Article Text

Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The Blastocystis genus has a high prevalence in human populations, and it is associated with higher microbial diversity, richness and microbial composition.
While often considered a human parasite, the Blastocystis genus might be a commensal, whose assumed potential pathogenicity derives from unsurveyed variables such as subtype diversity.
Nine out of the 17 known Blastocystis subtypes have been reported in human populations.
It has been shown that some subtypes have a distinct functional potential; yet, no study addresses potential associations between Blastocystis subtypes and microbiota profiles in the human gut at the population level.
What are the new findings?
In a non-clinical cohort, Blastocystis subtypes are strongly associated with microbiota community composition, with higher explanatory power than host characteristics.
Microbial richness and diversity are linked to Blastocystis prevalence and subtype variation.
Blastocystis prevalence is reduced in Flemish patients with IBD.
Host age is associated with Blastocystis subtype carrier status.
All Blastocystis subtypes detected in this cohort were found to be less prevalent in Bacteroides enterotyped samples.
There is an inverse correlation of subtype 3 and 4 with Akkermansia, suggesting differential associations between subtypes and host health.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
This result has potential implications on clinical practice in the foreseeable future. Current clinical consensus for faecal transplantation recommends the exclusion of donors of samples that are positive for Blastocystis.
This study shows that subtype 4 is associated with markers of a healthy gut microbiota, suggesting further investigations are needed to resolve the assumed pathogenic potential of Blastocystis.
As Blastocystis subtypes explanatory power outperforms previously reported microbiota covariates, clinical applications related to human gut microbiota findings should include the evaluation of individuals stratification based on Blastocystis subtypes.
Introduction
The human microbiota encompasses bacteria, archaea, viruses and single-cell eukaryotes (SCE).1 Current microbiota research efforts mostly focus on bacteria, archaea,2 their phages3 and viruses,4 leaving the eukaryotic fraction underexplored. Blastocystis, a genus commonly detected in the human GI tract, belonging to the SCE group stramenopiles,5 is an example of these understudied gut eukaryotes. While multiple studies have aimed to understand its biology, most of them did not address it within the context of the human gut microbiota diversity and structure. Historically, Blastocystis has been categorised as an intestinal parasite.6–8 Its detection in faecal samples of asymptomatic individuals has urged researchers to consider a commensal relationship between Blastocystis and its human host,5 9 10 challenging the assumption of a parasitic strategy previously assigned to the entire genus.7 8 Similar to Escherichia coli, Blastocystis commensalism or pathogenicity is highly dependent on the organism’s pan-genome (ie, coding for virulence factors and toxins). Therefore, only high-resolution-level analyses (at the strain/subtype level) can elucidate the pathogenic potential of Blastocystis subtypes or intra-subtypes. Still, little is known regarding the prevalence and phylogenetic variation of Blastocystis subtypes in healthy/IBD-affected populations and its association to bacterial and archaeal gut communities.
Blastocystis is a diverse genus comprising 17 characterised subtypes (ST1–ST17).11 Nine of these subtypes have been reported to occur in the human GI tract5 with varying prevalence across different populations around the world.12 Human gut-associated Blastocystis subtypes exhibit a substantial variation in gene repertoire.13 14 For example, the ST1 subtype has been reported to have gained genes involved in antibiotic synthesis, while ST3 appears more sensitive to oxidative stress and might have lost genes involved in mucus degradation.14 These findings suggest that different Blastocystis subtypes could thrive in divergent niches, which, in turn, could represent distinct potential interactions with particular human gut microbial ecologies. Although previous studies have suggested a potential role of Blastocystis in shaping the human gut ecosystem,13 15 16 to date, microbiota differentiation related to subtypes and intra-subtypes distribution remains unexplored.
The Flemish Gut Flora Project (FGFP), a population-wide microbiota monitoring effort, has generated one of the largest and best characterised faecal microbiota database currently available.17 To date, 1106 FGFP faecal samples have been analysed using amplicon sequencing. Extensive metadata collection efforts enabled the characterisation of participants in terms of their health and lifestyle, leading to the identification of 69 factors associated to microbiota variation (microbiota covariates hereafter). However, to this date, FGFP microbiome analyses have been limited to the bacterial and archaeal fractions of the gut microbiota.
Some studies regarding Blastocystis carrier status and their relation to the human microbiota have been conducted in clinical populations. To the best of our knowledge, we here present the first population-level study of the association of Blastocystis and Blastocystis subtypes with host parameters, microbiota diversity, composition and structure. Herein, we took advantage of the FGFP microbiota monitoring effort to investigate the prevalence of Blastocystis and its subtypes in a cross-sectional Western population cohort. Moreover, we assessed potential links among Blastocystis and established microbiota covariates and quantified microbiota differentiation in function of subtype relative abundances. Our analyses emphasise the need for large-scale high-resolution studies, at least at the subtype level, of gut-associated Blastocystis to clarify the putative pathogenicity and health impact of the genus.
Methods
Study population
Flemish Gut Flora Project
A subset of 616 stool samples from a random selection of participants in the FGFP was included in this study. A detailed description of the cohort and sample collection is available.17 18 Briefly, samples were collected in Flanders, the northern region of Belgium, from volunteers agreeing to participate in the study protocol that included a medical check-up with blood assessment and medical questionnaires, extensive questionnaires on lifestyle, dietary habits, well-being and childhood, and donation of a stool sample. Recruitment was done through popular media (television, newspapers, radio) and participants did not receive their personal results or any other compensation. As a result, the FGFP represents an average cohort from the Western European population for gut microbiota research.
IBD-L
A cohort of 107 patients with IBD recruited at the Leuven hospital were included in the analysis. The general information of this cohort is listed in online supplementary table S1.
