Article Text

Abstract
The spectrum of non-alcoholic fatty liver disease (NAFLD) ranges from simple hepatic steatosis, commonly associated with obesity, to non-alcoholic steatohepatitis, which can progress to fibrosis, cirrhosis and hepatocellular carcinoma. NAFLD pathophysiology involves environmental, genetic and metabolic factors, as well as changes in the intestinal microbiota and their products. Dysfunction of the intestinal barrier can contribute to NAFLD development and progression. Although there are technical limitations in assessing intestinal permeability in humans and the number of patients in these studies is rather small, fewer than half of the patients have increased intestinal permeability and translocation of bacterial products. Microbe-derived metabolites and the signalling pathways they affect might play more important roles in development of NAFLD. We review the microbial metabolites that contribute to the development of NAFLD, such as trimethylamine, bile acids, short-chain fatty acids and ethanol. We discuss the mechanisms by which metabolites produced by microbes might affect disease progression and/or serve as therapeutic targets or biomarkers for NAFLD.
- nonalcoholic steatohepatitis
- intestinal bacteria
- intestinal barrier function
Statistics from Altmetric.com
Key messages
Although genetic and environmental factors contribute to development of non-alcoholic fatty liver disease (NAFLD), changes in the intestinal microbiota are also involved.
Although there are technical limitations in assessing intestinal permeability in humans and the number of patients in these studies is rather small, fewer than half of the patients with NAFLD have evidence for disruption of the intestinal barrier.
Microbe-derived metabolites, such as trimethylamine, some secondary bile acids, short-chain fatty acids and ethanol contribute to the pathogenesis of NAFLD.
Succinate, phenylacetic acid and 3-(4-hydroxyphenyl) lactate, which can be produced by gut microbiota, might be used as markers for detection of NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of liver damage ranging from simple steatosis (or non-alcoholic fatty liver, NAFL) to non-alcoholic steatohepatitis (NASH) with the development of fibrosis and cirrhosis.1 Hepatocellular carcinoma (HCC) can develop in the absence of cirrhosis,2 and patients with NAFLD have an increased risk of HCC3 4. NASH is the second-leading indication for liver transplantation, but will likely become the leading cause in 2020.5
The pathogenesis of NAFLD involves environmental, genetic and metabolic factors, such as limited physical activity and a dysbalanced diet.6 7 Changes of intestinal microbiota are also associated with development of NAFLD.8 Multiple studies found that germ-free C57BL/6 mice gained less weight than conventional mice when given a sugar-rich and lipid-rich diet despite similar amounts of food consumption.9 10 This indicated that the absence of microbes protects mice from obesity and NAFLD induced by a Western-style diet. Interestingly, total body fat and liver triglyceride content increased following microbial colonisation of germ-free mice.9 Mice lacking the nod-like receptor pyrin domain-containing protein 3 (NLRP3) and NLRP6 inflammasomes have increased susceptibility to NASH, due to changes in the gut microbiota. Co-housing inflammasome-deficient mice with wild-type mice increased the susceptibility of wild-type mice to NASH, so fatty liver risk might be affected by the surrounding faecal microbiota.11 Moreover, epididymal fat weight, hepatic steatosis, multifocal necrosis and infiltration of liver by inflammatory cells were significantly increased in germ-free mice colonised with faeces from patients with NASH and then fed a high-fat diet (HFD).12 These results indicate that risk of NAFL and NASH can be transmitted, apparently by the faecal microbiota.
Gut microbiome
The intestinal microbiome has a strong mutualistic relationship with its host. Although the relative abundance and the composition of bacterial species is similar among healthy individuals, each person’s microbial profile is distinct. For instance, Bacteroidetes are higher in some Asian countries (30.2% and 36.6% in Korean and Japanese subjects, respectively) than in the USA (18.2%).13 Intestinal microbiota also shows high diversity along the gut. There are approximately 1011 microbial cells in the large intestine compared with 108 cells in the small intestine,14 with different microbial composition between faeces and rectal mucosa.15 16 Maternal microbiota shape the first microbial colonisation of newborns, then microbial diversity increases and converges toward an adult-like microbiota.17 After childhood, the composition of microbiota becomes a relative stable system throughout adulthood, although the diversity of microbiota can be altered by diet, exercise, medications (antibiotics), gastrointestinal infections and surgery, as well as geographical provenance.18–20 For example, HFD in mice altered the diversity of gut bacteria and caused a decrease and increase in some species of Ruminococcaceae and Rikenellaceae, respectively.21 Exercise training affects gut microbiota in a body mass index (BMI)-dependent manner, with Collinsella spp increased in the higher BMI group, Faecalibacterium spp decreased in higher BMI group but increased in the lower BMI group and Lachnospira spp increased in the lower BMI group.20 Bacterial genes outnumber human genes by approximately 100-fold.22 The bacterial metagenome has therefore a tremendous capacity to synthesise and secrete a large amount of metabolites, which are important not only for interbacterial communications, but also for bacterial–host interactions.
Gut–liver axis
Gut microbiota interact with the host in multiple different ways, with numerous links between the gut and other organs, such as brain, kidney and liver.23–25 The term ‘gut-liver axis’ was first described by Volta et al in 1978, when these researchers reported production of immunoglobulin A antibodies to dietary antigens in patients with liver cirrhosis,26 indicating interactions between gut and liver. The liver is unique in that it has a special blood supply—two sources of blood flowing into the liver, from the hepatic artery and portal vein. About 70%–75% of the liver blood supply comes from the portal vein, which drains blood from mesenteric veins of the intestinal tract.27 Nutrients such as monosaccharides and amino acids are absorbed by specialised transporters on enterocytes and can reach the liver through the portal vein, where many are taken up by hepatocytes and metabolised. If the gut barrier is disrupted, the liver is the first organ in the body that encounters the microbial products, toxins and microorganisms (such as bacteria and fungi) from the intestine. The liver therefore serves as a large collection base for compounds and substances originating from the intestine (figure 1).
