Article Text

Abstract
Objective Hepatocellular carcinoma (HCC) is heterogeneous, especially in multifocal tumours, which decreases the efficacy of clinical treatments. Understanding tumour heterogeneity is critical when developing novel treatment strategies. However, a comprehensive investigation of tumour heterogeneity in HCC is lacking, and the available evidence regarding tumour heterogeneity has not led to improvements in clinical practice.
Design We harvested 42 samples from eight HCC patients and evaluated tumour heterogeneity using whole-exome sequencing, RNA sequencing, mass spectrometry-based proteomics and metabolomics, cytometry by time-of-flight, and single-cell analysis. Immunohistochemistry and quantitative polymerase chain reactions were performed to confirm the expression levels of genes. Three independent cohorts were further used to validate the findings.
Results Tumour heterogeneity is considerable with regard to the genomes, transcriptomes, proteomes, and metabolomes of lesions and tumours. The immune status of the HCC microenvironment was relatively less heterogenous. Targeting local immunity could be a suitable intervention with balanced precision and practicability. By clustering immune cells in the HCC microenvironment, we identified three distinctive HCC subtypes with immunocompetent, immunodeficient, and immunosuppressive features. We further revealed the specific metabolic features and cytokine/chemokine expression levels of the different subtypes. Determining the expression levels of CD45 and Foxp3 using immunohistochemistry facilitated the correct classification of HCC patients and the prediction of their prognosis.
Conclusion There is comprehensive intratumoral and intertumoral heterogeneity in all dimensions of HCC. Based on the results, we propose a novel immunophenotypic classification of HCCs that facilitates prognostic prediction and may support decision making with regard to the choice of therapy.
- hepatocellular carcinoma
- cancer immunobiology
- immunogenetics
- immunotherapy
- energy metabolism
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Intratumoral heterogeneity of hepatocellular carcinoma (HCC) has been revealed in genomes, epigenomes, and transcriptomes; however, these results were untranslatable.
A comprehensive and multidimensional analysis on multilesional HCC is lacking.
The prognosis of HCC is dismal, and the efficacy of immunotherapy for HCC is unsatisfactory.
What are the new findings?
Using whole-exome sequencing, RNA sequencing, mass spectrum-based proteomics and metabolomics, and mass cytometry techniques, we demonstrated significant heterogeneity of tumour cells in all dimensions while the heterogeneity of immune statues of tumour microenvironment was acceptable.
We found local immunity is a suitable target for HCC treatment, and identified three subtypes of HCC with distinctive tumour microenvironment in regard of immune statues, chemokines/cytokines, and cell metabolism.
PI3K-Akt signalling and ketone body metabolism of tumour cells are associated with CD8+ T cell infiltration in HCC microenvironment.
The novel immunophenotypic classification can be recapitulated simply using immunohistochemistry by testing two markers (CD45 and Foxp3) with clear cut-off values, showing values of prognostic prediction and guidance of decision making for choice of therapies.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
The comprehensive exploration of tumour heterogeneity in HCC provides informative resources for understanding of HCC progression and enlightening of development of novel treatment strategies.
A clinically practical classification may be useful in the management and monitoring of HCC patients.
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of tumour-related death worldwide and mostly results from viral infection and liver fibrosis.1 2 Only surgical resection and liver transplantation are potential cures for HCC; however, these patients were suffered from rapid postoperative recurrence and shortage of liver donors. There are few effective nonsurgical treatments for HCC, and no mutations that are targeted by available drugs have been identified in HCC patients.3 Existing medicines such as sorafenib and immune checkpoint inhibitors (ICIs) show unsatisfactory efficacy.4–6 Therefore, novel interventions are urgently needed.
Intratumoral heterogeneity is one of the main reasons that current therapies are ineffective in most types of cancers including HCC.7 Multilesion forms of HCC are common and can be derived from concurrent carcinogenesis, intrahepatic metastases, or clonal spreading.8 Thus, the issue of intratumoral heterogeneity is particularly important in HCC. Lesions in the same tumour may have distinctive genomic alterations, biological behaviours, and local microenvironments and can respond differently to therapies. Even tumour cells in different regions of the same lesion can harbour different somatic mutations. It is thus inadequate to treat HCCs using targets that are only specific at cell or lesion level because removal of a small portion of the total tumour cells might be useless for achieving overall tumour control. An ideal intervention for multilesional HCC would be capable of controlling all lesions in a patient and should also consider cost and clinical feasibility. Therefore, the ideal intervention should be able to treat a group of HCC patients with similar characteristics. To this end, understanding the intralesional, interlesional, and intertumoral heterogeneity of HCC is essential.9
Tumour heterogeneity exists from molecular level to organism level. Previous investigations of the intratumoral heterogeneity of HCC have focused on genomes, epigenomes, and transcriptomes using multiregional or single-cell analyses, revealing astonishing intratumoral heterogeneity.10 However, these attempts are not directly translatable to clinical setting and have thus far failed to generate novel treatments. Therefore, a comprehensive interrogation of HCC at different levels of intervention is warranted to facilitate the development of novel treatment strategies.
To profile the heterogeneity of HCCs at all intervention levels and to identify the level best suited for the development of new therapies, in this study, we acquired 42 samples from eight HCC patients and determined the degree of heterogeneity in the genomes, transcriptomes, proteomes, and metabolomes as well as in the local immune microenvironment. By analysing the heterogeneity at the cell-cell, subclone-subclone, lesion-lesion, and patient-patient levels, we unveiled the characteristics of the heterogeneity in tumours and tumour microenvironments in HCC and suggested a new tumour-immunity-based classification of HCC that may be valuable in guiding the choice of immunotherapy in HCC patients.
Methods
Detailed methods are enclosed in the online supplementary materials.
