred between pregnant women and newborns.
Microbial shifts resulted in metabolic depletion in neonatal intestinal microbiota, and increased the prevalence of certain viruses in the meconium.
How might it impact on clinical practice in the foreseeable future?
GDM-associated variations of the neonatal microbiota highlight the importance of microbial inheritance during pregnancy, which encourages precise modulations of the initial microbial colonisation and interaction by maternal intervention to reduce the risk of adverse health outcomes.
Relative abundance of several most prevalent bacteria is significantly associated with GDM and the oral glucose tolerance test values, which may provide potential biomarkers for non-invasive GDM testing.
Introduction
The human body harbours numerous microbes that form a stable symbiotic relationship with the host and is indispensable for human health. A long list of diseases is associated with the imbalance of human microbiota.1 In recent years, extensive attention has been paid to the microbiota during pregnancy and early life, as they are very important to reproductive health.2 For example, there is a strong correlation between maternal vaginal microbiota and preterm birth.3 In addition, maternal and neonatal microbiota may affect the early development of the neonates,4 and may cause long-term physiological consequences in the future.5 Accordingly, efforts should be made to understand the effects of maternal health status during pregnancy on the initial microbiota of newborns and maternal contributions.
Gestational diabetes mellitus (GDM) is a disease of abnormal glucose tolerance that first occurs and recognised during pregnancy. The oral glucose tolerance test (OGTT) carried out between 24 and 28 weeks is the gold standard for diagnosing this disease.6 GDM is prevalent worldwide and its prevalence in some countries or areas even is higher than 20%.7 More seriously, the incidence of GDM continues to increase, which forms a great threat to maternal and neonatal health. It has been associated with a series of adverse outcomes including gestational hypertension, cardiovascular disease and pre-eclampsia in pregnant women,8 as well as fetal macrosomia, respiratory distress syndrome and type 2 diabetes in the offspring.9 Moreover, maternal diabetes status may cause microbiota dysbiosis in the meconium of newborns.10 Considering that the meconium is usually expelled and collected several hours after delivery, microbes may gain and reproduce during this period, the meconium microbiota was suspected to be the result of postnatal acquisition.11 However, sample types such as pharyngeal aspirates that are located deeply in the neonatal body and can be collected immediately at birth are ideal materials to examine the effects of GDM on initial microbiota.
In this study, we performed 16S rRNA gene and metagenomic sequencing on the specimens collected from multiple body sites of 486 pregnant Chinese women and neonates within seconds of delivery. The presence or absence of GDM was included to determine the variations of maternal and neonatal microbiota corresponding to the health status of the pregnant women, as well as to illustrate underlying microbial differentiations in early life and their implications for health outcomes.
Materials and methods
Subject recruitment
Pregnant women and neonates were recruited at Beijing Aviation General Hospital and Wenzhou People’s Hospital. Pregnant women with GDM (GDM+) were diagnosed by specialised doctors according to the results of OGTT and were recruited as a case group. Healthy pregnant women (GDM−) who had no history of other diseases, especially periodontitis, type 2 diabetes and bacterial vaginosis, served as the control group.
Sample collection
Sampling operations were executed by trained professionals under strict aseptic conditions and a uniform protocol. Amniotic fluid (5–10 mL per sample) was collected during delivery. About 0.5–1 mL of the newborn’s saliva and 2–3 mL of pharyngeal aspirate were sampled using sterile pipettes within 60 s of delivery before the umbilical cord was cut. Approximately 1 g meconium was sampled using a sterile plastic spoon within 24 hours as soon as the newborns passed their first intestinal discharge. Most neonatal specimens were collected from caesarean delivery neonates. Fresh saliva, stool and vaginal secretion were sampled from pregnant women 1–2 days before delivery. Vaginal specimens were collected from the posterior fornix under direct visualisation using three swabs. All specimens were placed in sterile tubes or vials, immediately frozen upon collection at –20°C, and then transported to the laboratory and stored at –80°C until total DNA extraction for latter sequencing.
DNA preparation
In a strictly controlled, separate and sterile workplace, approximately 0.2 mL saliva, pharyngeal aspirate and amniotic fluid samples were mixed with equal volumes of PBS and Qiagen’s AL buffer by pulse vortexing for 30 s. Total DNA was extracted in triplicate from the suspension of each sample type using QIAamp DNA Mini Kit (Qiagen, Valencia, CA). Faecal samples (~200 mg) were resuspended in Qiagen’s ASL buffer and homogenised for 2 min. Total faecal DNA was extracted from the supernatant using QIAamp DNA Stool Mini Kit (Qiagen). Two vaginal swabs were vortexed vigorously in 0.5 mL PBS and 0.5 mL Qiagen’s AL buffer for 2 min to resuspend and lyse the microbial cells. The resulting lysate was processed using the QIAamp DNA Mini Kit (Qiagen). Isolated DNA was eluted in 50 µL of distilled water. The DNA quality and concentration of each sample were measured by agarose gel electrophoresis and a Qubit 3.0 fluorometer (Life Technologies, Waltham, MA) and before downstream processing.
High-throughput sequencing
For each sample, we amplified variable regions 3 and 4 (V3-V4) of the 16S rRNA gene using modified 341F and 805R primers. Purified positive amplicons with different index sequences were pooled in equimolar amounts and then sequenced on a HiSeq2500 platform (Illumina, San Diego, CA) with v2 reagents and 2×250 bp cartridge. Metagenomic DNA of 50 meconium samples (~0.5 µg per sample) was sheared into ~300 bp fragments using a focused ultrasonicator (Covaris, Woburn, MA). Sequencing libraries were constructed with the sheared fragments using TruSeq Sample Preparation Kit v2 (Illumina). Insert length and integrity of the libraries were assessed by a Fragment Analyzer (Advanced Analytical Technologies) before paired-end (PE) sequencing (2×125 bp) on a HiSeq2500 platform.
16S rRNA sequence analysis
Raw sequencing reads of the 16S rRNA gene sequences were quality filtered and analysed using QIIME V.1.8.0.12 The operational taxonomic units (OTU) were classified taxonomically using the Greengenes 16S rRNA gene reference database.13 The taxonomic composition of microbial communities was visualised using Calypso.14 Community clustering was measured by unweighted UniFrac distance based on the normalised OTU table. Together with a data set of the neonatal skin microbiota retrieved from a recent study,15 Bray-Curtis dissimilarity between different sample types was calculated using the R package ecodist. Adult human microbial data sets including 121 anterior nares, 312 buccal mucosa, 73 mid-vagina, 102 saliva and 55 stool samples were downloaded from the Human Microbiome Project (HMP). Clustering analysis on the bacterial community structure was performed at the genus level.