Supplemental material
rRNA genes amplification for archaea, bacteria and eukaryotes identification
Abundance profiles for archaea, bacteria and eukaryotes were generated using amplicon-based sequencing of the V4 variable region in 16S rRNA and 18S rRNA genes, using the 515F/806R universal primer pair.19 Amplification, purification and sequencing conditions were performed as in a previously published protocol.20 Although the 515F/806R universal primer pair was designed to amplify the V4 archaeal and bacterial variable region of the 16S rRNA genes, in silico analysis showed the co-amplification of the V4 eukaryotic variable region of the 18S rRNA genes, from unicellular protists to multicellular mammals.19 Stramenopiles, such as Blastocystis, were identified in the data generated using this primer pair. Primers alignment against the most prevalent Blastocystis subtypes suggests these primers are suitable for amplification and subtyping of this genus in humans (online supplementary figure S8). Blastocystis subtypes could be identified based on the sequence of their 18S rRNA V4 region alone. This method allowed us to differentiate the 95% most prevalent Blastocystis subtypes in humans, namely subtypes ST1, ST2, ST3 and ST4,5 as well as other rarer subtypes, ST6, ST7, ST8 and ST9.
DNA extraction and amplicon sequencing
Extracted DNA from faecal samples, using a modified MoBio PowerMicrobiome DNA/RNA isolation kit,17 was quantified using a Qubit fluorometer. For each sample, we used 10 ng of DNA to amplify the V4 variable region of the 16S rRNA and 18S rRNA genes, using the 515F/806R primer pair (GTGYCAGCMGCCGCGGTAA/GGACTACNVGGGTWTCTAAT) modified to include Illumina adaptors and barcodes, in order to produce ready-to-sequence PCR products.20 Samples were amplified in triplicate and each reaction was visualised using agarose gel and SYBR Safe DNA Gel Stain. As a positive control, DNA from a previously characterised human stool sample was added; blanks were used as negative controls to monitor potential reagent and laboratory contamination. Amplicon concentration and fragment size distribution were analysed using a Fragment Analyser (Advanced Analytical Technologies) to produce equimolar pools. QIAquick PCR Purification Kit was used to purify and concentrate the pooled libraries and a Bioanalyzer (Agilent) was used to estimate the concentration and size distribution of the final pool. Size selection, before Illumina sequencing, was performed using Agencourt AMPure to remove fragments below 200 bases. Sequencing was performed using Illumina HiSeq 2500 (HiSeq-Rapid SBS kit V2 (500 cycles, 2×250 bp)) at the VIB Nucleomics core laboratory (Leuven, Belgium). Sequencing depth ranged from 10 493 to 109 649 observations per sample, producing a total of 193 genera, including bacteria, archaea and SCE reads (online supplementary table S1).
Analysis of 16S and 18S rRNA genes sequences