Supplemental material
Patients and samples
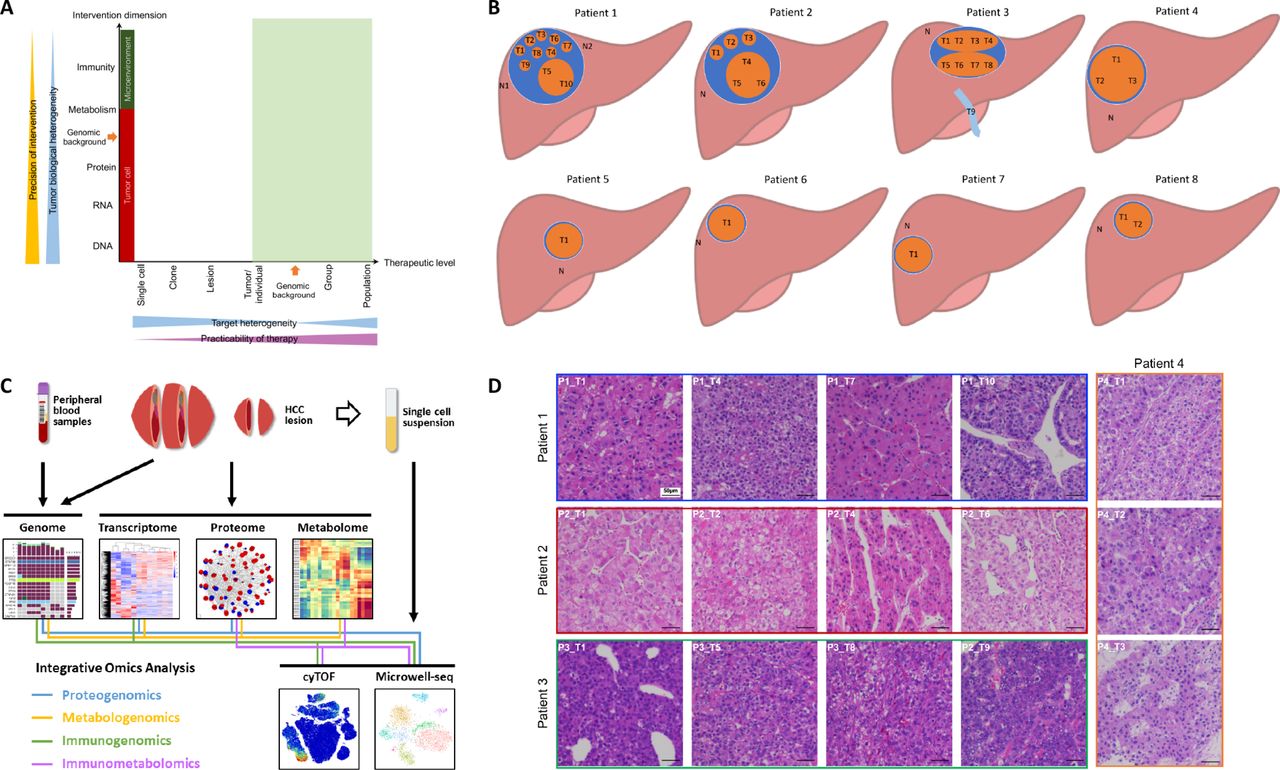
This study was approved by the ethics committee of the First Affiliated Hospital, Zhejiang University School of Medicine. Patients diagnosed with HCC who underwent operations during September 2018 and November 2018 were enrolled. All patients did not receive tumour-targeted therapy previously. The diagnosis of HCC was confirmed by pathological examinations. Samples with a volume of 5 mm ×5 mm×5 mm were collected from each site, cut into smaller pieces by scissors, and mixed thoroughly. A large part of each fresh sample was immediately placed in liquid nitrogen and then transferred to −80°C within 30 min for later whole-exome sequencing (WES), RNA sequencing (RNA-seq), and mass spectrum (MS) analyses. A small portion of the fresh sample was placed into a special tube for mass cytometry (cytometry by time-of-flight; CyTOF) analysis and kept at 4°C until use. For large tumours, equal volumes of samples from all sites were mixed together for single-cell analysis. Peripheral blood and normal liver tissue from 2 cm past the tumour edge were collected for controls.
Whole-exome sequencing
DNA was extracted from tissues using the DNeasy Blood & Tissue Kit (Qiagen, Venlo, Limburg, The Netherlands) following the manufacturer’s instructions. WES and library construction was performed using the Agilent SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA). After cluster generation, the DNA libraries were sequenced on an Illumina Hiseq XTEN platform (Illumina, San Diego, California, USA).
RNA-seq
After total RNA was extracted, mRNA was isolated by Oligo Magnetic Beads and cut into small fragments for cDNA synthesis. Libraries were generated using the NEBNext UltraTM RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) for the Illumina system following the manufacturer’s instructions. Sequencing was conducted using the Illumina Hiseq XTEN platform.
Quantitative proteomics by multiplexed tandem mass tag (TMT) MS
Proteins were extracted, digested with trypsin, and labelled with TMT reagents. The pooled peptides were separated into 15 fractions using a C18 column (Waters BEH C18 4.6×250 mm, 5 µm) on a Rigol L3000 HPLC.
Untargeted metabolomics by liquid chromatography (LC)-MS/MS
Metabolites were extracted from tissues, and LC-MS/MS analyses were performed using a Vanquish UHPLC system (Thermo Fisher Scientific) coupled with an Orbitrap Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific) in both positive and negative modes.