Partial least squares discriminant analysis and linear discriminant analysis
Partial least squares discriminant analysis (PLS-DA) and linear discriminant analysis (LDA) were used to identify taxonomic and functional changes in maternal and neonatal microbiota in response to GDM.16 17 PLS-DA was performed using the R package mixOmics (http://www.mixOmics.org). The discriminatory power of each taxon was quantified by means of variable importance for the projection (VIP) parameters. VIP scores were used to rank the ability of different taxa to discriminate between different groups. A taxon with a >1.0 VIP score was considered important in the discrimination. KEGG categories with a total relative abundance across all samples of <1% were removed. The effect sizes identified as significantly (p<0.05) different between groups were assessed to find discriminatory KEGG orthology (KO) by LDA using online Galaxy (https://huttenhower.sph.harvard.edu/galaxy). Only the functional categories with log LDA scores of >2.0 were considered as differential signatures that better discriminate between GDM+ and GDM− groups.
Concordance and co-occurrence analysis
OTU enrichment was calculated for each sample type by comparing both mean and median relative abundance between GDM+ and GDM− groups. For the co-occurrence network analysis, the bacterial correlations in the maternal and neonatal samples with and without GDM were computed respectively based on the relative abundance of each genus using SparCC with 100 bootstraps to estimate the p value.18 The correlation values with p value <0.05 were retained. The co-occurrence networks of maternal oral, intestinal and vaginal microbiota were visualised using igraph (http://igraph.org). Closeness and eigenvector of the nodes were calculated to measure node centralities in each network. The concordance of bacterial correlations between maternal and neonatal microbiota associated with GDM was inferred by counting the same bacterial correlations across sample types and was visualised with OmicCircos.19
Metabolic pathway reconstruction
KO functional profiling of the neonatal meconium microbiota was performed using assemblies derived from whole-genome shotgun (WGS) sequencing data. Low-quality reads were first trimmed from raw sequencing data using Trimmomatic.20 High-quality reads were assembled de novo to contigs using metaSPAdes and metaSort with default parameters.21 22 Next, we performed gene prediction using PROKKA V.1.11 for these scaffolds,23 and the predicted proteins were assigned to the KO using the KEGG Automatic Annotation Server.24 Trimmed high-quality reads locating on the given scaffolds were counted to calculate the abundance of KOs in each sample using Burrows-Wheeler Aligner (BWA).25 The matrix was normalised by dividing the absolute amount of each functional gene by the total number of reads that assigned to functional genes in each sample. Supervised analysis was conducted using LDA to elicit the microbial functional pathways that were differentially expressed in the gut microbiota of neonates delivered by women with different glucose level statuses.
Virome and subspecies analysis
After removing human DNA sequences, the taxonomy and abundance of virus and bacteriophage in the neonatal meconium microbiota were measured by aligning WGS sequencing reads to a custom database. This database was built by combining genomic sequences of viruses and phages from NCBI and PhAnToMe after removing duplicates as described in our previous study.26 Reads were BLASTX against this database with a threshold of E-value <10−5 using DIAMOND.27 The lowest common ancestor of all taxa in the collected BLAST hits was computed and used to determine taxonomic origin. Alpha diversities of the viral and phage communities were calculated using vegan (https://cran.r-project.org/web/packages/vegan). For the strain-level profiling of bacterial species from metagenomic sequencing data, genomes of 35 Escherichia and 69 Lactobacillus isolated from human oral cavity, gut or vagina were obtained from the HMP and were taken as references. The WGS reads of each meconium sample were mapped to these reference genomes using BWA.
Results
We enrolled 140 neonates and 346 pregnant women and acquired 1062 total samples, including saliva, pharyngeal aspirates, meconium and amniotic fluid from the neonates, and saliva, faeces and vaginal secretions from the pregnant women (online supplementary table S1). The vast majority of the neonatal samples, especially amniotic fluid, were collected from caesarean delivery (76 C-sections vs 17 vaginal deliveries) to avoid possible contaminations from the vaginal flora. For each sample, the 16S rRNA V3-V4 regions were sequenced, and 233 samples failed to generate data and were removed from the following analyses.
Supplementary file 1
The 16S rRNA gene sequencing of the eligible 248 neonatal (64 amniotic fluid, 20 oral, 81 pharyngeal and 83 meconium) and 581 maternal (175 oral, 147 intestinal and 259 vaginal) samples yielded 309 million PE reads (2×250 bp), with ~373 241 reads per sample. Each pair of PE reads was merged into one sequence by overlaps. The overlaps were 25–75 bp for each sample (online supplementary figure S1A), and more than 80% of PE reads were successfully merged into long sequences in 94.3% of samples (online supplementary figure S1B). In addition, according to the Good’s coverage and rarefaction curve estimation (online supplementary figure S1C,D), the number of sequences can well represent the microbial diversity of each community. There was little discrepancy when different methods were selected for OTU classification (online supplementary figure S1E). All the neonatal samples and half of the maternal samples (n=289) collected at the time of delivery were used to examine the possible microbial shift of multiple body sites suffering from GDM (online supplementary table S2). The remaining half of the maternal samples (95 oral, 98 intestinal and 99 vaginal samples) were served as validation data sets. A subset of the meconium samples was selected randomly for metagenomic sequencing, and 48 out of 50 samples (24 for GDM+ and 24 for GDM−) were sequenced successfully, which generated more than 1.5 billion PE reads (2×125 bp).