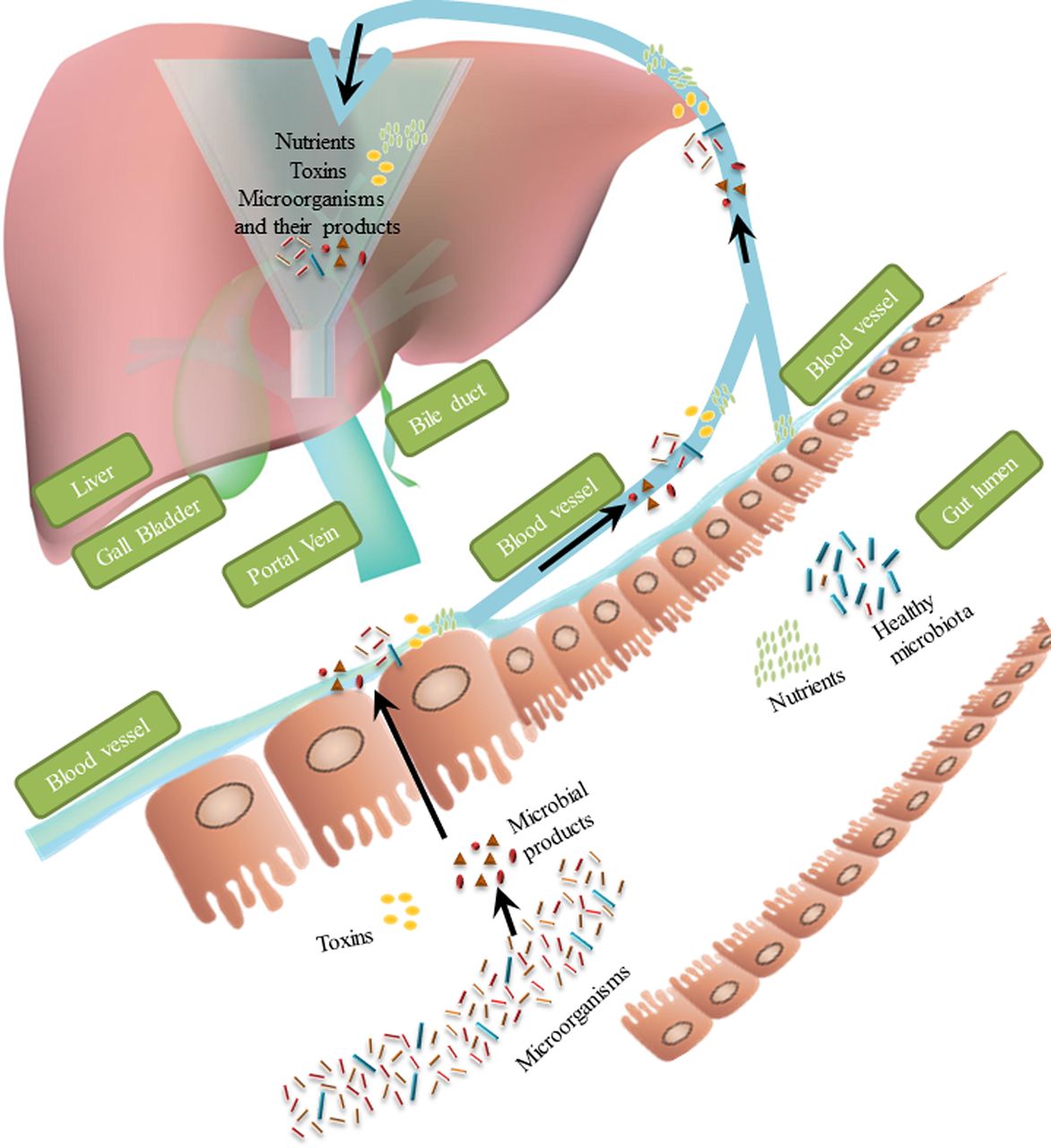
Nutrients and microbiota-related substances flow from intestine to liver via portal vein. Liver, acting as a collection base, is the first organ to receive substances that originate from intestine. Under normal conditions, nutrients absorbed by the enterocytes have access to the liver, whereas most of the toxins and microorganisms cannot pass through the gut barrier. When this barrier is disrupted, toxins and microorganisms (such as bacteria and fungi) with their secreted products can reach the liver via the portal blood.
Gut barrier dysfunction in patients with NAFLD
The gut barrier comprises a physical barrier, a biochemical barrier and an immune system barrier.28 The physical barrier is supported mainly by the mucus layer, microbes and a single layer of intestinal epithelial cells that are linked by tight junction proteins. Molecules with antimicrobial properties, such as bile acids and antimicrobial proteins (including α-defensins and β-defensins, C-type lectins, cathelicidin, lysozyme and intestinal alkaline phosphatase) support and mediate the biochemical barrier.29 Secretory immunoglobulin A and lymphoid follicles that contain a variety of immune cells, including B cells, T cells, dendritic cells and neutrophils, are the components of immune system barrier.28 Gut barrier dysfunction results in translocation of microbes, their secreted metabolites and other microbial products from the gut lumen into blood and lymphatics, allowing them to reach other tissues and organs including the liver.30 31
There are multiple ways to evaluate gut barrier function, such as assessing the flow of non-digestible markers from the intestinal lumen to extraintestinal space including sugars, radioisotopes (eg, chromium-51-labelled ethylenediamine tetra-acetic acid) and polyethylene glycols.32 Translocated microbial pathogen-associated molecular pattern molecules (PAMPs) such as lipopolysaccharides (LPS) can be measured in the systemic circulation, as surrogate markers of intestinal permeability. Despite all those different methods, techniques to evaluate intestinal permeability in humans still have several limitations and the ideal test has not been established yet.33 A combination of direct and indirect measures of increased intestinal permeability performed in the same patient might be the most accurate way to assess the integrity of the gut barrier and provide more conclusive answers of barrier dysfunction.