CyTOF analysis of immune cells
CyTOF analyses were performed by PLTTech Inc. (Hangzhou, China) according to the previously described protocol.11 In brief, tumour tissue was dissociated into single cells with DNAase, collagenase IV and hyaluronidase (Sigma-Aldrich, Saint Louis, MO, USA). Immune cells were enriched using Percoll density gradient media (Sigma-Aldrich), and red blood cells were removed using ACK Lysing Buffer (Sigma-Aldrich). Qualified samples were blocked and stained for 30 min with a surface antibody mix panel developed in-house (online table S1), followed by fixation overnight. Permeabilization buffer was applied, and the cells were incubated in an intracellular antibody mix. The cells were rinsed, and the signals were detected using a CyTOF system (Helios, Fluidigm, South San Francisco, CA, USA). The types of immune cells were identified via nonlinear dimensionality reduction [t-distributed stochastic neighbour embedding (tSNE)], followed by density clustering.12
Supplemental material
Statistical analysis
Two-sided Student’s t-tests or Mann-Whitney U tests were used to compare the differences between two groups, as appropriate. One-way ANOVA was used to compare the differences among three or four groups. Pearson’s correlation coefficient was used to evaluate the correlation matrices. Survival curves were generated using Kaplan-Meier estimates and compared using the log-rank test. P<0.05 was considered statistically significant.
Results
Comprehensive interrogation of HCC heterogeneity
According to systems biology, understanding the flow of information from the genome to the phenome using multiomic approaches is essential to identify the ‘Achilles’ heel’ of a disease.13 To determine a potential intervention target in HCC and to develop a clinically practicable therapy (figure 1A), we comprehensively investigated the HCC heterogeneity at different levels and across multiple dimensions. We obtained tumour samples from 21 lesions (including one vascular thrombus sample) in eight HCC patients (online table S2, figure 1B). We then used high-throughput technologies to acquire genomic, transcriptomic, proteomic, metabolomic, and phenomic (local immunity) data to assess tumour heterogeneity across various dimensions (figure 1C). To ensure that the sampling was reasonable and that the results would be minimally affected by the mutual interference of the presence of many types of cells in the same region, we performed single-cell transcriptomic analysis to compare with the results of RNA-seq from bulk tissue and single cells.

Comprehensive exploration of heterogeneity in HCC contributes to development of suitable therapeutic strategies. (A) Scheme showing the relation among precision of intervention, practicability of therapy, and tumour heterogeneity. Tumour biological heterogeneity decreases from genome to phenome, while target heterogeneity has a complicated change from single-cell level to a whole population due to the involvement of genomic background between individuals. Green shadow (individual level to population level) indicates practicable levels for developing therapies. (B) The locations of samples required in each patient the current study. (C) A scheme showing experiments and integrated analyses which were performed. (D) The morphological heterogeneity of typical samples under microscope.
Morphological heterogeneity was marked in large HCCs, especially in Patients 1, 2, and 3 (online figure S1). For instance, in Patient 1, T5 and T10 appeared soft and white, while other lesions looked yellow with cholestasis; in Patient 3, although the two lesions appeared similar in colour, one lesion (T5-8) was hard due to fibrosis. Microscopically, heterogeneity was clear among the lesions and even in different regions of the same lesion (figures 1D and S2). Intriguingly, Patient 1 had well-differentiated (T2 and T7), moderately differentiated (T1, T3, T6, T8, and T9), and poorly differentiated (T4, T5, and T10) lesions within the same tumour. Therefore, phenotypic heterogeneity is significant at the intralesional, interlesional, and intertumoral levels.
Proteogenomic analysis and copy number alteration
In total, we detected 11 266 nonsynonymous single neucleotide variants (SNVs) at the DNA level, 14 706 nonsynonymous SNVs at the RNA level, and 1875 single amino acid variants at the protein level in all samples (figure 2A). Mutant proteins were analysed by comparing the MS/MS spectra with the protein database transformed by the WES mutation data. Only 20.8% of the somatic mutations detected in the genome were expressed at the transcriptome. The number of confirmed protein-level variants in the genome and transcriptome was also low (9.32%). Although the verification rates of the RNA variants and DNA variants at the protein level were comparable (10.7% vs 10.4%), for somatic mutations, RNA variants were more likely than DNA variants to be confirmed by the proteome variants (6.48% vs 3.54%).

Effects of genomic alterations on RNA and protein levels. (A) Overlap of nonsynonymous single nucleotide variants/single amino acid variants detected in whole-exome sequencing, RNA-seq, and LC-MS/MS. (B) Correlation between copy number variation and mRNA expression in all tumour samples. Spearman’s correlation P (upper) and r values (lower) between copy number and gene expression (y axis) are shown. Genes are ordered by their chromosome positions (x axis). Black lines denote P = 0.05 (upper) and r = 0.3 (lower). (C) Heatmap of mutations, CNV, and their associations with RNA and protein expression of HCC-relevant genes across lesions and tumours. Tumour differentiation status is annotated.
The effects of copy number variations (CNVs) on mRNA abundance was evaluated, and we found that approximately the mRNA abundance values of 2150 (22.5%) genes were significantly correlated with the gene copy numbers (P<0.05 and Spearman’s r>0.3 or<−0.3; figure 2B). Frequent somatic mutations were detected in our study (online figure S4), which was consistent with the results of The Cancer Genome Atlas project regarding HCC.3 To illustrate the flow of information for a single gene, we analysed four frequently mutated genes (TP53, CTNNB1, NFE2L2, and ARD1A), one oncogene (RB1) and one tumour suppressor gene (TSC2) in HCC (figure 2C). Although the gene mutational status seemed to be significantly associated with the RNA level (online figure S4), it was more likely to be affected by the genomic background of the individual.
Different dimensions of tumour heterogeneity in HCC patients
We depicted the intralesional, interlesional/intratumoral, and intertumoral/interpatient heterogeneity using genomic, transcriptomic, proteomic, metabolomic, and immunologic analyses. In the genomic dimension, the mutation spectra varied among lesions in the same tumour, but the degree of interlesional/intratumoral heterogeneity was less than that of intertumoral heterogeneity (online figure S5). Notably, intralesional heterogeneity, as represented by the mutation spectrum, was also clear (eg, Patient 3). The numbers of SNVs and inserts and deletions varied significantly among lesions (figure 3A, online figure S6A, S7A, S8A). All lesions in the same tumour harboured both common and specific driver mutations (figure 3B, online figure S6B, S7B, S8B), demonstrating branch evolution during HCC progression as shown by the phylogenetic trees (figure 3C, online figure S6C, S7C, S8C). These driver mutations correlated with the tumour pathology and clearly showed the path of tumour evolution. For instance, in Patient 1, all poorly differentiated lesions had wild-type alleles of PDGFRB, ERC2, CTNNB1, FBN2, and IRS2, all of which were found to be altered in moderately or well differentiated lesions (figure 3B). However, an ARID1A mutation was detected in T5 but not in T10, suggesting the existence of subclones within the same lesion. The degree of diversity in CNVs (including number and position at chromosomes) among lesions was large (figure 3D and E, online figure S6D, S6E, S7D, S7E, S8D, S8E).