Altered maternal microbiota of multiple body sites in GDM
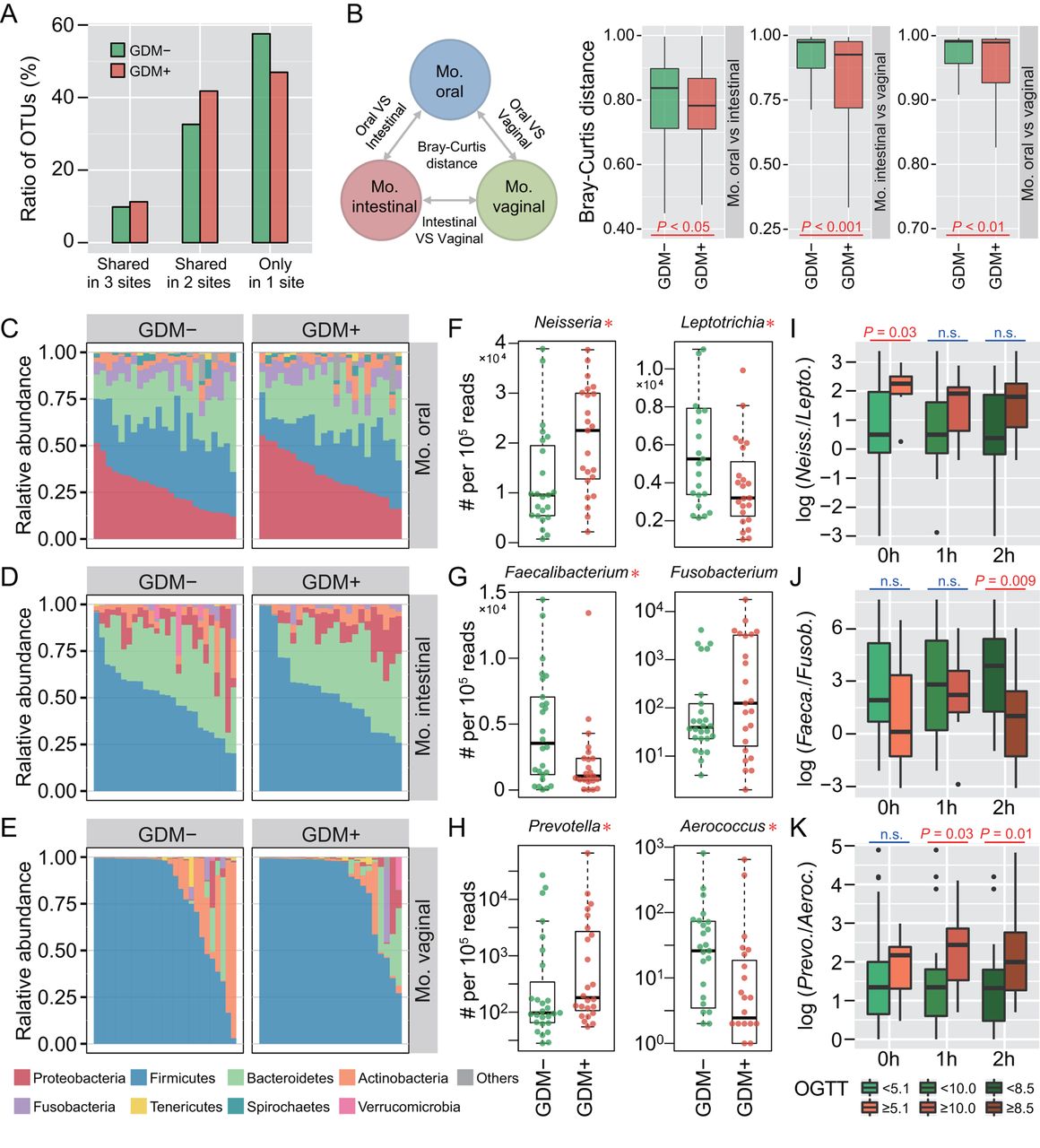
To investigate whether maternal microbiota can be driven by glucose levels, we first explored the microbial shift of pregnant women who were diagnosed suffering from GDM. Ratios of shared or unique OTUs in maternal oral, intestinal and vaginal samples were assessed to detect whether GDM has a similar effect on different body sites. We found that patients with GDM (GDM+) shared more OTUs in at least two body sites than non-GDM pregnant women (GDM−), with approximately 57.6% OTUs appearing in only one body site of GDM− compared with 47.0% in GDM+ (figure 1A). Additionally, we calculated Bray-Curtis distances using normalised OTU abundance. The distances between samples of any two body sites were significantly smaller in GDM+ than in GDM− (p<0.05, Mann-Whitney test) (figure 1B). These results indicate that microbial shift in maternal microbiota of different body sites may be associated with GDM during pregnancy.
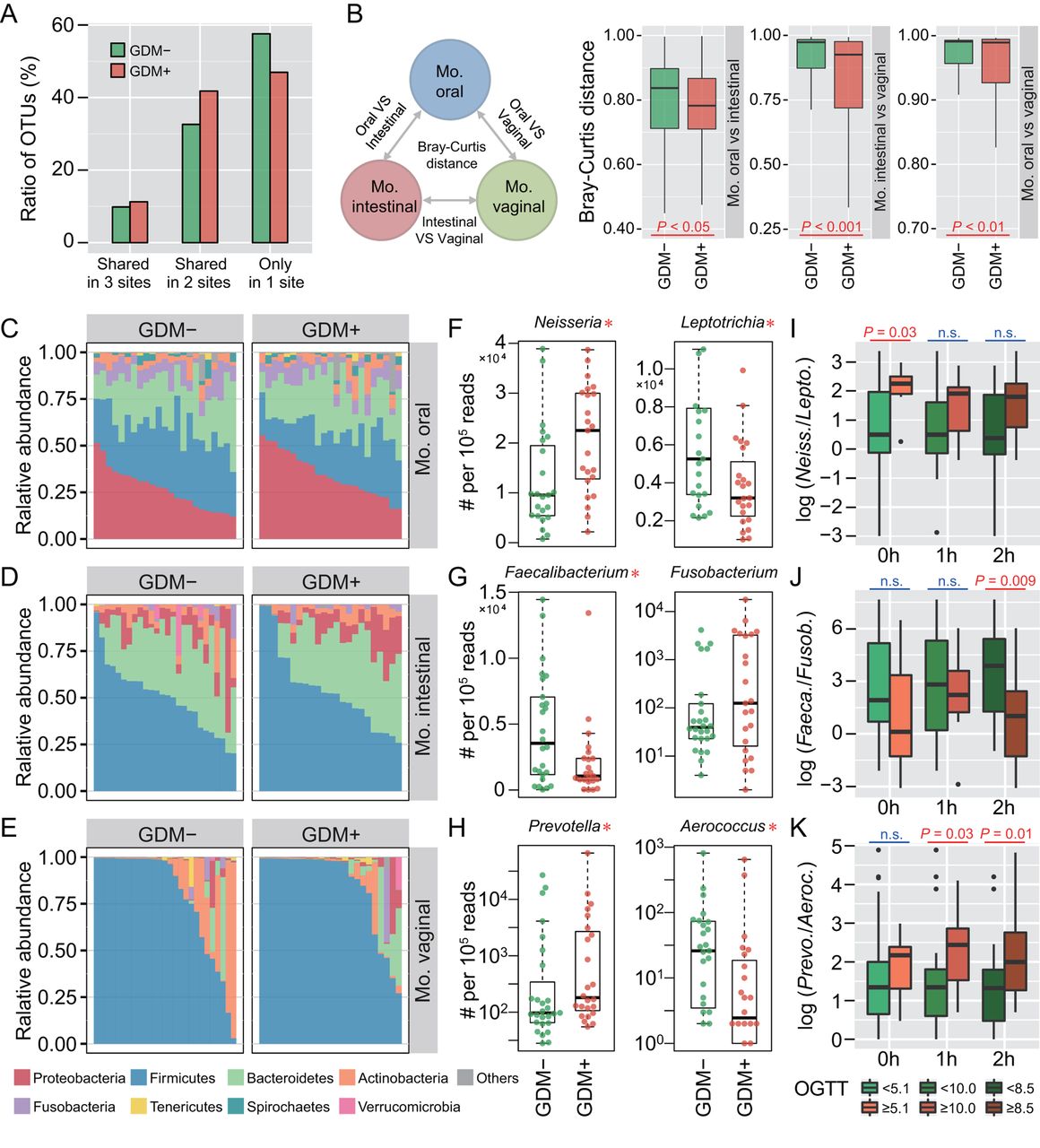
Microbial variations of the pregnant women suffering from gestational diabetes mellitus (GDM). (A) Proportions of shared operational taxonomic units (OTU) among maternal oral, intestinal and vaginal microbiota. Shared OTU denotes that a certain OTU was detectable in two or three body sites. (B) Bacterial community dissimilarities between any two body sites of maternal samples. Bray-Curtis distances were independently calculated for GDM+ and GDM− groups. VS represents versus. Statistical significance was determined by the Mann-Whitney test. (C–E) Relative abundance of bacterial phyla in the oral, intestinal and vaginal microbiota of pregnant women. (F–H) Bacterial genera with significant (Mann-Whitney test with false discovery rate (FDR) correction; *p<0.1) differences between GDM+ and GDM− groups. Genus abundance in each sample was normalised to 100 000 reads. (I–K) Correlations between significantly differential bacteria and oral glucose tolerance test (OGTT) values. The labels 0 hour, 1 hour and 2 hours under box plots represent the OGTT testing time points. A pair of boxes indicates the bacterial ratios corresponding to the blood glucose levels below (left box) or above (right box) the threshold values (5.1 at 0 hour, 10.0 at 1 hour and 8.5 at 2 hours during OGTT). A significant difference (Mann-Whitney test, p<0.05) is indicated by the character in red colour, while ns in blue colour denotes a non-significant difference (Mann-Whitney test, p≥0.05).
To further demonstrate these variations corresponding to GDM in different body sites, we compared the bacterial abundance between groups at the phylum level. The largest changes occurred in the oral cavity, with more Proteobacteria (p<0.05, Mann-Whitney test with false discovery rate (FDR) correction using the Benjamini-Hochberg method) but less Firmicutes (p<0.01) found in GDM+ (figure 1C). No obvious variation in either intestinal or vaginal microbiota was shown (figure 1D,E). PLS-DA was then conducted to improve the resolution of GDM flora. Seven to nine genera with a VIP value of >1 were detected from three body sites (online supplementary figure S2A–F). To validate the microbial variations corresponding to GDM, 95 oral, 98 intestinal and 99 vaginal samples collected from a second cohort of pregnant women were used. About 77.8% (7 out of 9), 88.9% (8 out of 9) and 71.4% (5 out of 7) VIP genera identified in data sets of the first batch could be validated in the second batch, and most of them exhibited consistent enrichment or depletion in the two cohorts (figure 1F–H and online supplementary figure S2G–I). To further examine the relationship between these genera and GDM, we associated their abundance with the results of OGTT. The threshold values (5.1 at 0 hour, 10.0 at 1 hour and 8.5 at 2 hours during OGTT) are based on the diagnostic criteria recommended by the International Association of the Diabetes and Pregnancy Study Groups in 2011. As shown in figure 1I–K, the relative abundance of these bacteria was highly correlated to OGTT values (p<0.05, Mann-Whitney test).