Numerous studies have shown that patients with alcoholic liver disease or cirrhosis have increased intestinal permeability, which might be an important contributor to disease progression.34–36 Patients with NAFL or NASH are believed to have gut barrier dysfunction, with increased translocation of microbial products to the liver. Although there have been many reports of increased intestinal permeability in mice with fatty liver following an HFD or steatohepatitis following a Western-like diet,11 37 these results were not confirmed in all studies.38 Intestinal permeability has been most frequently evaluated by measuring translocated microbial products in the systemic circulation of rodents. Although these measurements indicate gut barrier function, these experiments are technically challenging.
More importantly, serum endotoxin was increased in only 42.1% (8/19) patients with NASH39 and a meta-analysis found that only 39.1% of patients with NAFLD (n=128) had increased intestinal permeability.40 Most of the studies included in the meta-analysis used ratios of lactulose:mannitol or lactulose:rhamnose in urine to assess intestinal permeability. Although there are technical limitations in assessing intestinal permeability in humans and the number of patients in these studies is rather small, only a subset of patients with NAFLD have increased intestinal permeability.
Binding of microbial products to pathogen pattern receptors promotes liver inflammation and injury. Toll-like receptor (TLR)-4 is the cell receptor for LPS and changes in its signalling contribute to development of fatty liver in rodents.41 However, a recent clinical trial showed that inhibition of TLR4 by JKB-121 did not reduce hepatic fatty content or fibrosis in patients with NASH compared with patients with placebo.42 So, gut barrier dysfunction with subsequent translocation of microbial products might make only a small contribution to development or progression of fatty liver disease, or only in a subset of patients. In animal models, fatty liver development appears to require gut microbiota, but other microbiota-associated factors might be required for pathogenesis of NAFLD.
Metabolomic studies of NAFLD
Recent technological advances in untargeted metabolomic analyses, with nuclear magnetic resonance, gas chromatography-mass spectrometry and liquid chromatography-mass spectrometry, led to identification of differentially regulated metabolites in patients with NAFLD. A careful comparison of published metabolomic data (faeces, serum, plasma, urine or liver) from healthy individuals versus those with NAFL or NASH is provided in the online supplementary tables 1–4.
Supplemental material
The severity of fibrosis in NAFLD associates with changes of metabolites in serum.43–49 Inosine and hypoxanthine (associated with nucleoside metabolism) are enriched in serum samples from patients with mild or moderate NAFLD (stage 0–2 fibrosis), whereas succinate, malate, α-ketoglutarate, glutamine, serine, fumarate, α-ketobutyrate, glutamate and lactate (associated with amino acid and carbon metabolism) are enriched in serum samples from patients with advanced fibrosis (stages 3–4).44 A recent study also showed that severity of liver steatosis correlates with phenylacetic acid in humans.50 Levels of some metabolites, such as 3-(4-hydroxyphenyl) lactate, N-formylmethionine, phenyllactate, mannitol, allantoine and N-(2-furoyl) glycine, are heritable and have a shared-gene effect with hepatic steatosis and fibrosis.49
None of these metabolites is exclusively synthesised by microbes—these metabolites can also be synthesised by the host and/or come from the diet.51 For example, amino acids can be obtained from food, but faecal levels correlate with Alistipes and correlate inversely with Enterococcus and Bacteroides in mice.52 However, there are some metabolites that cannot be produced without bacterial fermentation, such as the choline metabolite trimethylamine (TMA), the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA), and short chain fatty acids (SCFAs).53–55 Preclinical studies demonstrate that faecal choline levels correlate with Bacteroides and inversely with Prevotella and Clostridium 52, while the secondary bile acid DCA correlates with Bacteroides, Erysipelotrichaceae incertae sedis and Enterococcus, but inversely with Prevotella, Barnesiella and Alistipes in mice.52 In contrast, faecal SCFAs showed a completely opposite pattern; they correlate with Prevotella, Barnesiella and Alistipes, and inversely with Bacteroides, Erysipelotrichaceae incertae sedis and Enterococcus in mice.52
To identify metabolites that are only derived from gut microbiota, studies of germ-free mice or mice given antibiotics are used. Mice without bacteria, fungi or viruses lack metabolites derived from gut microbiota. Therefore, comparisons of metabolite profiles of germ-free mice versus conventional mice should identify metabolites of microbial origin. Conventional mice whose diet are supplemented with choline have increased plasma levels of TMA N-oxide (TMAO), compared with mice on control chow diet. However, choline supplementation did not increase levels of TMA (and thereby TMAO) in germ-free mice.54 Similarly, caecal contents from germ-free mice do not contain DCA and LCA, because Clostridium scindens is required for 7α-dehydroxylation of primary bile acids.53 SCFAs such as acetate, propionate and butyrate were detected in caecal contents of conventional but not germ-free mice.55 These metabolites can therefore be considered bona fide metabolites derived from microbes. It will be important to study their effects in the pathogenesis of NAFL and NASH.