Data analysis was performed with the pipeline LotuS1.56521 for Operational Taxonomic Units (OTUs) definition using default settings: quality score above 25, UCHIME for chimaera removal, UPARSE, as part of usearch9.0.2132 for linux32,22 and FastTree V.2.1.723 for tree building. For taxonomic annotation, we used PR2 (Protist Ribosomal Reference database)24 for eukaryotes and RDP Classifier V.2.1225 for archaea and bacteria (scripts and settings are presented in online supplementary material).
SCE and validation of Blastocystis genus detection
SCE were identified from OTU sequence clusters generated by LotuS and PR2 (online supplementary table S3). An orthologous method was used to confirm the presence or absence of Blastocystis; from the same DNA samples used for amplicon sequencing, 20 ng of DNA was amplified in duplicates, using the Blastocystis-specific primer combination RD5. All/ATCTGGTTGATCCTGCCAGT, BhRDr. All/GAGCTTTTTAACTGCAACAACG and a 40-cycle reaction.26 Amplicons were visualised using agarose gel and SYBR Safe DNA Gel Stain. Samples without amplicons were considered negatives, and samples with amplicon bands between 550 and 600 bp were considered positives.27 SCE-relative abundance was calculated as the quotient of SCE over bacteria, archaea and SCE together. Blastocystis subtypes were identified using BLAST searches against a non-redundant nucleotide database28 and an alignment of 181 bases fragments from the most abundant sequence of each subtype was performed29 (figure 1C). In addition to Blastocystis, other SCE were confirmed using the same BLAST approach (online supplementary table S2). Samples containing additional SCE (Pentatrichomonas and Entamoeba) and rare Blastocystis subtypes were removed before performing further analysis (online supplementary table S3).
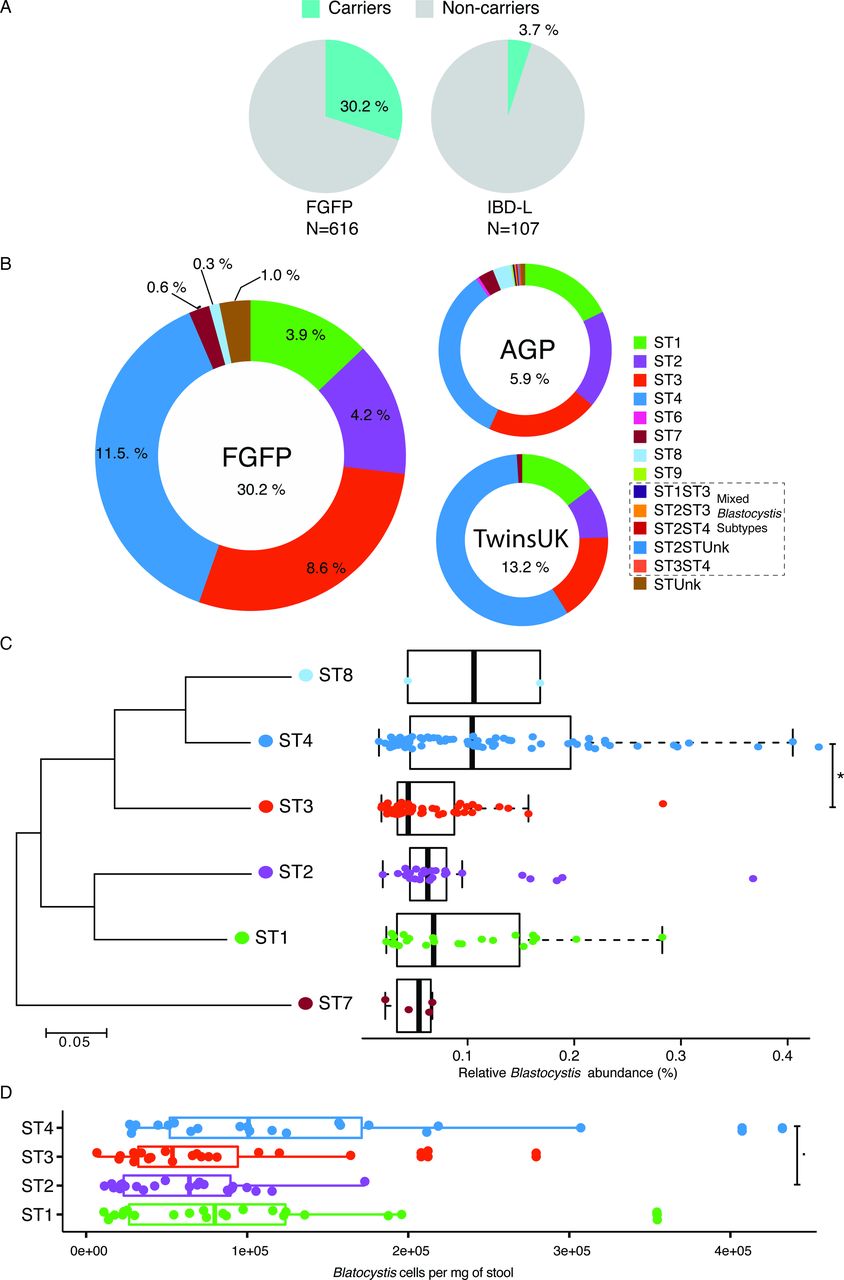
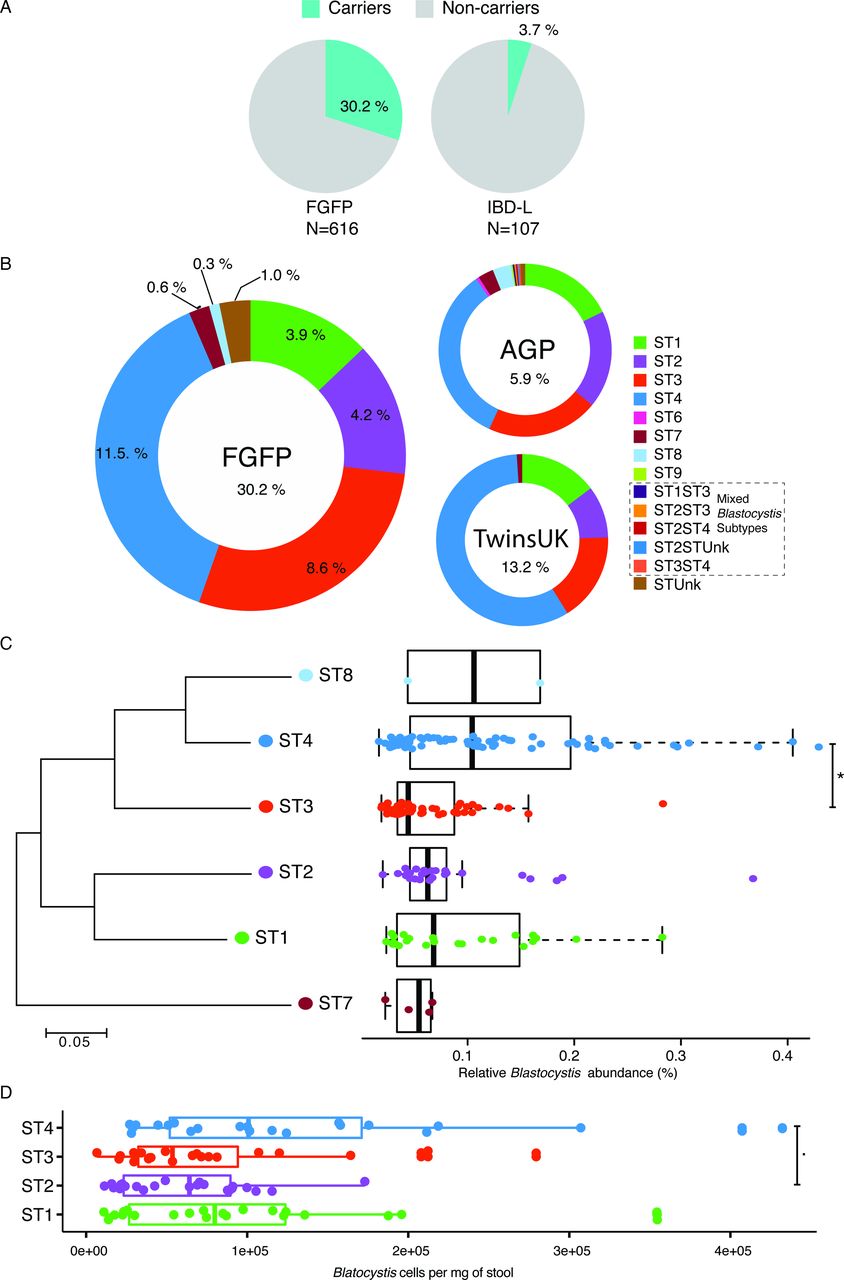
Prevalence, distribution and cell density of Blastocystis subtypes. (A) Prevalence of Blastocystis subtypes in the Flemish Gut Flora Project (FGFP) and IBD-L cohorts. IBD-affected individuals present lower prevalence of Blastocystis. (B) Prevalence of Blastocystis subtypes in the FGFP, TwinsUK (n=1045) and American gut project (AGP) (n=7567). (C) Neighbour-joining 18S rRNA gene phylogenetic tree of FGFP Blastocystis subtypes based on an alignment of 181 bases fragments from the most abundant sequence of each subtype29 and boxplot of their relative abundances in Blastocystis-positive individuals. Two extreme outliers were omitted from the visualisation. (D) Boxplot of Blastocystis cells per milligram of stool in the four more prevalent ST in the FGFP. The body of the boxplots represents the first and third quartiles of the distribution, and the median line. The whiskers extend from the quartiles to the last data point within 1.5×IQR, with outliers beyond. Dunn’s multiple-comparison test, false discovery rate (FDR): *FDR<0.01 and .FDR<0.05.