Microbiota of neonates whose mothers suffered from GDM differs significantly from that of controls
To explore the possible effects of GDM on neonatal microbiota, we next investigated the community composition and structure of the newborns. In the principal coordinate analysis (PCoA) of unweighted UniFrac distances, samples were clustered primarily by body sites, with distinct maternal oral, intestinal and vaginal clusters, and neonatal pharyngeal, meconium and amniotic fluid clusters (figure 2A). Body site-specific clustering was observed in the neonatal samples from different body sites although the separation was not as remarkable as that of the mothers’. In addition, we calculated the Bray-Curtis distance within or across body sites to quantify community similarity of the same sample type. Community distances within samples obtained from the same body site, as expected, were smaller than those between different body sites (figure 2B), which further confirmed the clustering results by PCoA. We calculated Bray-Curtis distances between neonatal and maternal samples. As a result, with the only exception of a few outlier samples of vaginally delivered babies that obviously originated from the mother’s vaginal community, mode of delivery had only a very slight effect on neonatal microbiota (figure 2C). Moreover, the neonatal microbiota exhibited higher alpha diversities than those of the vaginal microbiota (figure 2D). Species richness of the pharyngeal and amniotic fluid samples was even comparable to the maternal oral and intestinal communities, whose microbial diversities were rather high (online supplementary figure S3). The neonatal microbiota of each body site harboured a unique community structure (figure 2E). By contrast, the maternal communities of those three body sites showed a typical structure of the adult human microbiota (online supplementary figure S4).

Colonisation and body site specificity of the neonatal microbiota at birth. (A) Principal coordinate analysis (PCoA) of the unweighted UniFrac distances for maternal oral, intestinal and vaginal samples, and neonatal amniotic fluid, oral, pharyngeal and meconium samples. Ellipses represent a 95% CI. (B) Bacterial community dissimilarities within and between body sites. The labels Phar, Inte, Amni and Vagi represent pharyngeal, intestinal, amniotic fluid and vaginal microbiota, respectively. The average Bray-Curtis distance for each group of pairwise comparisons is provided. Red and blue shadows indicate the smallest and the largest values in each column, respectively. (C) Effect of delivery modes on neonatal bacterial communities. Three edges of a triangle represent maternal oral (brown), intestinal (yellow) and vaginal (green) microbiota. Each hollow point represents a neonatal sample, and its vertical distance to three edges of the triangle indicates the average Bray-Curtis distance between this sample and the maternal samples. Points closer to the edges denote that such neonatal samples are more similar in bacterial community to the indicated maternal microbiota. (D) Alpha diversities of neonatal and maternal microbiota. The violin with box plot shows the median and IQR of the diversity indices of a sample type, and the width of the violin represents the density distribution of the indices. (E) Taxonomic profiling of neonatal pharyngeal, intestinal and amniotic fluid microbiota. The average relative abundance of each taxonomic category is represented by the thickness of a branch. Each node in the phylogenetic tree represents one microbial taxon.