Microbiota-derived metabolites in NAFLD
Choline and choline-related metabolites
Choline, considered an essential nutrient by the Institute of Medicine,56 can influence multiple physiological processes in the liver such as lipid and cholesterol metabolism, signalling through lipid second messengers and enterohepatic circulation of bile acids.57 Choline is mainly obtained from diet, although the liver can produce some choline. Preclinical studies demonstrate that decreased levels of choline can lead to fatty liver disease by reducing the efflux of very-low-density lipoproteins (VLDL) from hepatocytes.58 59 A choline-deficient diet has been used to induce steatohepatitis in rodents for decades.60
TMA is a product of choline metabolism by the intestinal microbiota.61 It reaches the liver via portal vein where it is oxidised by hepatic flavin-containing monooxygenases to form TMAO. TMAO is then released into the circulation.62 Conversion of choline into TMA by microbiota in 129S6 mice on an HFD led to a reduction in bioavailability of choline, mimicking the effect of choline-deficient diet.63 TMA was proposed to be hepatotoxic,63 although direct evidence for promoting NAFLD is currently lacking. TMAO might contribute to the development of NAFLD via different mechanisms. TMAO modulates glucose metabolism and increases insulin resistance in mice on an HFD.64 In addition, TMAO promotes inflammation in adipose tissue, which can promote insulin resistance by increasing the serum level of inflammatory cytokine C-C motif chemokine ligand 2.64 TMAO also affects lipid absorption and cholesterol homeostasis by reducing conversion of cholesterol into bile acids.65 Levels of the bile acid synthesis enzymes cytochrome P450 family 7 subfamily A member 1 (CYP7A1) and CYP27A1 were reduced in livers of mice fed a TMAO-containing diet.65
Clinical evidence suggests that subjects placed on choline-deficient diet developed fatty liver in MRI studies, but only if they carried a single-nucleotide polymorphism in the promoter region of PEMT (rs12325817). This polymorphism affects de novo synthesis of phosphatidylcholine66. Serum levels of TMAO are significantly higher in patients with NAFLD than in healthy people and correlate with the severity of steatosis (mean level in patients with moderate or severe NAFLD, 0.434+0.073 µM; mean level in patients with mild NAFLD, 0.160+0.038 µM; mean level in healthy people, 0.104+0.040 µM).43
Apart from NAFLD, other metabolic diseases are also affected by TMAO. TMAO promotes atherosclerosis, increases risk of cardiovascular diseases and is associated with type 2 diabetes mellitus.62 67 Atherosclerosis, cardiovascular diseases and type 2 diabetes mellitus are closely correlated with NAFLD progression, so it is possible that TMAO indirectly affects NAFLD, by increasing the risk of atherosclerosis, cardiovascular disease and/or type 2 diabetes mellitus in patients. Or these disorders might share pathways of pathogenesis.
In summary, preclinical and clinical evidence indicates that intestinal choline metabolism into TMA and TMAO can contribute to development of fatty liver disease (figure 2). Strategies to reduce TMA and/or TMAO might be developed for treatment or prevention of NAFLD. For example, 3,3-dimethyl-1-butanol, a structural analogue of choline that inhibits TMA production from microbes by inhibiting microbial TMA lyases and thereby reducing levels of TMAO, reduced atherosclerosis in mice fed a high-choline or L-carnitine diet68. Each person’s microbial profile is distinct, so 3, 3-dimethyl-1-butanol is likely to have effects on fatty liver in only a subset of patients—effects are likely to depend on genetic, environmental and lifestyle determinants of choline metabolism by the intestinal microbiota. Thus, additional characterisation of NAFLD patient cohorts is required to determine whether all or only a subset of patients with NAFLD have increased systemic TMAO level.

Choline and its metabolites (TMA and TMAO). Choline is converted to TMA by intestinal microbiota and results in choline deficiency (downward arrow), which reduces efflux of VLDL (downward arrow) from hepatocytes and promotes inflammation. Higher levels of TMA lead to increased production of TMAO and promote development of NAFLD by increasing insulin resistance and reducing glucose tolerance. NAFLD, non-alcoholic fatty liver disease; TMA, trimethylamine;TMAO, trimethylamine N-oxide; VLDL, very-low-density lipoprotein.
Bile acids
Bile acids and their metabolites help maintain hepatic glucose, cholesterol and triglyceride homeostasis. Preclinical studies suggest that bile acids can contribute to development of NAFL and NASH by altering nuclear bile acid receptor farnesoid X receptor (FXR) signalling.69 70 The primary bile acid chenodeoxycholic acid (CDCA) activates FXR signalling, whereas the secondary bile acid DCA inhibits this activation and counteracts the effects of CDCA.71