The capability of our protocol to detect Blastocystis subtypes from amplicon sequencing data was tested using published data sets. Demultiplexed fastq files from two large cohorts, the American gut project (AGP) (n=7567)30 and the TwinsUK (n=1045)31 studies, were analysed using the LotuS pipeline and taxonomic annotation was performed using PR2, in the same manner as we analysed our own data. Blastocystis subtypes and other SCE were confirmed using the BLAST approach described above (online supplementary table S4 and S5). To evaluate Blastocystis prevalence, non-carriers were defined as individuals with more than 10 000 sequences, none of which corresponded to Blastocystis.
Blastocystis cell density estimation
Blastocystis density was estimated using qPCR of the 18S rRNA ribosomal genes. We followed a published protocol32 and conducted duplicated reactions using primers BL18SPPF1 and BL18SR2PP in a subset of 90 Blastocystis carrier samples of the four most prevalent subtypes (ST1=22, ST2=23, ST3=23 and ST4=22) from the FGFP. Estimate number of cell per milligram of stool was calculated using the values of total number of 18S rRNA copies from qPCR and assuming 100 copies of 18S rRNA gene per Blastocystis cell.33
Bacteria and archaea
Analysis of bacteria and archaea was performed from the BIOM file generated using RDP classifier from the LotuS pipeline. Sequences annotated to the class Chloroplast, family mitochondria or unknown bacteria were removed prior to the analyses. The sequencing data was rarefied to 10 000 observations to calculate inter-individual and intra-individual microbial diversity. All further analyses were performed at the genus level.
Statistical analyses
Faecal microbiota derived features, such as observed richness, Shannon diversity index and Bray-Curtis dissimilarity index, were calculated in a final data set of 598 samples, after removing samples containing other SCE (Entamoeba and Pentatrichomonas) and rarer subtypes of Blastocystis. All statistical analyses were performed using R, and packages phyloseq34 and vegan.35
The contribution of metadata variables to microbiota community variation (effect size) of each of the 76 variables was determined by distance-based redundancy analysis (dbRDA) on genus-level Bray-Curtis dissimilarity with the capscale function in the vegan package.35 We used 69 variables, previously reported to be associated to gut microbiota variation,17 and added 7 variables related to Blastocystis (presence, Blastocystis subtypes and Blastocystis subtypes relative abundance). Enterotyping (or community typing) of the samples, based on the Dirichlet Multinomial Mixtures (DMM), was obtained from a previous publication on this cohort.17
For microbiota and metadata association analysis, we excluded taxa unclassified at the genus level, and taxa present in <20% of samples. Correlations between bacteria and archaea genera and Blastocystis subtypes were performed using biweight midcorrelation in the WGCNA and microbiome R packages and using non-parametric Spearman tests. For metadata and carrier status (ST1, ST2, ST3, ST4 and non-carrier) association analysis, Wilcoxon test and Kruskal-Wallis test (non-parametric analysis of variance) were performed on the variables of the metadata. Pearson’s χ2 test and Fisher’s exact test for count data were used for testing statistical independence on categorical data. Associations between genera relative abundances and carrier status were analysed using Kruskal-Wallis test and post hoc Dunn test for pairwise comparisons. Correction for multiple testing using Benjamini-Hochberg’s procedure (false discovery rate (FDR))36 was performed when applicable; unless specified differently, significance was defined at FDR<5%.
Network inference
The abundance matrix was filtered to keep OTUs present in at least 20 samples and only samples positive for Blastocystis. Blastocystis subtypes—bacteria and archaea genera co-occurrence patterns were inferred using CoNet,37 with Spearman and Kullback-Leibler dissimilarity as measures of similarity. Briefly, for each similarity measure, a distribution of all pairwise scores was computed and thresholds were selected such that the initial network contained 45 positive and negative edges. For each measure and each edge, 1000 permutations and bootstrap scores were generated, following the ReBoot routine, which mitigates compositional bias. The measure-specific p value was then computed as the probability of the null value (represented by the mean of the null distribution) under a Gauss curve generated from the mean and SD of the bootstrap distribution. Edges with original edge scores outside the 95% CI, provided by the bootstrap distribution, were discarded. P values were then merged using Brown’s method38 and corrected for multiple testing using Benjamini-Hochberg’s procedure.36 Edges with a p value >0.05 were discarded.
Results
Blastocystis, a common constituent of the human gut microbiota
First, we assessed the prevalence and abundance of Blastocystis in a random subset of the FGFP cohort.17 A total of 616 samples were screened for Blastocystis via sequencing and sample carrier status was subsequently confirmed using targeted PCR. Out of the 616 samples profiled, 186 (30%) were found positive for Blastocystis (figure 1) with a 100% concordance between both detection methods (no discordant cases were observed). Relative Blastocystis abundances in the FGFP, as observed in sequencing data of carrier samples, ranged between 0.02% and 1.08% (figure 1C; online supplementary table S1). No association between sequencing depth and Blastocystis presence was observed (Pearson, r = −0.03, p value=0.49; online supplementary figure S1). Six distinct Blastocystis subtypes were identified among FGFP samples (figure 1B), with ST1, ST2, ST3 and ST4 being most prevalent (13%, 14%, 29% and 38% of carriers, respectively). In terms of relative abundance, highest values were observed for ST4, with ST3 being present in significantly lower proportions (Dunn’s multiple comparison, FDR=1.2e-04; figure 1C).
In order to assess absolute abundance of Blastocystis, qPCR was conducted in a subset of carriers of the four most prevalent subtypes (ST1=22, ST2=23, ST3=23 and ST4=22) from the FGFP. Blastocystis loads of ST3 and ST4 carriers were significantly different in the FGFP cohort. Quantitative qPCR analysis of 18S copy number revealed that ST4 carriers harbour a larger load of Blastocystis than that observed in ST1, ST2 and ST3 (decreasing order) (figure 1D).
Although the distribution of Blastocystis subtypes in the FGFP was consistent with the prevalence we observed in both the AGP and TwinsUK cohorts (figure 1B), the global prevalence of Blastocystis is higher in the FGFP cohort, which in contrast to the AGP does not include diabetes-affected participants.