Considering that the neonatal microbiota may have been established at birth, we wondered if such microbial community would be shaped by the health status of gestational women during pregnancy. Accordingly, Bray-Curtis distances were calculated using the OTU abundance of the microbiome from amniotic fluid, pharyngeal and meconium samples to assess the effects of GDM on neonatal microbiota. We found that the distances were significantly smaller among the three sample types in GDM+ than those in GDM− (p<0.001, Mann-Whitney test) (figure 3A), which implied that GDM might influence neonatal microbiota and therefore result in community homogeneity across multiple sample types. This variation tendency in community distance was consistent with that which occurred in the maternal oral, intestinal and vaginal microbiota (figure 1B). PCoA illustrated the minimal dissimilarities between the health and disease status of the amniotic fluid (Adonis p<0.001, R 2=0.069), pharyngeal (Adonis p<0.001, R 2=0.050) and meconium (Adonis p<0.05, R 2=0.026) microbiota (figure 3B–D). The samples were clustered primarily to GDM+ and GDM− assemblages. These results indicate that the microbial composition and variation of both mother and newborn could be driven by the health status of the pregnant woman. The effects of GDM on microbes in pregnancy might be vertically transmitted to the baby during pregnancy.

Neonatal microbiota associated with gestational diabetes mellitus (GDM). (A) Bacterial community dissimilarities between neonatal microbiota. Bray-Curtis distances were calculated for GDM+ and GDM− samples independently. Statistical significance was determined by the Mann-Whitney test. (B–D) Principal coordinate analysis (PCoA) of the unweighted UniFrac distances for amniotic fluid, pharyngeal and meconium microbiota. Ellipses represent a 95% CI. (E–G) The most abundant genera with significant difference between GDM+ and GDM− groups. Genus abundance in each sample was normalised to 100 000 reads. Mann-Whitney test with false discovery rate (FDR) correction: none. *p<0.1; *p<0.05; **p<0.01; ***p<0.001. A pair of taxa was highlighted in dark colour because their relative abundance was the largest among the significantly enriched and depleted genera of GDM+ in each sample type. (H–J) Correlation between significantly differential bacteria of neonatal microbiota and oral glucose tolerance test (OGTT) values. The labels of 0 hour, 1 hour and 2 hours represent three testing time points of the OGTT. A pair of boxes shows the bacterial ratios corresponding to the blood glucose levels below (left box) or above (right box) the threshold values (5.1 at 0 hour, 10.0 at 1 hour and 8.5 at 2 hours during OGTT). Mann-Whitney test; *p<0.05; **p<0.01; ***p<0.001; ns p≥0.05.
Bacterial abundance between GDM+ and GDM− groups was further compared at the genus level to clarify the body site-specific variations associated with GDM in the neonatal microbiota. As shown in figure 3E–G, amniotic fluid samples possessed more genera with significant intergroup differences than either the pharyngeal or the meconium samples (p<0.1, Mann-Whitney test with FDR correction) in the top 30 most abundant genera, indicating a greater microbial differentiation in the amniotic fluid (figure 3B–D). To examine the correlation between discriminatory bacteria and GDM, we selected the most abundant enriched or depleted genera, and calculated the correlation of their abundance to the corresponding maternal OGTT values. As a result, strong correlations were observed in several groups (figure 3H–J and online supplementary figure S5). We also performed PLS-DA analysis in the neonatal samples to identify differential signatures between health and disease groups (online supplementary figure S6). VIP genera such as Prevotella, Streptococcus, Bacteroides and Lactobacillus were prevalent in multiple sample types of maternal and neonatal microbiota, reflecting their possible significance to GDM response across generations and body sites.
Concordance of microbial variation between mothers and neonates suffering from GDM
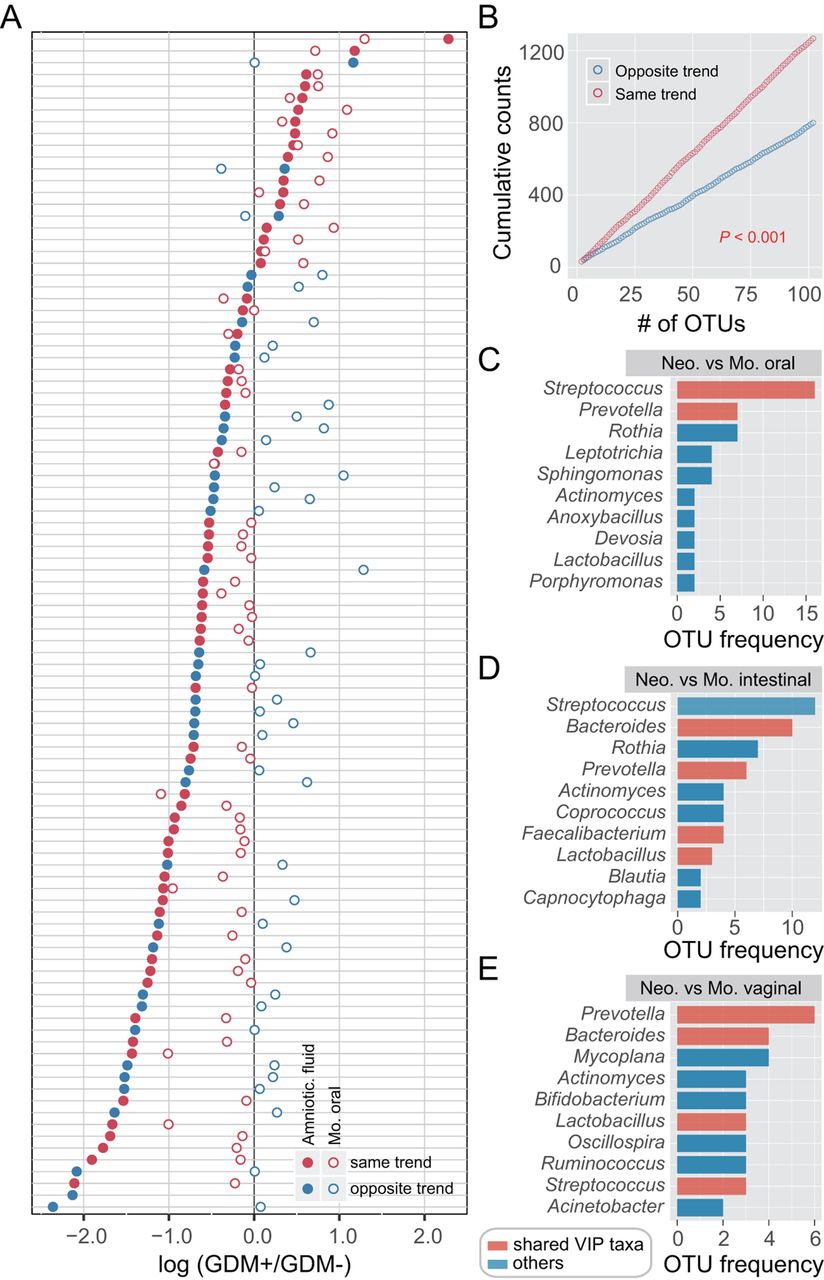
Given that both maternal and neonatal microbiomes of multiple body sites were notably altered with GDM, we sought to investigate the concordance of microbial variation between mothers and neonates. We first calculated the number of OTUs enriched or depleted in neonates with GDM and then focused on those shared in maternal samples. An OTU that was enriched (or depleted) in both maternal and neonatal microbiota of GDM+ was defined as varying by the same trend. In contrast, an OTU that was enriched in the neonatal microbiota but depleted in the maternal microbiota of GDM+ was defined as the opposite trend, and vice versa. Accordingly, the taxonomy and number of OTUs varying by the same trend across maternal and neonatal microbiota were counted, and a large number of high abundant OTUs were found to vary with the same trend. The most remarkable concordance in microbial variation was shown between the amniotic fluid and maternal oral microbiota (figure 4A,B and online supplementary figure S7), in which the majority of the most abundant OTUs varied with the same trend. In addition, nearly 100 OTUs were found to vary with the same trend in one maternal and three neonatal sample types. Prevotella, Streptococcus and Bacteroides were the most prevalent taxa showing the concordance of microbial variation between mothers and neonates (online supplementary figures S8–S11). Together with Lactobacillus and Faecalibacterium (figure 4C–E), these bacteria were consistent with the shared VIP genera of maternal and neonatal sample types (online supplementary figures S2 and S6), likewise reflecting their importance to GDM response. These observations proved that, although there was body site-specific variation, GDM may have similar effects on both maternal and neonatal microbiota.