Heterogeneity of lesions within a tumour in various dimensions (Patient 1). (A) The differences of SNV, INDEL, and driven mutations in all HCC lesions. (B) Frequent driver mutations were shown in each lesion. (C) A phylogenic tree showing the genomic similarity of all HCC lesions. (D and E) Comparison of copy number variations. (F) Comparison of differentially expressed RNAs. Up- and down-regulation as compared to normal tissue. (G) A flower plot showing common and specific differentially expressed RNAs. (H) KEGG pathways differentially expressed RNAs enriched in different lesions. (I) Fusion genes detected in these lesions. (J) A flower plot showing common and specific differentially expressed proteins. (K) A flower plot showing common and specific differentially detected metabolites. (L) Comparison of frequencies of some key types of immune cells in HCC lesions.
In the transcriptome dimension, more RNAs were downregulated (compared with the levels in normal tissue) than upregulated. The numbers of these differentially expressed RNAs varied by as much as approximately one-fold among lesions in the same tumour (figure 3F, online figure S6F, S7F, S8F). Remarkably, only a small portion of these differentially expressed RNAs were shared by all lesions (figure 3G, online figure S6G, S7G, S8G). Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway enrichment of these RNAs showed significant differences (figure 3H, online figure S6H, S7H, S8H), suggesting distinct functions involving these RNAs. The fusion genes were also analysed based on RNA-seq (figure 3I, online figure S6I, S7I, S8I), and a high degree of diversity was observed among lesions (online figure S9). Single-cell RNA-seq clearly identified three or four HCC clusters in each tumour, regardless of the extent of morphological heterogeneity (online figure S10). Although the results of the two methods were in accord with each other, single-cell analysis revealed further heterogeneity among cells in the same lesion.
Similarly, in the proteome and metabolome dimensions, significant differences were found among lesions in the same tumour (online figures S11-13), and only a few differentially expressed proteins and differentially expressed metabolites (compared with normal tissue) were common to all samples from the same patients (figure 3J and K, online figure S6J, S6K, S7J, S7K, S8J, S8K). The types of infiltrating immune cells also varied markedly among lesions and tumours (figure 3L, online figure S6L, S7L, S8L). However, in samples acquired from the same lesion that were spatially far apart (Patient 4), more common RNAs, proteins, metabolites, and infiltrating immune cells were found (online figure S8G,J,K and L), indicating that the degree of intralesional heterogeneity was less than that of interlesional heterogeneity.
Comparison among interlesional, intratumoral, and intertumoral heterogeneity
Based on these multiomic data, we aimed to determine the degrees of interlesional, interlesional/intratumoral, and intertumoral/interpatient heterogeneity. Determining these degrees of heterogeneity is critical to develop novel antitumour strategies against the background of absolute heterogeneity among HCC cells.10 14 15 To assess the degree of genomic heterogeneity, we used Jaccard similarity coefficients,16 which showed that the degree of intralesional heterogeneity was less than the degree of interlesional/intratumoral heterogeneity, which in turn was far less than the degree of intertumoral/interpatient heterogeneity (figure 4A). The clustering of all HCC samples based on differentially expressed RNAs suggested that in the transcriptome dimension, intralesional similarity was not necessarily greater than interlesional or intertumoral/interpatient similarity (figure 4B), and no clear clusters could be generated. However, clustering based on differentially expressed proteins and differential metabolites showed that all patients were independent and unique in the proteome and metabolome dimensions (figure 4C and D).

Comparison of heterogeneity in different dimensions and various levels. (A) Jaccard scoring showed genomic difference between lesions and between patients. (B) Hierarchical clustering differentially expressed RNAs of all lesions. (C) Hierarchical clustering differentially expressed proteins of all lesions. (D) Hierarchical clustering differentially detected metabolites of all HCC lesions. (E) Flower plots showed common and specific enriched KEGG pathways using differentially expressed RNAs, proteins, and metabolites. (F) plotDIABLO shows the correlation between transcriptome, proteome, metabolome, and immunome. Three patients with more than five samples tested were analyzed. (G) A high enrichment score was associated with a high level of tumor infiltrating T cells in samples from Patient 3 and other patients. *, P < 0.05; **, P < 0.01. (H) Scheme showing main components of the PI3K-Akt signaling pathway. (I-K) The negative correlations between protein levels of PIK3CA (I), IRS1 (J), IRS2 (K) and infiltrating T cell level. Protein levels were derived from the proteomic data.
We further investigated the dysregulated functions that were shared by all lesions in the same tumour and that were caused by those differentially expressed molecules. Interestingly, there were more enriched KEGG pathways in the proteome dimension than in the transcriptome dimension (figure 4E), suggesting a decrease in tumour cells heterogeneity from the transcriptome dimension to the proteome dimension. Unexpectedly, shared enriched KEGG pathways were rare in the metabolome dimension (figure 4E and online figure S14), suggesting that it would be difficult to treat HCC by targeting tumour cell metabolism. Using the multiblock integration analysis, we demonstrated that the correlation decreased along the ‘central dogma’ and that tumour local immune state is best correlated with metabolome (figure 4F). Altogether, these results suggested that the genome, transcriptome, proteome, and metabolome dimensions might be unsuitable targets for HCC interventions due to significant interlesional heterogeneity (genome and transcriptome) or patient specificity (proteome and metabolome).