Another significant difference between the population cohorts analysed is the fact that ST4 is more widespread in the TwinsUK cohort (Pearson’s χ2, p value=1.6e-03). Remarkably, in both the FGFP and the TwinsUK cohorts, carriers solely harboured a single subtype: no mixed populations were observed (online supplementary table S5), suggesting Blastocystis occupies a very specific niche within the human gut microbiome. Among 445 AGP carrier samples, 5 samples were identified as harbouring multiple Blastocystis subtypes (online supplementary table S4). Overall, our results confirm that Blastocystis is indeed a common constituent of the human gut microbiota in Western populations.
In order to assess whether Blastocystis (or any of its subtypes) displays hints of opportunistic pathogenic behaviour, we evaluated its prevalence in a cohort where clear dysbiosis is expected: a Flemish cohort of 107 patients affected by active IBD.
The IBD-L, included 31 patients with UC and 76 with Crohn’s disease, 9 females and 98 males, with a mean of age of 42.8 (SD ±16), body mass index (BMI) of 24.3 (SD ±4.3) and disease activity of 122 months (SD ±125) (online supplementary table S1). In contrast with the prevalence of Blastocystis in the FGFP, the IBD-L exhibited a much lower prevalence, with only 4 (4%) out of the 107 samples found positive for Blastocystis, including three ST4 and one ST1 (online supplementary table S1).
While Blastocystis is common in the FGFP, it is rare among IBD-L cohort (30% vs 4%, respectively; Fisher’s exact test, p value=0.00001) (figure 1A). Consistent with previous reports,39 the gut microbiota of patients with IBD-L is distinct than non-IBD individuals (online supplementary figure S2).
Blastocystis prevalence is associated with enterotype differentiation
Next, we explored potential associations among Blastocystis prevalence, host phenotype and gut microbiota variation in the FGFP cohort. With the exception of age, carrier status in FGFP samples was not related to any of 68 others previously identified phenotypic microbiota covariates (Wilcoxon test, FDR=0.02; table 1; online supplementary table S6).17 At the subtype level, this association with age was mostly driven by the ST4 distribution, with ST4 carriers being significantly older than Blastocystis non-carriers (pairwise Wilcoxon test, FDR=7.1e-05; table 1; online supplementary figure S3; online supplementary table S7). Among the 69 microbiota covariates included in our analysis, IBS status (as noted by the participants’ general practitioners during anamnesis) was previously linked to the presence of Blastocystis in stool samples.40 41 Here, performing the largest screening to date in a single Western cohort, we did not detect an association between Blastocystis prevalence and IBS status (Fisher’s exact test, p value=0.6652). Among carriers, Blastocystis relative abundances did not differ significantly between IBS and non-IBS individuals (Wilcoxon test, r=0.0220, p value=0.50; online supplementary figure S4), nor could a particular subtype be linked to the IBS status reported (Kruskal-Wallis, p value=0.78).
Descriptive statistics of Flemish Gut Flora Project individuals grouped by Blastocystis carrier status and subtype
A principal coordinate analysis of FGFP microbiota community variation based on bacteria and archaea genus-level Bray-Curtis dissimilarity distances revealed that Blastocystis carriers and non-carriers tended to cluster separately (adonis, r2=0.0362, p<1.0e-03; figure 2A). We found Blastocystis presence to be associated with higher observed richness and Shannon diversity (Wilcoxon test, p value<2e-16 for both indices). Beyond this association, among Blastocystis carriers, the highest diversity was observed in ST2 and ST4 samples compared with ST3 (Wilcoxon test, FDR<0.05; figure 2B). Focusing on community types, Blastocystis prevalence was found to be unevenly distributed over DMM-characterised enterotypes (figure 2C), with the Bacteroides 1 and 2 clusters comprising significantly less carriers compared with Ruminococcaceae and Prevotella (Pearson’s χ2, p value<2.2e-16). However, in spite of the entero-subtype associations observed, subtype relative distributions among carriers were not significantly different in most pairwise comparisons. The only comparison with significant difference was between ST3 and ST4, with ST4 being more prevalent in Ruminococcaceae enterotyped samples (Fisher’s exact test, p value=0.04925). In addition, ST2 was more prevalent than ST3 in Ruminococcaceae compared with Prevotella enterotyped samples (Fisher’s exact test, p value=0.05847).