FXR regulates glucose and lipid metabolism via different mechanisms, such as increasing insulin sensitivity, repressing hepatic gluconeogenic genes and increasing hepatic glycogen synthesis.72 73 FXR can also induce the expression and secretion of hepatic fibroblast growth factor 21 (FGF21), a regulator of metabolism that stimulates the uptake of glucose in adipose tissue74. Additionally, FXR activation inhibits lipogenesis and promotes fatty acid oxidation and also affects cholesterol transportation.75 76 Moreover, FXR can induce expression of FGF15 in the ileum of mice (FGF19 in humans)—this protein binds to FGF receptor 4 (FGFR4) and klotho beta (KLB) on hepatocytes, which inhibits expression of CYP7A1 and bile acid synthesis.77
Bile acids can also bind to G-protein-coupled receptor TGR5.69 TGR5 is expressed in non-parenchymal liver cells, enteroendocrine L cells, brown adipose tissue, white adipose tissue, skeletal muscle, gallbladder and some areas of the central nervous system.78 Bile acids can inhibit LPS-induced production of cytokines such as interleukin 1 alpha (IL1A), IL1B, IL6 and tumour necrosis factor (TNF) by Kupffer cells via TGR5-cAMP dependent pathways,79 so TGR5 regulates inflammation in liver. TGR5 also regulates glucose homeostasis, by inducing expression of glucagon-like peptide-1 (GLP-1) and inhibiting activation of the NLRP3 inflammasome. Activation of TGR5 results in more energy consumption and less body weight gain.80 81 Administration of TGR5 or FXR agonists to mice reduced NAFLD by inhibiting lipogenesis, improving hypercholesterolaemia, inducing energy expenditure and decreasing hepatic inflammation.82 83
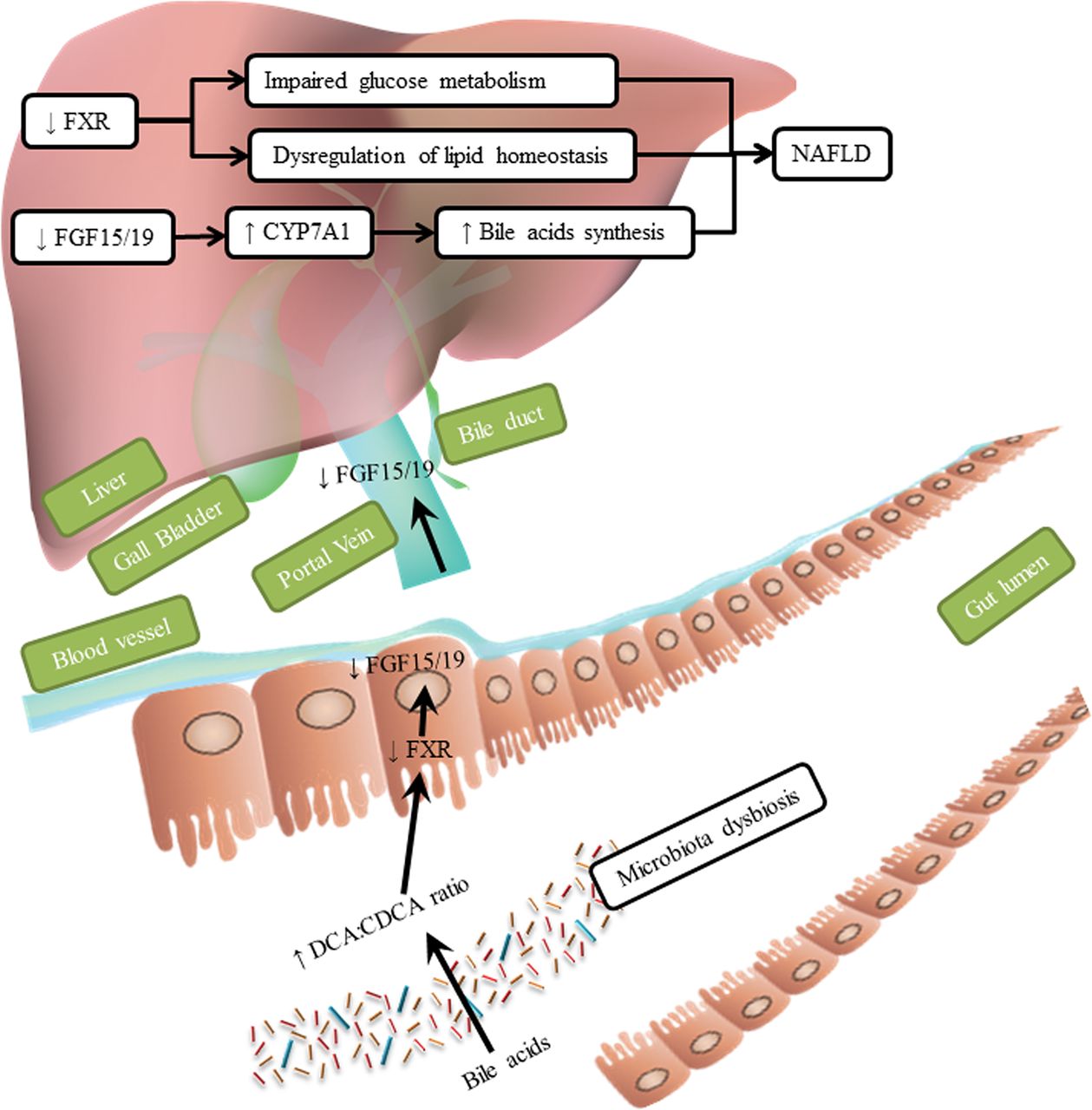
Clinical studies indicate that dysregulation of bile acid homeostasis and its related signalling pathways is associated with the occurrence of NAFLD. Concentrations of bile acids such as CDCA, DCA and ursodeoxycholic acid are increased in serum and urine samples of patients with NASH compared with healthy subjects.84 Levels of cholic acid are increased in liver tissues from patients with NASH.85 Interestingly, another research group found that liver levels of cholic acid and DCA were significantly decreased in patients with NASH.86 In addition, plasma level of glycocholate, taurocholate, glycochenodeoxycholate, taurochenodeoxycholate and ursodeoxycholic acid were increased in patients with NASH compared with patients with NAFL.87 Levels of taurolithocholic acid, glycocholate and taurocholate correlated with severity of portal inflammation, lobular inflammation, steatosis and hepatocyte ballooning, respectively.87 The ratio of DCA to CDCA was significantly increased in patients with NASH,70 84 85 and levels of FXR were lower in liver tissues from paediatric patients with NAFLD compared with healthy children.88 Serum or plasma levels of FGF19 are reduced in patients with NAFLD, reducing hepatic FGFR4 signalling to promote NAFLD.88 Decreased FGF19 is indicative of an intestinal bile acid profile that has a lower affinity for FXR in intestinal epithelial cells.
Agents that activate FXR or TGR5 signalling are being developed for treatment of NAFLD. 6-ethylchenodeoxycholic acid (obeticholic acid), a potent activator of FXR, reduced histological features of NASH in liver samples from patients, compared with placebo.89 Furthermore, an engineered analogue of FGF19 named NGM282 rapidly reduced absolute liver fat content in patients with NASH.90 Taken together, changes in bile acid metabolism affect development of NAFL and NASH via FXR and TGR5 signalling (figure 3). Positive outcomes in clinical trials demonstrate how microbiota-mediated changes in bile acids can be used as therapeutic target.

Effects of bile acids. Dysbiosis of the intestinal microbiota changes the composition of bile acids. A higher ratio of DCA:CDCA reduces activation of FXR, leading to lower expression of FGF15/FGF19. A lower level of FXR in the liver impairs glucose metabolism and dysregulates lipid homeostasis. Reduced levels of FGF15/FGF19 cause increased expression of CYP7A1, increasing bile acids synthesis and promote the development of NAFLD. CDCA, chenodeoxycholic acid; CYP7A1, cytochrome P450 family 7 subfamily A member 1; DCA, deoxycholic acid; FGF, fibroblast growth factor; FXR, farnesoid X receptor; NAFLD, non-alcoholic fatty liver disease.
Short-chain fatty acids
SCFAs include formate, acetate, propionate and butyrate; they are generated by gut microbe fermentation of non-digestible carbohydrates such as non-starch polysaccharides, resistant starch and miscellaneous low-digestible saccharides.55 SCFAs provide not only important sources of nutrients and energy from the intestinal epithelium, but also precursors for lipogenesis and gluconeogenesis (a metabolic pathway that results in the generation of glucose from non-carbohydrate carbon substrates such as lactate, glycerol and glucogenic amino acids). Gluconeogenesis is involved in the development of fatty liver disease, after absorption and delivery of nutrients to the liver via the portal vein.