PI3K-Akt signalling pathway and ketone body metabolism are associated with antitumoural immunity
To understand the underlying mechasnims behind tumour heterogenetiy and find potential relationship between tumour cells and local immunity, we developed an enrichment approach and generated an integration map using genomic, transcriptomic, and metabolomic data. Two KEGG pathways (ie, PI3K-Akt signalling pathway, synthesis and degradation of ketone bodies) were robustly correlated with the abundance of tumor infiltrating T cells in HCC microenvironment (online figure S15). A high enrichment score of PI3K-Akt signalling pathway was associated with increased infiltration of T cells (figure 4G). In aggrement, the expression level of most proteins (from proteomic data) involved in this pathway were negatively and significantly correlated with T cell abundance (figures 4H-K and online figure S16). Similar findings were observed for ketone body metabolism (online figure S17), which is the characteristic metabolism of HCC.17 These data provide molecular insight in tumour heterogenetiy and suggest novel mechsnisms by which tumour cells affect anti-tumoral immunity in HCC.
Landscape of local immunity in the HCC microenvironment
Intratumoral heterogeneity contains tumour cell heterogeneity and microenvironment heterogeneity. In tumour tissue, the genomic, transcriptomic, proteomic, and metabolomic analyses revealed the features of tumour cells. Because the degrees of heterogeneity were significant in all these dimensions, we then tested the microenvironment heterogeneity by CyTOF. More than 4 million immune cells were analysed, and 40 clusters were identified (figure 5A and B). In general, the HCC immune landscape was significantly altered compared with that in normal liver tissue, especially with regard to macrophages, CD8+ T cells, regulatory T (Treg) cells, and B cells (figure 5C-E). The differences among the eight patients were also marked (figure 5F and G), as were the differences among lesions within the same tumour (online figure S18).

The detailed heterogeneity in the local immunity of HCC lesions. (A) A tSNE plot compares the difference in local immunity of the tumour and normal tissue. (B) A total of 40 clusters were identified, and were shown in a tSNE plot. (C) tSNE plots color-coded for expression of marker genes for eight main types of immune cells. (D) A heatmap showing the differential expression of 42 immune markers in the 40 cell clusters. Certain clusters were identified as known cell types according to typically expressed markers. (E) Frequencies of the 40 clusters in tumour and normal tissue. (F) Frequencies of the 40 clusters in different patients. (G) tSNE plots showing distinct immune landscape of tumours in different patients. (H) Correlation between different cell clusters.
Among the 40 cell clusters identified, we detected 94 pairs of clusters that showed strong correlations with each other (figure 5H). Noticeably, Treg cells, regulatory B (Breg) cells, and PD-L1- macrophages were positively correlated with PD-1+CD8+ T cells (online figure S19), which are intrinsically less responsive to antigenic stimulation.18 PD-L1 +macrophages were positively correlated with PD-1+CD4+ T cells, while dendritic cells (DCs) showed positive correlations with CD11b+CD4+ T cells and CD11b+CD8+ T cells. These results suggest that Treg cells, Breg cells, DCs, and macrophages play important roles in the regulation of the immune microenvironment in HCC.
Novel immunophenotypic classification of HCCs
We then compared the similarities among all samples and conducted hierarchical clustering using the CyTOF data. With few exceptions, all tumour samples formed three clusters according to specific marker gene expression levels (figure 6A). The three HCC subtypes presented distinct immune cell profiles (figure 6B and C and online figure S20). Briefly, subtype 2 samples were characterised by reduced infiltration of lymphocytes, with consequent high frequencies of DCs and natural killer (NK) cells (figures 6C-E and online figure S21). Subtype 3 samples showed high frequencies of Treg cells, Breg cells, and M2-polarised macrophages, all of which are immunosuppressive cells. Subtype 1 samples showed relatively normal T cell infiltration levels, but there were fewer infiltrating B cells, especially Breg cells (figure 6C and D). Consistent with these findings, the expression levels of immunosuppressive molecules including PD-1, PD-L1, Tim-3, and CTLA-4 were significantly upregulated in subtype 3 HCC samples (figures 6F and online figure S22). In detail, Breg cells showed enhanced expression of PD-L1 and Ki-67; CD8+ T cells expressed more CTLA-4, TIM-3, and PD-1; macrophages demonstrated higher expression levels of PD-L1 and IL-1β; and Treg cells overexpressed 4-1BB, ICOS, and Ki-67, suggesting more functional and proliferative Treg cells in subtype 3 HCCs. In parallel with the increased frequency of myeloid cells in subtype 2 samples, these monocytes also overexpressed IL-1β (figure 6F). Moreover, the RNA-seq data revealed that the expression levels of all immune suppressive marker genes (eg, LAG3, PDCD1, HAVCR2, TIGIT, and TNFRSF9) and functional genes (eg, GZMA and GZMB) were upregulated (figure 6G). Based on these characteristics, we classified HCC subtypes 1, 2, and 3 as the immunocompetent subtype, immunodeficient subtype, and immunosuppressive subtype, respectively. Importantly, these subtypes were applicable to nearly all HCC lesions from the same patient and from multiple patients, suggesting that interlesional heterogeneity did not alter the classification.

The novel immunophenotypic classification of HCCs and the characteristics of different subtypes. (A) Hierarchical clustering of all HCC lesions according to the 40 clusters resulted in three obvious subtypes. (B) The difference in immune landscape of the three HCC subtypes. (C) tSNE plots color-coded for expression of marker genes for several main types of immune cells in the three HCC subtypes and normal tissue. Red boxes indicate main types of immune cells. (D) Frequencies of eight main types of immune cells in the three HCC subtypes. (E) Immunohistochemistry showing the differential expression of marker genes for immune cells. (F) Expression of functional genes in five key types of immune cells. Data derived from CyTOF. (G) Expression of functional genes in the three HCC subtypes. Data derived from RNA-seq. *P< 0.05; **P<0.01; ***P<0.001.