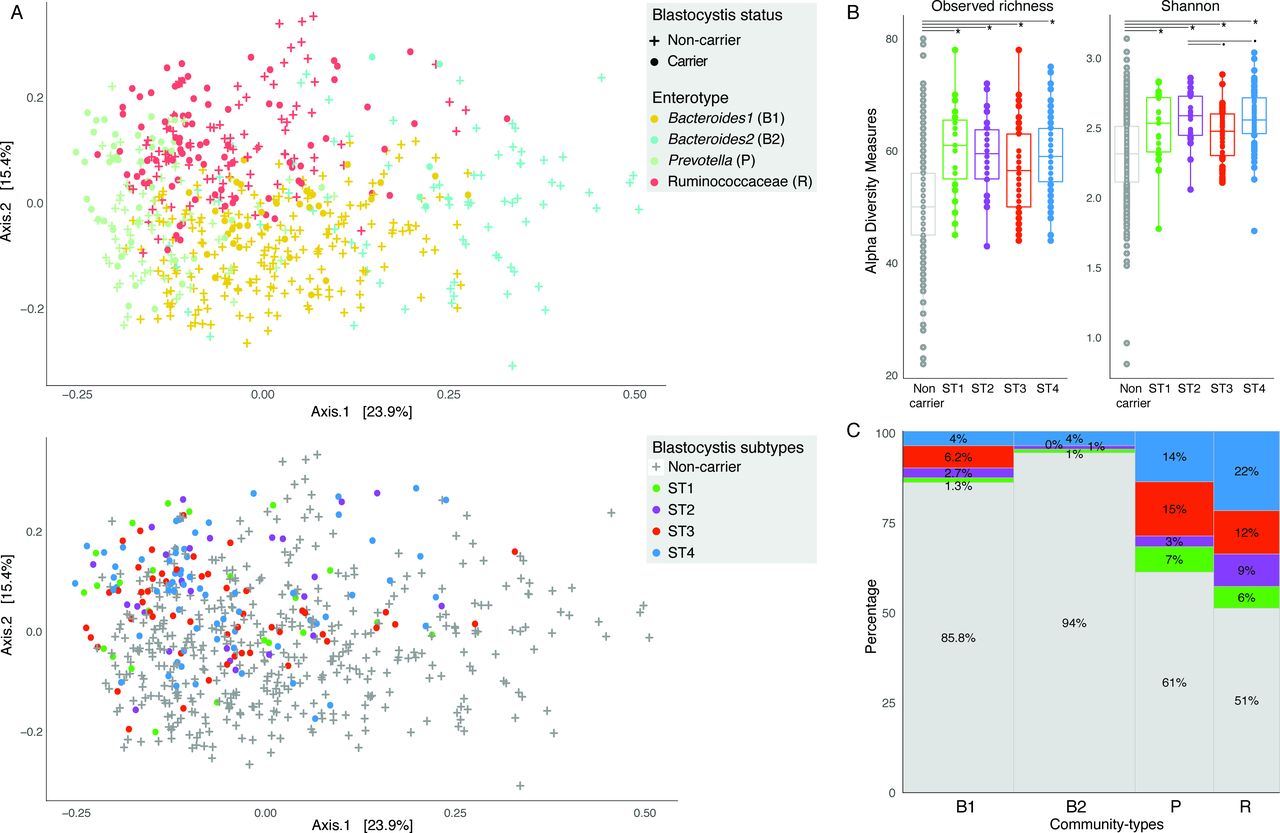
Microbial community variation associated to enterotypes and Blastocystis subtypes in the Flemish Gut Flora Project (FGFP) cohort. (A) Blastocystis carrier and non-carrier individuals (top panel) and subtype carriers (bottom panel) position in microbiota community space, represented by principal coordinates analysis (PCoA) of genus-level Bray-Curtis dissimilarity of the bacterial and archaeal fraction. The percentage of variation explained by the two first PCoA dimensions is reported on the axes. (B) Observed genus richness and Shannon diversity index (SDI) across Blastocystis non-carriers and carriers grouped by subtype. Presence of Blastocystis is associated to higher richness and SDI. Subtypes ST2 and ST4 have higher SDI compared with ST3. Wilcoxon test false discovery rate (FDR): *FDR<0.01; .FDR<0.05. (C) Distribution of Blastocystis non-carrier and subtypes carriers with respect to enterotypes, using Dirichlet Multinomial Mixtures community typing (Falony et al 2016). The Ruminococcaceae and Prevotella enterotypes have a higher proportion of Blastocystis carriers than Bacteroides1 and especially Bacteroides2. The body of the box plot represents the first and third quartiles of the distribution, and the median line. The whiskers extend from the quartiles to the last data point within 1.5×IQR, with outliers beyond.
Blastocystis subtypes correlate differentially with Akkermansia relative abundance
Third, we assessed potential interactions among Blastocystis subtypes and both bacterial and archaeal gut microbiota constituents. Out of 60 bacterial and archaeal genera present in a least 20% of FGFP samples, we identified 38 taxa with distinct relative abundance distributions between carrier and non-carrier samples (Wilcoxon test, FDR<0.05; online supplementary figure S5; online supplementary table S8). Four genera were found differentially associated with Blastocystis subtypes (Kruskal-Wallis test; pairwise Wilcoxon test; FDR<0.05; figure 3; online supplementary table S9), notably Akkermansia was more abundant in ST2 and ST4 samples than in ST3 carriers. Akkermansia is a faecal isolate of clinical interest that has not only been linked to the response of obese individuals to restriction diets,42 but also reported to prevent the onset of obesity and associated metabolic disorders in murine models.43 44

Blastocystis subtype carriers are strongly linked to microbial community composition. (A) Correlation between bacteria and archaea genera and Blastocystis subtypes. Heatmap of correlations between bacteria and archaea genera and Blastocystis subtypes is indicated by colours, positive (red) and negative (blue). The significant correlations (false discovery rate (FDR) <0.05) are indicated by ‘+’; only bacteria and archaea genera and Blastocystis subtypes with at least one significant correlation are shown. (B, C) The four genera with significant differences in relative abundances between the most prevalent Blastocystis subtypes carriers. (B) Akkermansia-relative abundances correlate negatively with abundances of Blastocystis ST3 and positively with ST4. (C) Differences in relative abundances of Anaerotruncus, Coprobacter and Enterococcus between the most prevalent Blastocystis subtypes carriers. Kruskal-Wallis with post hoc Wilcoxon test with multiple testing correction (FDR). *FDR<0.05 and Spearman correlation, FDR<0.05. The body of the boxplots represents the first and third quartiles of the distribution, and the median line. The whiskers extend from the quartiles to the last data point within 1.5×IQR, with outliers beyond.
Relative abundances of nine genera correlated with those of Blastocystis subtypes—four among them associated with ST4. We observed ST3 and ST4 Blastocystis to show opposite (negative and positive, respectively) correlations to Akkermansia abundances (figure 3A, B). In addition, this inverse association of ST3 and ST4 with Akkermansia was confirmed by reconstructing an ensemble co-occurrence network combining Spearman correlation and Kullback-Leibler similarity37 (online supplementary table S10). Of note, throughout all subtype-level analyses, ST4 was also shown to be positively associated with Methanobrevibacter relative abundances (figure 3A; online supplementary figure S6 D; online supplementary table S10). However, while increased relative abundances of both Akkermansia and Methanobrevibacter have been shown to be linked to hard stools,45 we did not detect a significant correlation between ST4 and stool consistency, potentially due to sample size and stool score distribution (online supplementary figure S7).