Preclinical studies indicate that SCFAs activate the G-protein-coupled receptors (GPCRs) GPR41 and GPR43, which are expressed on adipocytes and gut enteroendocrine L cells. Conventionally raised Gpr41–/– mice had lower body weight than their wild-type littermates.91 Consistent with these results, Gpr43–/– mice did not become obese, have increases in serum triglycerides or develop insulin resistance when placed on an HFD.92 Activation of these GPCRs therefore appears to contribute to obesity and metabolic disease, and might participate in the pathogenesis of fatty liver disease via multiple mechanisms.
Activation of GPR41 and GPR43 stimulates secretion of peptide-YY, inhibits gut motility and slows intestinal transit, thereby enhancing nutrient absorption, increasing energy harvest from the diet and promoting hepatic lipogenesis.91 93 Additionally, activation of GPR41 and GPR43 promotes secretion of GLP-1, which not only inhibits gastric emptying and food intake, but also activates genes in hepatocytes that regulate fatty acid β-oxidation and insulin sensitivity.94 95 Finally, activation of GPR43 in adipocytes inhibits lipolysis and decreases plasma fatty acids.96
Apart from their activation of GPR41 and GPR43, SCFAs enter the liver through the portal vein, where they contribute to lipid and glucose metabolism.97 Increased acetate in the liver can cause triglyceride accumulation because acetate is an important substrate for fatty acid synthesis.98 Increased levels of propionate promote gluconeogenesis in the liver, since propionate is a precursor for gluconeogenesis.97 99 Triglyceride accumulation and gluconeogenesis have each been associated with development of NAFLD.
Although SCFAs directly or via binding to GPRs appear to promote NAFLD, other effects of SCFAs might be beneficial for NAFLD. For example, butyrate might modulate development of NAFL and NASH by activating AMP-activated protein kinase (AMPK). AMPK inhibits oxidative stress and inflammation,100 regulates energy homeostasis by affecting glucose and lipid metabolism,101 controls fatty acid oxidation by regulating mitochondrial biogenesis and suppresses expression of lipogenic genes by reducing the activity of the sterol regulatory element-binding transcription factor 1 c (SREBP-1c).102 Butyrate activates AMPK in liver, which reduces HFD-induced obesity and insulin resistance in mice.103 Consistently, in vitro experiments showed that butyrate increased AMPK activity and accelerated the assembly of tight junction proteins in the colonic epithelial cell line Caco-2.104
Furthermore, SCFAs appear to inhibit development of NAFL and NASH at the epigenetic level. Histone deacetylases (HDACs) prevent gene transcription by removing histone-bound acetyl groups. Inhibitors of class I and II HDACs (such as SCFAs) block this epigenetic regulation to allow transcriptional activation—their activity has been associated with development of different diseases. Butyrate inhibits HDAC activity, increasing acetylation of histone and non-histone proteins at genes and their transcription. Butyrate also inhibits LPS-induced activation of the SRC proto-oncogene, which encodes an enzyme that phosphorylates focal adhesion kinase; butyrate thereby reduces macrophage motility and inflammation.105 HDAC inhibitors also activate transcription of the forkhead box P3 gene to increase numbers of regulatory T cells, which suppresses the immune response and reduces inflammation.106
SCFAs therefore appear to promote and also prevent development of NAFL and NASH depending on the signalling pathway or mechanism they activate, which include GPR41 and GPR43, AMPK, HDAC inhibition and/or directly promoting liver triglyceride accumulation and gluconeogenesis (figure 4). Supplementation studies allow to directly assess the net effect of SCFAs. Treatment of wild-type mice with tributyrin, a butyrate prodrug, protects against diet-induced obesity, hepatic steatosis and insulin resistance.107 Supplementation of acetate or propionate protects against diet-induced weight gain, insulin resistance and hepatic steatosis in mice by reducing hepatic fatty acid uptake and suppressing lipogenesis in the liver.108 Based on these preclinical data SCFA supplementation appears to exert overall beneficial metabolic and antisteatotic hepatic effects.

Effects of SCFAs. SCFAs are generated by gut microbiota digestion of dietary fibres. They enter the liver through the portal vein. In the liver, acetate and propionate promote accumulation of TGs and gluconeogenesis, respectively. SCFAs also activate GPR41 and GPR43 on enteroendocrine L cells, leading to secretion of PYY and increased nutrient uptake, more energy intake and increased hepatic lipogenesis. On the other hand, butyrate can activate AMPK and induce epigenetic changes, which in turn can reduce hepatic steatosis. AMPK, AMP-activated protein kinase; GPR41, G‑protein receptor 41; PYY, peptide-YY;SCFA, short-chain fatty acid; TG, triglyceride.
Clinical studies demonstrate that formate and acetate are enriched in faecal samples from adults with advanced fibrosis, whereas butyrate and propionate are enriched in faecal samples from adults with mild or moderate NAFLD.44 It therefore appears that levels and subtypes of SCFAs correlate with the severity of fatty liver disease. However, another study reported that some SCFAs, especially formate, acetate and valerate, were less abundant in faecal samples from children with NAFLD.109 These apparent differences might be due to differences in patient age, but could also be related to differences in diet, environmental factors and technical issues. SCFAs are volatile substances and immediate processing is required for accurate measurement.
In summary, whether altered production of SFCAs by the intestinal microbiota contributes to liver disease in patients with NAFL or NASH, deserves further investigations. Larger studies are required to demonstrate whether systemic and faecal levels of SCFAs are consistently altered in patients with NAFLD.
Ethanol
Clinical studies demonstrate that low levels of endogenous ethanol are detected in blood samples from subjects after intake of alcohol-free food110—endogenous ethanol might be generated by the intestinal microbiota. Non-alcoholic paediatric patients with NASH had higher serum levels of ethanol than obese and healthy children without NASH.111 112 Endogenous ethanol might therefore contribute to the pathogenesis of NAFL and NASH. Alcoholic steatohepatitis and NASH have similar histological features.113 It is conceivable that production of ethanol by the gut microbiota increases the severity of liver disease in patients with NAFL or NASH.