Similar microbial shifts between maternal and neonatal microbiota associated with gestational diabetes mellitus (GDM). Operational taxonomic unit (OTU) abundance in each sample was normalised to 100 000 reads. (A) Concordancy of OTU variations between amniotic fluid and maternal oral microbiota. The average relative abundance of the top 100 most abundant OTUs was compared between GDM+ and GDM− groups. Solid points represent OTUs of amniotic fluid microbiota, and hollow points represent maternal oral microbiota. Red points denote OTUs varying by the same trend, while blue points denote the opposite trend. (B) Cumulative counts of OTUs varying by the same trend or the opposite trend. p<0.001, t-test. (C–E) The most prevalent taxa showing the concordance of microbial variation between mothers and neonates. Each colour bar indicates the OTU frequency of a certain genus. The bar in red represents a variable importance for the projection (VIP) genus associated with GDM that is shared among maternal and neonatal sample types.
To investigate the concordance of microbial co-occurrence between mothers and neonates, we next computed the correlations between bacterial genera in the maternal and neonatal samples with and without GDM. Approximately 47–62 genera (nodes) and 60–134 connections (edges) were retained under different correlation cut-offs (0.4–0.5) (online supplementary figure S12A,B) in the co-occurrence networks of the maternal oral, intestinal and vaginal microbiota. When comparing the networks of GDM+ with those of GDM−, their components and topographies were quite distinct, and the most notable difference comes from the VIP genera’s connections to other bacteria (figure 5A–C). To quantify such difference, we counted the number of edges and the centrality of nodes in the maternal microbial networks under different GDM status. Despite having a few overlapped edges, dozens of edges were specific to GDM+ or GDM− (figure 5D), and the closeness and eigenvector of shared nodes between GDM+ and GDM− were also quite different (figure 5E and online supplementary figure S12C). Furthermore, an intragroup difference was always evident in each correlation value interval (online supplementary figure S12D–F). As for neonates, the bacterial correlations were also distinct between GDM+ and GDM− (figure 5F,G and online supplementary figure S12G). For example, in the pharyngeal microbiota, about 69.2% correlations with a >0.4 correlation value only existed in GDM+, while ~18.7% were only present in GDM− (figure 5F). Similar findings were also observed in meconium, with 76.9% and 15.3% unique correlations present in GDM+ and GDM−, respectively (figure 5G). Remarkably, the bacterial correlations were highly conserved between maternal and neonatal samples associated with GDM (figure 5H and online supplementary figure S12H). About 88.8% of all bacterial connections had the same co-occurrence trend between generations, and 69.1% were only detected in GDM+ but not in GDM−. Among them, positive correlation between Neisseria and Haemophilus, and negative correlations between Lactobacillus and several VIP bacteria (including Porphyromonas and Bacteroides) were present in almost all maternal and neonatal body sites (online supplementary figure S12I). In addition, the connections containing Blautia, Coprococcus, Roseburia and Sutterella showed high frequencies of co-occurrence across multiple sample types, suggesting their universal attributions to microbiota associated with GDM. According to these results, it can be assumed that the consistency of microbial variation across mothers and neonates suffering from GDM has happened in bacterial abundance and in their interactions.