Blastocystis subtypes explains more microbiota variation than host covariates
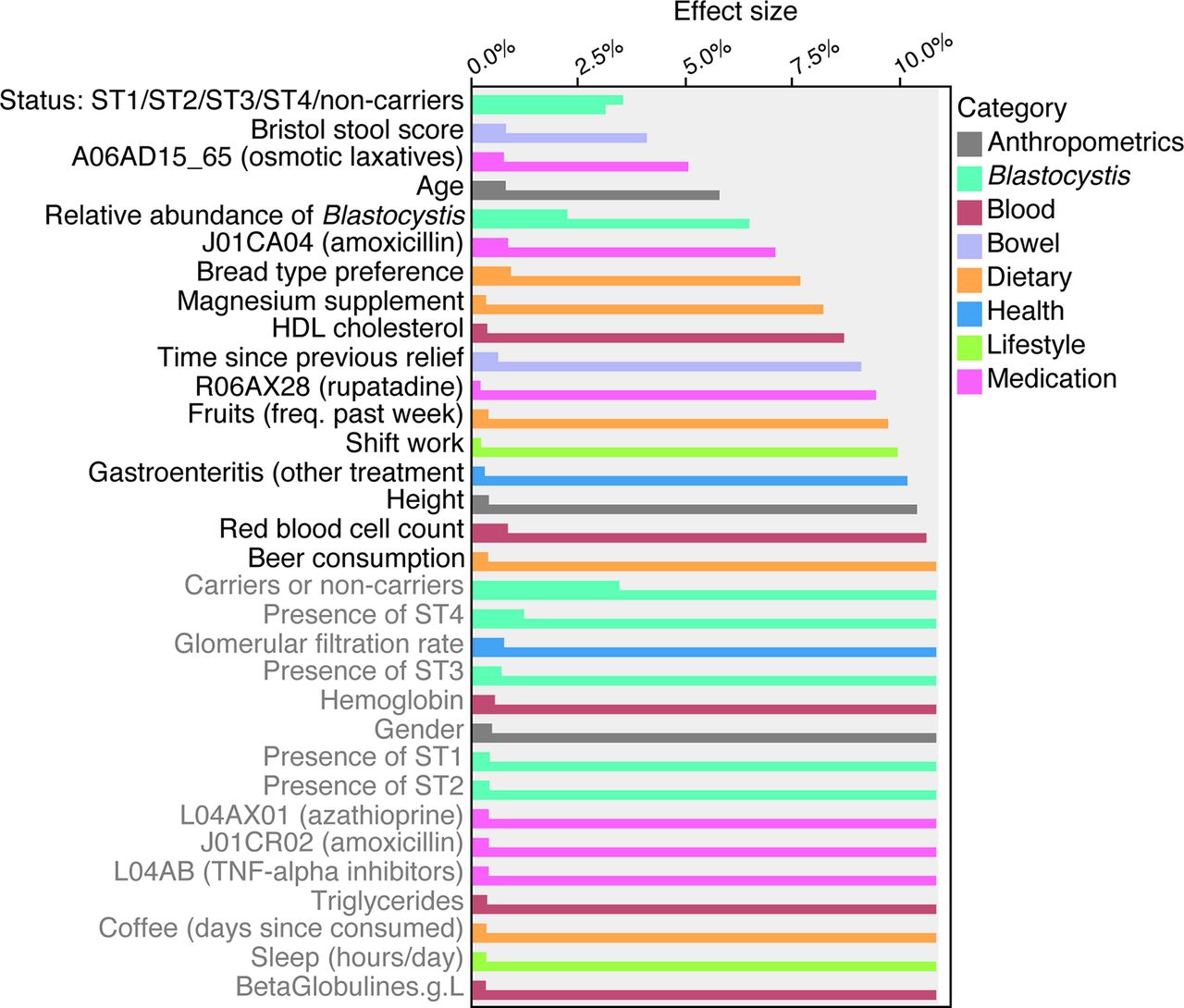
The presence of Blastocystis subtypes appeared to have a substantial impact on gut microbiota ecology. In order to quantify the strength of the interactions between Blastocystis presence and microbiota composition, we computed the effect size of Blastocystis as a microbiota composition covariate in the FGFP population cohort. To do so, we applied a strategy previously described.17 To the list of 69 microbiota covariates, we added 7 Blastocystis-associated variables: genus carrier status, genus relative abundance and subtype carrier status, and subtype abundances (per subtype) (online supplementary table S11). Next, we performed a dbRDA on bacteria and archaea genus-level Bray-Curtis dissimilarity matrix. In the subset of 616 FGFP samples analysed, subtype carrier status displayed a non-redundant effect size of 3.5%, outclassing stool consistency as top covariate of microbiota variation. Blastocystis-relative abundance, or proportion of Blastocystis sequences over bacteria, archaea and SCE sequences, was also found to contribute significantly to bacterial and archaeal microbiota variation with an additional contribution of 2.2% (figure 4; online supplementary table S11). These observations hint towards Blastocystis playing a putative key role in shaping the large intestinal ecosystem.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Explanatory power of Blastocystis carrier status and its total or subtypes relative abundances on microbiota community variation (bacterial and archaeal fraction), compared with the microbiota covariates reported in Falony et al.17 Variables are ordered according to the cumulative effect size of non-redundant covariates selected by stepwise redundancy analysis analysis (right bars) compared with individual effect sizes assuming independence (left bars) (false discovery rate<0.1). Variables labelled in grey did not contribute to additional explanatory power in the cumulative model. The full list is available in online supplementary table S11. Individuals carrying both Blastocystis and other single-cell eukaryotes were excluded from the analysis. HDL, high-density lipoprotein; TNF, tumour necrosis factor.