Preclinical studies have established that ethanol consumption increases intestinal permeability by well-characterised mechanisms, such as increasing intestinal expression of inflammatory cytokines. The ethanol metabolite acetaldehyde also appears to affect the function of tight junction proteins.114 Further studies are needed to explore whether a subgroup of patients with NAFLD with increased intestinal permeability have increased ethanol production by the microbiota.
Microbe PAMPs, which translocate to the liver through disrupted tight junctions, increase hepatic expression of TNF and levels of lipogenesis-related factors such as acetyl-CoA carboxylase, fatty acid synthase and SREBP-1c.115 Endogenous ethanol inhibits the tricarboxylic acid cycle, thus increases levels of acetate, thereby promoting triglyceride accumulation in hepatocytes.98 Ethanol can also increase the activity of CYP2E1116 to catalyse ethanol oxidation and produce free radicals that might promote liver inflammation.117 Moreover, acetaldehyde has oxidant-dependent metabolic and cytotoxic effects, which can further enhance liver injury.118
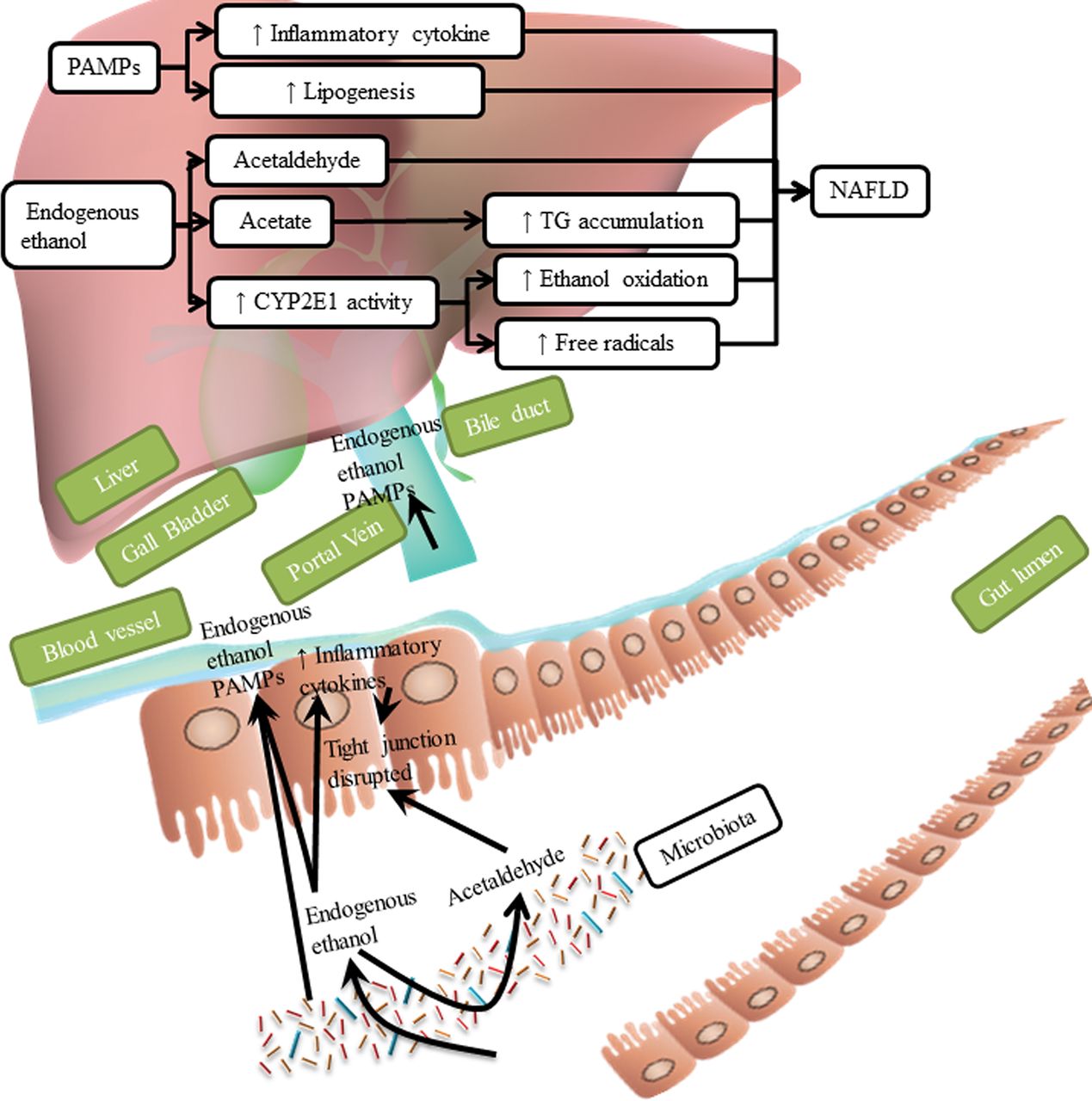
In summary, endogenous ethanol might contribute to the pathogenesis of NAFLD by increasing intestinal permeability, levels of acetate and formation of acetaldehyde, which promotes liver injury (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of ethanol. Endogenous ethanol increases intestinal permeability by activating production of inflammatory cytokines and acetaldehyde. This leads to translocation of PAMPs into the liver, further increasing production of inflammatory cytokine and lipogenesis. Acetaldehyde produced from endogenous ethanol is cytotoxic to hepatocytes, and acetate increases accumulation of TGs. Endogenous ethanol increases the activity of CYP2E1, which catalyses oxidation of endogenous ethanol to produce free radicals, which causes liver inflammation. CYP2E1, cytochrome P450 family 2 subfamily E member 1; PAMP, pathogen-associated molecular pattern molecule; TG, triglyceride.