Underlying rationales of the novel immunophenotypic classification
We thus tried to understand the underlying mechanisms by which such distinctive local immune statuses are established in the HCC microenvironment. Because our data suggested that the immunome was better correlated with metabolome than transcriptome and proteome (figure 4F), and the differentiation and function of immune cells are largely determined by cell metabolism,19 we first explored the metabolomic variations among the three HCC subtypes. HCC subtype 3 showed inhibited glycolysis and enhanced mitochondrial respiration, supported by the reduced lactic acid level and increased abundance of metabolites involved in the tricarboxylic acid (TCA) cycle (figure 7A). HCC subtype 2 showed increased activity of nucleotide biosynthesis, while subtype 1 was characterised by upregulated flow through the urea cycle. The altered expression levels of enzymes that mediated these metabolic processes were consistent with the changes in the levels of the metabolites. The clustering of differentially expressed RNAs based on the immunophenotypic classification showed marked changes in genes involved in metabolic regulation (figure 7B and C). Notably, the enriched KEGG pathways, such as metabolic pathways, oxidative phosphorylation, glyoxylate and dicarboxylate metabolism, and the TCA cycle, were largely consistent with the metabolomic findings.

Differences in tumour cell metabolism and cytokine/chemokine expression among the three HCC subtypes. (A) Metabolomic analysis revealed featured alterations of TCA cycle, glycolysis, urea cycle, and nucleotide biosynthesis pathway in different HCC subtypes. (B) Hierarchical clustering of differentially expressed genes in the three HCC subtypes. (C) KEGG pathway enrichment of RNA- seq data showed significant changes in metabolism among the three HCC subtypes. Arrows indicate metabolism-related pathways. (D) Expression of important chemokines, cytokines, and other secretory biomaterials among the three HCC subtypes is shown. Data derived from RNA-seq. *P<0.05; **P< 0.01; ***P< 0.001.
Infiltrating immune cells are recruited by chemokines and cytokines. We therefore investigated whether there was a characteristic chemokine/cytokine microenvironment in each HCC subtype. The RNA-seq data revealed that the expression levels of all chemokines and cytokines were quite low except that of CCL14 (which is able to recruit monocytes and macrophages) in HCC subtype 2, which is consistent with its immunodeficient nature (figure 7D). The overexpression of VEGFA and many other genes encoding tumour suppressive chemokines/cytokines (eg, TGFB1, CCL8 and IL10) was observed in HCC subtype 3. Enhanced expression levels of T-cell recruiting molecules such as CXCL9, CXCL10, CXCL11 and CXCL16 were found in both subtypes 1 and 3, in agreement with their abundant T cell infiltration. Thus, the characteristics of the cytokine/chemokine microenvironment were strongly associated with the immune landscape in each HCC subtype.
The prognostic role of immunophenotypic classification
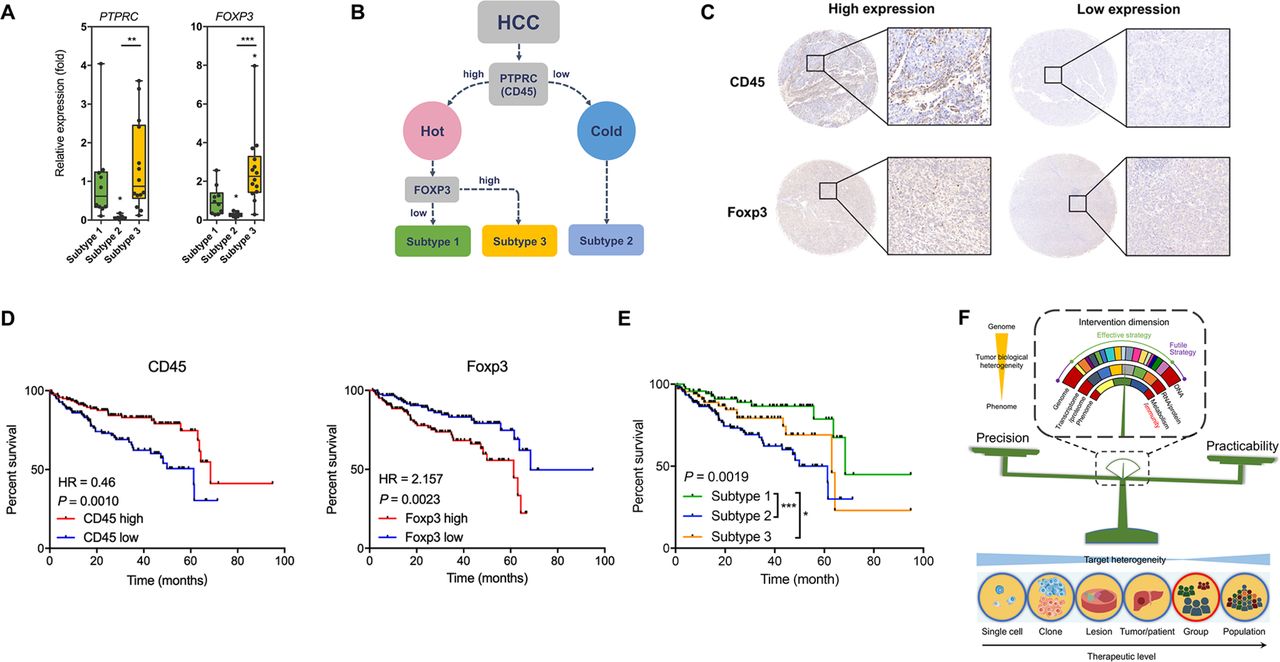
We then asked whether this immunophenotypic classification of HCC had clinical value and could be applied in a population. For this purpose, we enrolled another 30 HCC patients to confirm the immunophenotypic classification. Clearly three sample clusters were idenetified (online figure S23, online table S3). Careful analysis of the density of infiltrating immune cells and the chracterisitc cell clusters in these sample clusters showed that HCC subtype 1 contained antitumoural T cells including CD8+CD127+ T cells, CD4+CD127+ T cells, and γδ T cells, HCC subtype 3 was featured with Treg cells and immunosuppressive CD4+ T cells (CD4+CD25+CD127-Foxp3+ T cells),20 and HCC subtype2 was characterised by lack of CD45+ cells (around 40% less compared with the other two subtypes). Thus, the cohort generally confirmed our immunophenotypic classification. To make this system more clinically practicable, we tried many immune markers and found that CD45 and FOXP3 were promising. qPCR assays confirmed the characteristic expression levels of PTPRC (CD45) and FOXP3 in the three subtypes (figure 8A). We attempted to use PTPRC and FOXP3 to differentiate HCCs into the three subtypes (figure 8B). A density of 100 positive cells/mm2 for CD45 and a density of 25 positive cells/mm2 for Foxp3 were found with best performance as potential cut-off values for high and low expression of the proteins (figure 8C). Using tissue microarray, we confirmed that enhanced expression levels of CD45 and Foxp3 were protective and harmful factors for overall survival of HCC patients, respectively (figure 8D), and the three subtypes showed distinctive prognoses (figure 8E, online table S4). Using another independent HCC cohort, qPCR confirmed these results (online figure S24), suggesting that our immunophenotypic classification was reliable, practicable, and clinically valuable.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The clinical value of the novel immunophenotypic classification of HCC. (A) Expression of PTPRC (CD45) and FOXP3 in the three HCC subtypes. Data derived from RNA-seq of the eight patients. (B) Scheme showing the classification of HCCs according to marker genes PTPRC and FOXP3. (C) Representative of IHC staining for subtype analysis using HCC tissue microarray. For CD45, the cut-off value for high and low expression was a density of 100 cells/mm2. For Foxp3, the cut-off value for high and low expression was a density of 25 cells/mm2. (D) Survival curves showing the predictive role of marker genes for patients’ prognosis. Data derived from a tissue microarray containing a cohort of 298 HCC patients. (E) Survival curves showing the distinctive prognosis of different HCC subtypes. Data derived from the tissue microarray. (F) Scheme showing that immunity would be a suitable invention dimension to treat HCC by balancing precision and practicability. *P<0.05; **P<0.01; ***P<0.001.