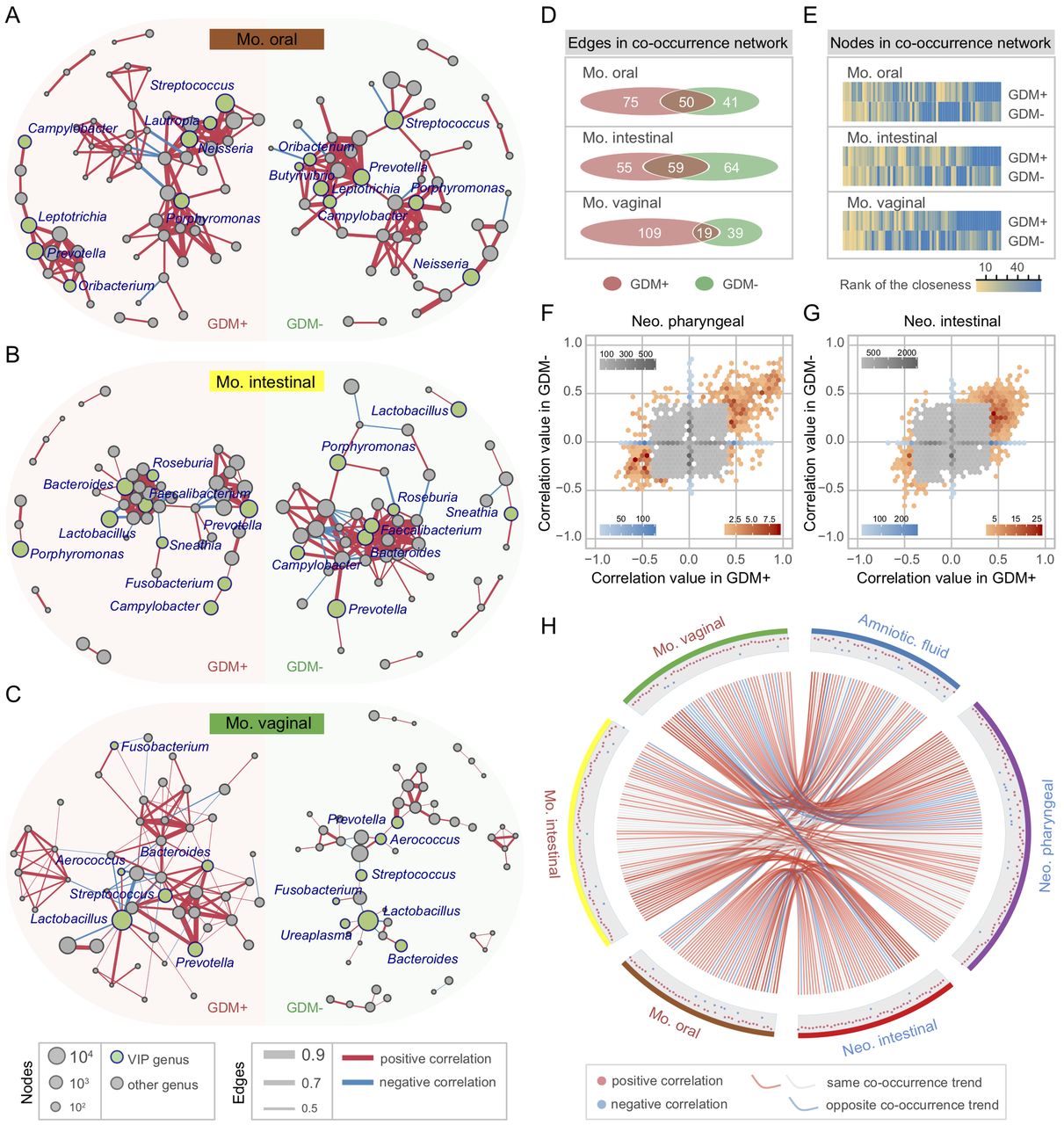
Bacterial co-occurrence network and concordance between maternal and neonatal microbiota associated with gestational diabetes mellitus (GDM). (A–C) Co-occurrence networks of maternal oral, intestinal and vaginal microbiota of GDM+ (left panel) and GDM− (right panel). The co-occurrence network was inferred for each maternal sample type by a pairwise correlation of relative abundance (normalised to 100 000) for all genera. Each node in the network indicates a bacterial genus. Node size represents the average relative abundance of one genus in each maternal sample type. Nodes in green colour show variable importance for the projection (VIP) genera associated with GDM. Only the bacterial connections (edges) larger than cut-offs (correlation values >0.4, 0.45 and 0.5 in the three networks, respectively) are retained. Edge width represents the correlation value supporting this connection. Edge colour shows positive (red) and negative (blue) correlations, respectively. (D, E) Discrepancies of the bacterial co-occurrence networks between maternal GDM+ and GDM−. The number of unique and shared edges, and centralities (rank of the closeness) and discrepancies of nodes in GDM+ and GDM− co-occurrence networks were counted, respectively. (F–G) Discrepancies of the bacterial correlations between neonatal GDM+ and GDM− microbiota. Each bin shows the number of the same pair of bacterial correlations occurred in both GDM+ and GDM−, with the colour changes representing weak (grey) or strong (blue and red) correlations, respectively. (H) Concordance of bacterial correlations between maternal and neonatal microbiota associated with GDM. The concordance was inferred by counting the same bacterial correlations (cut-off >0.4) across different sample types. Each point in the outer cycle represents one connection of two correlated bacteria (at least one is VIP genus). The curve in red (occurred only in GDM+) and grey (occurred in both GDM+ and GDM−) denotes the same co-occurrence trend of such connection between maternal and neonatal microbiota, while the curve in blue denotes the opposite trend.
Altered microbial metabolism and increased viral load in GDM+ meconium
Neonatal intestinal microbiota is important for the metabolism and immunity of developing infants and adults.28–30 Therefore, we attempted to assess the potential risks of microbial shift to neonates by profiling the meconium microbiota, and thus a subset of the meconium samples was selected for WGS sequencing. Forty-eight samples were sequenced successfully, which yielded more than 375 Gb WGS sequencing data. Metagenomic taxonomic profiling on WGS sequencing data of meconium samples was performed using MetaPhlAn.31 Compared with 16S rRNA-based taxonomic profiles of meconium, metagenomic sequencing generates a quite similar bacterial community structure (online supplementary figure S13). We looked for metabolic differences between the intestinal microbiota of neonates delivered by women with and without GDM. As a result, smaller Pielou indices of KOs were shown in the GDM+ group (figure 6A), which means that the neonatal microbiota of this group had lower evenness than in GDM−. LDA was used to identify intergroup differences in KOs, and a large number of KOs were depleted in GDM+, denoting the decline in metabolic capabilities compared with the gut microbiota of neonates delivered by women without GDM (figure 6B and online supplementary table S3). The lack of such metabolic pathways may have adverse effects on nutrition absorption or certain metabolic capacity of the neonates.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Microbial and functional variation in meconium microbiota associated with gestational diabetes mellitus (GDM). (A) Evenness of the KEGG orthology (KO). (B) The number of enriched KOs. (C, D) Distribution of viral richness and evenness. (E, F) Relative abundance of herpesvirus and poxvirus. Viral abundance in each sample was normalised to 100 000 reads. (G, H) Prevalence of mastadenovirus and papillomavirus in meconium. (I, J) Significant differentiation of Escherichia and Lactobacillus strains between GDM+ and GDM−. Genomes of Escherichia and Lactobacillus strains isolated from the human oral cavity, gut or vagina were downloaded from the Human Microbiome Project (HMP) and were taken as references. Sequencing reads were mapped to these references, and strain abundance in each sample was normalised to 100 000 reads.
We also compared the WGS data against known viral genomes to detect possible alterations in the meconium virome. Although the higher richness (number of viral species observed) of eukaryotic viruses in meconium of GDM+ was not significant (figure 6C), both the viral evenness (Pielou index in each sample) and alpha diversity indices were significantly lower than those of GDM− (p<0.05, Mann-Whitney test) (figure 6D and online supplementary figure S14A–D). In contrast, there was no obvious intergroup difference in the alpha diversities of bacteriophages (online supplementary figure S14E–H). Further investigation estimated the relative abundance of recognisable viral taxa and identified four types of viruses that varied by the health status of pregnant women, including herpesvirus, poxvirus, mastadenovirus and papillomavirus. Among them, herpesvirus and poxvirus were more abundant in the GDM+ group (figure 6E,F), and high-abundance mastadenovirus and papillomavirus were also more prevalent in the samples of this group (figure 6G,H). These findings suggested that pregnant women suffering from GDM increase the prevalence of viruses in the intestinal microbiota of neonates, and may give rise to a population highly vulnerable to expose to these viruses.