Changes of other metabolites in NAFLD
Levels of other metabolites differ between individuals with versus without NAFLD. For instance, levels of some alcohols and ketones are higher in faecal samples from patients with NAFLD, as well as some esters and lactates (L-lactate and D-lactate).44 109 119 120 Blood (plasma and/or serum) samples from patients with NAFLD had higher levels of glucose, some lipids (monounsaturated fatty acids) and branched-chain amino acids (leucine, valine and isoleucine),50 121 122 while urine samples also had higher levels of glucose and branched-chain amino acids.47 50 123 Liver levels of butyryl carnitine (C4) and lauryl carnitine (C12) are increased in patients with NAFLD, whereas the level of linoleic acid is decreased.86
Although these metabolite changes seem diverse, it appears that succinate,44 124 glucose,47 49 86 122 123 branched-chain amino acids50 and some non-essential amino acids (tyrosine and cysteine)46 47 86 123 were increased in multiple types of samples from patients with NAFLD. This might be due to lifestyle features of these patients, such as a sugar-rich diet. However, these consistent findings may provide an opportunity to develop a diagnostic biomarker for NAFLD. Succinate, which was increased in faecal, serum and liver samples from patients with NAFLD, is an extracellular signalling molecule as well as an intermediate in the citric acid cycle. It might promote progression of NAFLD by causing production of α-smooth muscle actin, a biomarker of fibrogenic response, by activating GPR91 in hepatic stellate cells.125 Succinate is an important metabolite that is produced by mammalian cells and by intestinal microbes such as Bacteroidaceae and Prevotella 126 127—these microbes are increased in faecal samples of patients with NASH.45 111
A recent study showed that plasma phenylacetic acid correlates with hepatic steatosis and NAFLD activity score in humans.50 It is a product from metapleural gland secretions in ant species and an oxidation product of phenethylamine in humans.128 129 Gut bacteria can also produce phenylacetic acid.50 130 Phenylacetic acid promotes development of NAFLD via inducing triglyceride accumulation in human primary hepatocytes and promoting the expression of lipid metabolism genes (LPL and FASN).50 It reduces AKT phosphorylation, therefore leading to insulin resistance.50
Levels of 3-(4-hydroxyphenyl) lactate are also increased in serum samples from patients with NAFLD compared with controls. It is synthesised by bacterial enzymes such as D-hydrogenase and cinnamoyl-CoA:phenyllactate CoA transferase, and many intestinal microbes are capable of producing it.49 3-(4-hydroxyphenyl) lactate has a shared-gene effect with hepatic steatosis and fibrosis.49 So, risks of NAFLD might involve genetic factors, features of the intestinal microbiota and derived metabolites such as 3-(4-hydroxyphenyl) lactate.
Succinate, phenylacetic acid and 3-(4-hydroxyphenyl) lactate are intestinal microbe metabolites that promote NAFLD development and might be developed as diagnostic markers. Researchers might also develop strategies to reduce microbe production of succinate, phenylacetic acid and 3-(4-hydroxyphenyl) lactate, thereby reducing development or progression of NAFLD. These are important molecules for further study.
Conclusion
Many preclinical studies have shown that altering gut microbiota can promote development of fatty liver. Although it is commonly thought that translocated microbial products contribute to NAFLD, fewer than half of the patients with NAFLD have evidence for increased intestinal permeability. However, this is based on a relatively small number of patients and technically challenging techniques. Future studies with higher number of patients with NAFLD are required to evaluate gut barrier dysfunction. It might be more accurate to perform several assays in the same patients, for example, faecal albumin, systemic LPS and a sugar absorption test.
On the other hand, other microbial factors, such as microbiota-related metabolites, seem important for NAFLD disease progression. Metabolomic studies are challenging, the identity of many detected metabolites is unknown and the microbial origin is reported for a few metabolites only. There are inconsistent or even conflicting results in metabolomic profiles in patients with NAFLD, which might be a result from the relative small number of patients, lack of uniform methodology and standardised protocols. However, such differences, if reproducible, might offer opportunities for categorising NAFLD into several subtypes. As not all patients with NAFLD have a disrupted gut barrier, different NAFLD subtypes might have different metabolites as driver for disease progression. Carefully designed metabolomic studies, larger scale clinical studies, and different ethnic background with uniform methodology to monitor metabolites in different samples (stool, serum/plasma, urine and liver tissue) over time will provide insights into mechanisms of fatty liver and steatohepatitis development, and lead to new diagnostic and therapeutic approaches. Such approaches might confirm metabolic NAFLD subtypes and open the opportunity for personalised treatment approaches. Moving towards metabolic NAFLD subtypes requires clinical studies to not only report the mean or average of a certain metabolite, but also report the percentage of patients with normal and elevated/lower levels.
Agents are being developed to target some of these metabolites (activate, block or alter production of) for treatment of patients. For example, microbial choline conversion into TMA can be blocked using an inhibitor of the bacterial enzyme 3,3-dimethyl-1-butanol. Such a precision-microbiome therapy might be suitable for a choline-deficient metabolic NAFLD subtype (if existent) and might be used as personalised medicine approach. Multiple preclinical studies are trying to develop agents that target some of these metabolites (activate, block or alter production of) to treat NAFLD. Well-designed clinical studies are required to effectively testing the effects of metabolites in humans.
Taken together, identification of microbial metabolites causing or driving NAFLD, the field is currently in its infancy. With additional technological advances and better designed clinical studies, we will be able to categorise patients with NAFLD into different metabolic subtypes and design personalised or subtype specific therapies.
Acknowledgments
The authors appreciate the numerous support from the research team at UCSD and the critical feedback of the manuscript from Dr Xiaohua Hou at Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. The authors also appreciate the financial support for HC from Union Hospital, Tongji Medical College, Huazhong University of Science and Technology.
References
Footnotes
Contributors HC, YD and BS: contributed to the conception and design of the work. All authors wrote and critically reviewed and edited the manuscript; approved the final version of the manuscript.
Funding This work was supported by National Natural Science Foundation of China (No.81570530, 81370550; to LY) and NIH grants R01 AA020703, R01 AA024726, U01 AA021856 (to BS).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.