Discussion
The HCC ecosystem, which is mainly composed of tumour cells and immune cells, is complicated and varies dynamically. The heterogeneity at all levels from the single cell to the lesion reduces the efficacy of treatment due to drug resistance or immune escape.9 Many efforts have been made in the past decade to investigate intratumoral heterogeneity using multiregional sampling and high-throughput analyses.14 15 21 These studies unveiled the features of clonal evolution at the genomic level as being non-Darwinian and mostly branched.22 Most investigations focused on the genetic changes in HCC cells; however, a large proportion of genetic alterations may not be reflected in the protein dimension and do not necessarily influence cell behaviour due to epigenetic modification, post-transcriptional/post-translational regulations, and the feedback regulation of cell signalling pathways. To the best of our knowledge, the current study is the first attempt to comprehensively investigate the heterogeneity of HCC from the genome to the phenome and from the single-cell level to the organism level. In this study, we attempted to apply the concepts and research methods of systems biology to HCC and aimed to identify a reasonable therapy for HCC. Our results confirmed that although the flow of information from the genome to the metabolome seemed ineffective, the heterogeneity of tumour cells was clear in all dimensions. One reason for this phenomenon is the complicated regulation according to the ‘central dogma’, and the other important reason is the ‘dilution effect’ of the genomic background (wild-type genes). In spite of this, the degrees of interlesional heterogeneity in the proteome and metabolome dimensions were significant. Furthermore, we found that proteomes and metabolomes were highly specific to individuals. These findings suggested that the heterogeneity of tumour cells is significant in all dimensions and that targeting tumour cells themselves is unlikely to result in the development of an ideal treatment with balanced precision and practicability. In contrast, the local immune status of HCC is less heterogeneous, likely because of the ‘dilution effect’ of other microenvironmental factors such as angiogenesis and extracellular matrix.9 Therefore, manipulating the local immune status of the HCC microenvironment may be an appropriate strategy because such treatments may be able to affect all lesions in an individual and may be applicable to a group of patients as well (figure 8F). More importantly, many new tools for immunotherapy have been developed and improved. Furthermore, by integrated analysis, we observed that some signalling pathways of tumour cells were significantly associated with local antitumour immunity, providing novel understanding of immunometabolism of HCC23 and suggesting possible targets to regulate antitumoural capacity.
People gradually realise that enhancement of host immunity may be an effective strategy to cure cancer. Unfortunately, we know little about the local immune characteristics of tumour microenvironment. Single-cell sequencing and CyTOF technique provide new approaches to characterising the phenotypes and functions of tumour-infiltrating immune cells. In HCC, the landscape of infiltrating T cells has been characterised, revealing the enrichment of Treg cells and the depletion of CD8+ T cells.24 CyTOF analysis confirmed the enrichment of immunosuppressive cells in patients with HCC.12 25 In this study, we investigated the infiltrating immune cells in multiple regions of HCCs for the first time using CyTOF analysis, revealing significant diversity among lesions in the same tumour and even between subclones/clones in the same lesion. Despite this, the degree of intratumoral heterogeneity in the local immunity microenvironment was acceptable, and almost all HCC samples could be clustered into three clear groups, suggesting the possibility of treating HCC by regulating local immunity. The similarity of local immune status of the HCC microenvironment among lesions within a large HCC not only supports the development of practicable treatments but is also convenient for classification. In this case, an HCC patient can be correctly classified into one of the three HCC subtypes according to our novel classification scheme simply by biopsy without interference by interlesional/intratumoral heterogeneity. More importantly, we used an independent HCC patient cohort to confirm the classification using CyTOF analysis. Although the significant inter-patient heterogeneity of local immune status, the three subtypes of HCC were nicely identified, suggesting that the classification can be applied in a population of HCC patients.