Finally, to investigate the intraspecific diversity of certain neonatal intestinal bacteria, we aligned WGS sequencing data of meconium samples to currently available Escherichia and Lactobacillus reference genomes, which include genomes of 35 Escherichia and 69 Lactobacillus isolated from the human oral cavity, gut and vagina retrieved from the HMP (figure 6I,J). Only the uniquely mapped reads to these genomes were taken into account. A large number of reads could be assigned to the genomes of reference strains isolated from the human oral and gut besides vagina, which further corroborated the observation that neonatal microbiota is not just derived from the vagina. In addition, we observed that both Escherichia and Lactobacillus showed strain-level differences between neonates delivered by mothers with and without GDM. Notably, Lactobacillus iners strains were significantly more abundant in GDM+ (p<0.05, Mann-Whitney test with FDR correction) (figure 6J). These findings demonstrated that Lactobacillus spp and their subspecies in newborns may be shaped by the GDM status of their mothers.
Discussion
Most of the neonatal samples in our study were collected within seconds of C-section delivery, which allowed us to eliminate the possibility of microbial exposure and colonisation in vitro. Microbes present in these samples denote the establishment of the fetal microbiota, unlike the detection of microbes in meconium, which was doubted to be the result of postnatal acquisition.11 The 16S rRNA gene sequencing detected bacterial signals from every neonatal body site, and some body sites (such as the pharyngeal and amniotic fluid microbiota) even have relatively high species richness. Coupled with the remarkable difference in taxonomic composition and community structure between the neonatal and maternal microbiota, these results reveal that the microbial nucleic acids detected in the amniotic fluid and neonatal samples are not all contaminants from the mother, as previously suspected.32 Additionally, as shown in the analyses of PCoA and Bray-Curtis dissimilarity, it is very clear that neonatal samples were clustered according to sample types. There would be no observable clustering across the different sample types if their microbes came from potential maternal or reagent contamination. Moreover, living bacteria have been successfully cultured from amniotic fluid of both GDM+ and GDM− pregnant women (data not shown). Despite that one review used germ-free mice as an example to refuse the hypothesis of microbial colonisation in utero,11 we speculate that the differences in placental cellular structures and gestation length between mice and humans may be the reason. Two layers of syncytiotrophoblasts and one complete layer of cytotrophoblasts in a mouse placenta provide strong barriers to prevent vertical microbial transmission from mother to offspring,33 and the short gestation length (19–21 days) also reduces the chance of bacteria entering the mouse uterus. In contrast, humans have only one layer of syncytiotrophoblasts in the placenta, and the longer gestation gives the bacteria more chances to enter the uterus. Taxonomic profiling of both 16S rRNA and metagenomic sequencing demonstrated that Enterobacteriaceae and Escherichia are the most abundant taxa in meconium samples. It is consistent with previous studies on the microbiota of either placenta34 or meconium.15 These results suggest that Escherichia is prevalent in utero and during early life.
Combined with the results of previous studies,10 35 we believe that GDM can affect the microbiota of both pregnant women and neonates at multiple body sites during pregnancy. Although OGTT was taken as a gold standard for diagnosing GDM, the monitoring needs blood test and is only conducted between 24 and 28 weeks during gestation.6 There is a strong correlation between several discriminatory bacteria and the OGTT values. For example, in the maternal microbiota, oral Neisseria/Leptotrichia ratios were positively correlated to blood glucose values, especially fasting blood glucose concentrations, which reflect the daily secretory capacity of basal insulin. Low intestinal Faecalibacterium/Fusobacterium ratios corresponded to high blood glucose values, where the correlation was highly significant between the bacterial ratios and 2-hour blood glucose levels, representing the regulatory and recovery capability after sugar intake. In addition, high vaginal Prevotella/Aerococcus ratios were also correlated to high blood glucose values, particularly to the 1 and 2-hour values of the OGTT. Although maternal samples collected before OGTT are needed, it may provide a promising approach to develop GDM biomarkers, especially for the oral microbes which are very convenient for sampling and have the largest microbial variations associated with GDM.
In both maternal and neonatal microbiota, microbial variations tended to be convergent across body sites in GDM+, which exhibited more similar community structure than those in GDM−. Microbes varying by the same trend between the maternal and neonatal microbiota were also observed in our study, revealing the intergenerational concordance of microbial variations associated with GDM. These results suggest that GDM may have similar effects on the microbiota across generations and body sites. Such microbial shifts likely increase the risk of gestational complications or undesirable outcomes. Lower evenness but more depletion of KOs, as well as higher abundance or prevalence of some eukaryotic viruses may lead to declined metabolic capacities and more viral exposures to neonates with than without GDM. Additionally, L. iners is considered to be more conducive to the occurrence of abnormal microbiota, which may result in adverse health outcomes.36 37 L. iners has the smallest genome size among the Lactobacilli and lacks the genes necessary to synthesise amino acids de novo.38 A relatively high ratio of this strain may decrease the efficiency of the microbial amino acid metabolism in newborn’s digestive tract.
Here we describe the effects of maternal health status on microbial vertical transmission and early-life colonisation. The links between microbial dysbiosis during early life and a large number of diseases may note the long-term impacts of the initial microbiota on human health.39–41 Our study focuses on microbial contact and colonisation in early life as well as the shape of the neonatal microbiome. These observations will contribute to better understanding the origin of the human microbiota and microbial vertical transmission from mothers to babies. This study sheds light on another form of inheritance and highlights the importance of understanding early-life microbiome formation, which offers a promising approach of modulating initial microbial colonisation and interaction by maternal intervention to reduce the risk of adverse health outcomes.
References
Footnotes
JW, JZ and WS contributed equally.
Contributors FZ conceived the study. FZ and JW designed the study and prepared the manuscript. JW, JZ, XX, ZJ, YW, ZZ and HZ collected the samples and conducted the experiments. JW, WS, ND, YZ, PJ and FZ analysed the data. All authors approved the final version of the manuscript.
Funding This work was supported by grants from the National Natural Science Foundation of China (31722031, 31670119), the National Key R&D Program (2016YFC1200800) and the Key Research Program of the Chinese Academy of Sciences (KFZD-SW-219).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Wenzhou People’s Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.