Discussion
To our knowledge, the present study is to date the largest screening effort to survey Blastocystis subtype prevalence and relative abundance in a Western population. We identified 30% of individuals in the FGFP population cohort as Blastocystis carriers. This observation questions the historical classification of this SCE as a pathogen6–8 since intestinal discomfort was not associated with presence of Blastocystis (online supplementary table S6). Moreover, the association between Blastocystis and gut health was additionally backed up by its lowered prevalence (4%) in an IBD cohort, in line with a previous report of a Danish/Spanish active ulcerative colitis cohort.15 Our findings suggest that the current ambiguity regarding the role of Blastocystis in human health could be related to subtype13 or even intra-subtype diversity within this prevalent genus.12
Out of the nine human gut-associated Blastocystis subtypes, we identified four as being commonly present in the FGFP cohort. Among them, ST4 was the most prevalent, in accordance with both our reanalyses of the TwinsUK and AGP cohorts, as well as with previous studies both in clinical32 46 47 and non-clinical settings.13 Our results also confirm previous reports of Blastocystis being associated with higher microbiota richness and diversity.15 16 Although questioned,45 48 microbiota richness is generally considered an indicator of ecosystem stability49 and human health.50 Regarding community types, we observed reduced prevalence of Blastocystis in both Bacteroides enterotypes, consistent with previous reports.51 The Bacteroides community type has been linked to obesity, low-grade inflammation52 and reduced microbiota resilience.49 The Bacteroides2 enterotype has been shown to present reduced microbial cell counts and to be more prevalent among patients with Crohn’s disease.53 While the high richness and enterotype distribution observed in Blastocystis carriers indicate that it is a common member of a stable, resilient and health-associated microbiota, a subtype-level analysis revealed important differences within the genus. The most striking disparity is in the inverse associations found between subtypes ST3, ST4 and Akkermansia-relative abundance. Akkermansia is generally considered a health-promoting GI isolate. In murine models, administration of Akkermansia muciniphila has been shown to reduce weight gain and suppress development of low-grade inflammation.54 Studies regarding the effect of Akkermansia administration on metabolic syndrome occurring in obese human hosts are ongoing (ClinicalTrials.gov NCT02637115). The negative correlation between ST3 and Akkermansia could potentially indicate an association of this Blastocystis subtype with a less healthy host phenotype. Of note, ST3 was previously identified as more prevalent among IBS patients40, a finding that we were, however, unable to replicate in the FGFP cohort.
Importantly, our results question current consensus protocols for faecal microbiota transfer (FMT) that classify Blastocystis in stool as ‘potentially transmittable disease-causing agents’ and advise that the presence of Blastocystis in a donor microbiota results in the immediate exclusion of the sample.8 55 Although more research is needed to unveil the relation between Blastocystis subtypes and both microbiota composition and host health, our study underlines that protocols should be refined at the subtype level. Given the co-occurrence of Akkermansia and ST4, strict application of the current consensus protocol would result in discarding those faecal samples that might prove to be most valuable when, for example, applying FMT as treatment of metabolic syndrome.44 Thus, when selecting optimal donors, at least in the Flemish population, samples should be screened for bacterial and archaea diversity, rather than direct exclusion of Blastocystis carriers, which, as demonstrated here, should not be considered a marker of unhealthy gut microbiota.
We identified and analysed Blastocystis subtype abundance based on the sequence of their 18S rRNA V4 region. ST5, ST6 and ST9 were not detected in our data set. As we managed to identify ST6 and ST9 with the exact same method in published data sets, we consider this is a true finding and not a limitation of our method. If other subtypes were present but unidentified in our study sample, they must have been at a much lower relative abundance than the subtypes already assigned, virtually undetectable at the sequencing depth used for the study. Similarly, subjects in our cohort were identified to be carriers of a unique type of Blastocystis, but samples from the AGP were identified as carriers of multiple Blastocystis subtypes. Its currently unclear whether this difference is biological or methodological in nature.
The explanatory power of Blastocystis subtype presence on microbiota variation exceeds the impact of stool consistency, previously identified as a top microbiota covariate.45 Hence, future studies on gut microbiota should consider the stratification of populations based on subtyping of Blastocystis, one of the most abundant SCE in the human gut. We cannot exclude that other currently overlooked SCE display a similar effect on community structure, further emphasising the importance of surveying the gut ecosystem across domains. Only the ecological consideration of all gut lineages and their interactions will allow us to facilitate the translation of human gut microbiota research into clinical practice. Surveying and controlling our forgotten symbionts are crucial to move the field towards generalisable conclusions with the potential to impact human life as a whole.
Conclusions
Performing the largest screening of Blastocystis subtype prevalence and relative abundance in a Western population to date, we confirmed the genus as a common constituent of a human gut microbiota, present in faecal samples of 30% of individuals in a healthy population cohort compared with 4% prevalence in patients with active IBD. Out of 69 established microbiota covariates, only age was associated with Blastocystis carrier status—with carriers of the most prevalent ST4 subtype being older than non-carriers. All four Blastocystis subtypes commonly present in FGFP samples were found to be less prevalent in Bacteroides enterotyped microbiota. However, the inverse correlations of ST3 and direct correlation of ST4 with Akkermansia suggest differential associations of subtypes with host health. Overall, given the significant association of subtype presence and abundance of microbiota variation, we recommend further characterisation of the link among Blastocystis and both microbiota community structure and host health at the subtype resolution, with special consideration of current FMT consensus protocols that might need to be adjusted accordingly.
Acknowledgments
The authors thank all members of the Raes lab for lively discussions and feedback. They acknowledge the contribution of Flemish GPs and pharmacists to data and sample collection. Finally, the authors thank all FGFP volunteers for participating in the project.
References
Footnotes
Contributors Conception and design of the study: RYT and JR; data collection: RYT, CC, MJ, SC, JW, SV-S, GF, YD, LR and CV; data preparation: RYT, MJ, CC, JW, SVS, FH and YD; statistical analysis, data analysis and interpretation: RYT, MJ, SC, SVS, GF, GL-M, JW, FH, PB, SV and JR; writing and critical revision of the manuscript: RYT, MJ, SC, GF, SV-S and JR. All authors approved the final version for publication.
Funding The FGFP was funded with support of the Flemish government (IWT130359), the Research Fund–Flanders (FWO) Odysseus program (G.0924.09), the King Baudouin Foundation (2012-J80000-004), FP7 METACARDIS HEALTH-F4-2012-305312, VIB, the Rega institute for Medical Research, and KU Leuven. SV-S is supported by Marie Curie Actions FP7 People COFUND–Proposal 267139 (acronym OMICS@VIB). MJ, SV-S, GL-M, SC and JW are supported by postdoctoral (five) fellowships, from the FWO.
Competing interests None declared.
Patient consent Not required.
Ethics approval GFP procedures were approved by the medical ethics committee of the University of Brussels–Brussels University Hospital (approval 143201215505, 5/12/2012). The local ethical committee approved the study (B322201213950/S53684).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The datasets generated and analysed during the current study are available upon reasonable request to the corresponding author. In line with the ethical protocol, data will be made available after a data transfer agreement has been signed.