The three HCC subtypes suggested by our classification represent the clinical situations in human patients. Patients with HCC subtype 1 have relatively competent antitumoural immunity and thus have good prognoses, supporting by the normal levels of cytokines and chemokines. The characteristic urea cycle metabolism in HCC subtype 1 also provided more arginine, which can boost immunity by enhancing T cell survival and its antitumoural function.26 However, the release of cytotoxic molecules was relatively low; thus, the antitumoural function of infiltrating T cells in this subtype could be further activated. The combined use of T cell response activating agents (eg, IL-12, CpG oligonucleotide) and ICIs may be reasonable.27–29 HCC subtype 3 has normal lymphocyte infiltration, but the frequencies of immunosuppressive cells were significantly increased. However, it is noteworthy that although the overexpression of TIM3, PDCD1, CTLA4 and LAG3 was detected at both RNA and protein levels, the upregulation of genes with cytotoxic functions (eg, members of the granzyme family and IFNG) indicated that exhausted cytotoxic T cells were induced on activation rather than by intrinsic defects.24 In this case, ICIs monotherapy may sustain or recover the anti-tumoral potency of these infiltrating T cells. HCC cells of subtype 3 tended to use mitochondrial respiration rather than glycolysis and produced more ATP, which also functions as an immunosuppressive factor when released to the tumour microenvironment.30 HCC subtype 2 is more like the so-called ‘cold’ tumour with few infiltrating lymphocytes, which limits the efficacy of ICIs. Thus, combination of ICIs with oncolytic viruses (eg, JX-594) or peptides (eg, LTX-315), which can significantly enhance lymphocyte infiltration,31–33 is promising for this subtype of HCCs. Elaborate regulation of tumour angiogenesis (eg, low dose of sorafenib or bevacizumab) to enahce T cell trafficking or to inhibit the suppressive MDSCs may also boost the efficacy of ICIs in HCC subtype 2.34 35Therefore, our immunophenotypic classification may provide guidance for the selection of treatment. Currently, people commonly use TNM staging, tumour differentiation grading, and microvascular invasion for the guidance of therapy for HCC patients.36 However, these classifications are hardly relevant to local immune status and are thus unable to suggest immunotherapy. PD-L1 expression and tumour mutational burden are now used to identify HCCs which can potentially benefit from anti-PD-1/PD-L1 immunotherapy.37 Our classification, however, provides a more comprehensive perspective of local immune status in HCC microenvironment and can be more helpful for the choice of many other immune-related therapies such as oncolytic viruses, antiangiogenic agents, and radiotherapy. Hence, our classification can improve current HCC classification for better management of HCCs.
Although other immune-related classifications of HCC have been suggested previously, they were based on mRNA levels or multiplex immunohistochemistry.38–40 While such analyses were relatively stable, they could not comprehensively reflect the immune status of HCC. Compared with those classifications based on the abundance of infiltrating immune cells,38 39 the rationale of our classification includes both the abundance and function of these cells and thus is more clinically relevant. In addition, for the purpose of clinical practice, we confirmed that only two markers tested by immunohistochemistry with clear cut-off values were sufficient for classification, much simpler than the previously reported combinations of multiple markers.38 40 Logically, the density of CD45+ and Foxp3+ cells actually represents an immunosuppressive gradient across HCC patients, similar to the one that first proposed by Chew et al across the HCC micronenvrionment, normal liver microenvironment, and peripheral blood.25 Our attempts on other immune markers failed to show better performance, suggesting an essential role of such immunosuppressive gradient in the prognosis of HCC patients.
There are some limitations to our study. First, although we demonstrated the intratumoral heterogeneity in multiple dimensions, the correlations of these dimensions were not deeply analysed because there is not a well-developed workflow for such extensive analyses ranging from the genome to the metabolome. Second, we included only eight patients in the study. Although similar studies also had an average sample size of nine (ranging from 1 to 23),41–47 the relatively small number of patients may have reduced the reliability of our conclusions. To minimise this weakness, we used two independent HCC cohorts and two detection techniques to verify our classification, but further investigations using prospective cohorts are important. Third, the number of samples from each patient varied, and all samples were pooled for analysis. This analytic manipulation was also adopted by similar studies.15 48 Additionally, we logically proposed that our immunophenotypic classification contains information for clinical decision making, but this was not proven by experiments or in clinical practice. Because establishing animal models for such purposes is difficult, clinical trials may provide valuable information.
In conclusion, this comprehensive study of tumour heterogeneity using multiomics approaches revealed the landscape of heterogeneity in both tumour cells and tumour microenvironment. Based on the findings, we believe that local immunity might be a suitable target for the treatment of HCC and propose a clinically practical HCC classification scheme with prognostic and potential decision-making value. Further studies using large cohorts are warranted to verify the clinical value of this classification.
Acknowledgments
We appreciate Mr Pengpeng Li, Mr Fei Teng, Mrs Yue Wang and Mrs Xiaoxiao Zhang from Novogene, and Mr Zongbao Gao from PLTTECK for their great support in resource arrangement and data analysis. We also appreciate Mr Jianfeng Wang for specimen collection.
References
Footnotes
QZ, YL and JY contributed equally.
Contributors Study concept and design: QZ, TL, XB; Acquisition of data: QZ, YL, JY, JW, QF, MY, XZ, HG, QC, JW, JW, YC, TW; Data analysis: QZ, YL, JY, CM, JF, JW, LW, XH, YZ, CY, YX; Obtained funding: QZ, TL, XB; Study supervision: GJ, TL, XB; Drafting of manuscript: QZ, YL, YZ; Critical revision of manuscript: JY, XF, GJ, TL, XB.
Funding This work was supported by Key Program of the National Natural Science Foundation of China (81830089), the National Natural Science Foundation of China (81871320, 81472212), the Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.