Article Text

Abstract
Objective ARID1A is commonly mutated in pancreatic ductal adenocarcinoma (PDAC), but the functional effects of ARID1A mutations in the pancreas are unclear. Understanding the molecular mechanisms that drive PDAC formation may lead to novel therapies.
Design Concurrent conditional Arid1a deletion and Kras activation mutations were modelled in mice. Small-interfering RNA (siRNA) and CRISPR/Cas9 were used to abrogate ARID1A in human pancreatic ductal epithelial cells.
Results We found that pancreas-specific Arid1a loss in mice was sufficient to induce inflammation, pancreatic intraepithelial neoplasia (PanIN) and mucinous cysts. Concurrent Kras activation accelerated the development of cysts that resembled intraductal papillary mucinous neoplasm. Lineage-specific Arid1a deletion confirmed compartment-specific tumour-suppressive effects. Duct-specific Arid1a loss promoted dilated ducts with occasional cyst and PDAC formation. Heterozygous acinar-specific Arid1a loss resulted in accelerated PanIN and PDAC formation with worse survival. RNA-seq showed that Arid1a loss induced gene networks associated with Myc activity and protein translation. ARID1A knockdown in human pancreatic ductal epithelial cells induced increased MYC expression and protein synthesis that was abrogated with MYC knockdown. ChIP-seq against H3K27ac demonstrated an increase in activated enhancers/promoters.
Conclusions Arid1a suppresses pancreatic neoplasia in a compartment-specific manner. In duct cells, this process appears to be associated with MYC-facilitated protein synthesis.
- pancreatic cancer
- pancreatic tumours
- cancer genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Intraductal papillary mucinous neoplasms (IPMN) and pancreatic intraepithelial neoplasia (PanIN) are precursors to pancreatic ductal adenocarcinoma (PDAC).
Understanding the molecular mechanisms that drive precursor formation and progression can improve PDAC outcomes.
ARID1A is frequently mutated in PDAC.
Arid1a loss in mice leads to IPMN formation that may be mediated by Sox9 and the mTor (mammalian target of rapamycin) pathway.
What are the new findings?
Duct-specific Arid1a loss induces duct enlargement and tumourigenesis.
Acinar-specific Arid1a heterozygous deletion results in accelerated PanIN formation and worse outcomes.
ARID1A loss in cultured pancreatic duct cells leads to MYC overexpression and increased protein synthesis.
ARID1A deletion in cultured pancreatic duct cells leads to broadly increased enhancer/promoter histone marks.
How might it impact on clinical practice in the foreseeable future?
Insights into the mechanisms that drive premalignant PDAC lesions may lead to novel therapeutic strategies against this lethal disease.
Background
Pancreatic ductal adenocarcinomas (PDAC) can arise out of multiple types of precursor lesions such as pancreatic intraepithelial neoplasia (PanIN) or intraductal papillary mucinous neoplasms (IPMNs).1 Large-scale sequencing studies have defined the genomic profile of PDAC being dominated by mutations in KRAS, TP53, SMAD4 and CDKN2A followed by a ‘long tail’ of the remaining mutations that occur in 10% or less of samples.2 3 The most common of these mutations include ARID1A, which is also altered in 10% of IPMN.4 5
ARID1A is part of the SWI/SNF complex, which is a multisubunit complex that remodels chromatin by shifting, inserting or evicting nucleosomes in an ATP-dependent manner.6–8 Combinatorial assembly of subunits provides the SWI/SNF complex tissue and temporal specificity in gene regulatory functions that may be activating or repressive.8 ARID1A is thought to modulate the chromatin remodelling activity of the SWI/SNF complex through direct interactions with DNA and by recruiting and binding transcriptional cofactors.9 While it is presumed that ARID1A is a tumour suppressor gene because of widespread loss-of-function mutations found in human cancer samples,10 the effects of ARID1A loss in the pancreas and PDAC have generally not been well-characterised, with the exception of a recent study showing that pancreatic Arid1a loss led to IPMN formation which may be mediated through Sox9 expression and the mTor (mammalian target of rapamycin) pathway.11
Protein synthesis is increased by almost all oncogenic signalling pathways including RAS, WNT–β‐catenin and PI3K–mTOR.12 Aberrantly increased translation supports the transformation of normal cells and the acquisition of various hallmark features, such as angiogenesis, altered metabolism and proliferation. The MYC pathway also increases translation. Previous studies have shown that protein synthesis driven by MYC can be oncogenic, and mitigation of the aberrant translation suppresses MYC’s oncogenic activities.13
In this study, we characterised the effects of pancreas-specific Arid1a loss in mice. We found that pan-pancreatic Arid1a deletion synergised with an activating Kras mutation to induce IPMN. Using lineage-specific Cre expression systems, we found that Arid1a loss in pancreatic duct cells resulted in cystic dilation of ducts and PDAC, while heterozygous Arid1a deletion in the acinar compartment accelerated PanIN and PDAC formation. Arid1a loss in murine pancreata induced gene signatures associated with Myc activity and protein translation. Using human pancreatic ductal epithelial (HPDE) cells, we confirmed that ARID1A loss led to elevated protein expression mediated by MYC overexpression and a global increase in H3K27ac marks. Finally, we tested EZH2 inhibition as a way to blunt Arid1a loss-induced IPMN.
Results
Arid1a loss is sufficient to initiate inflammation, PanIN and mucinous cysts
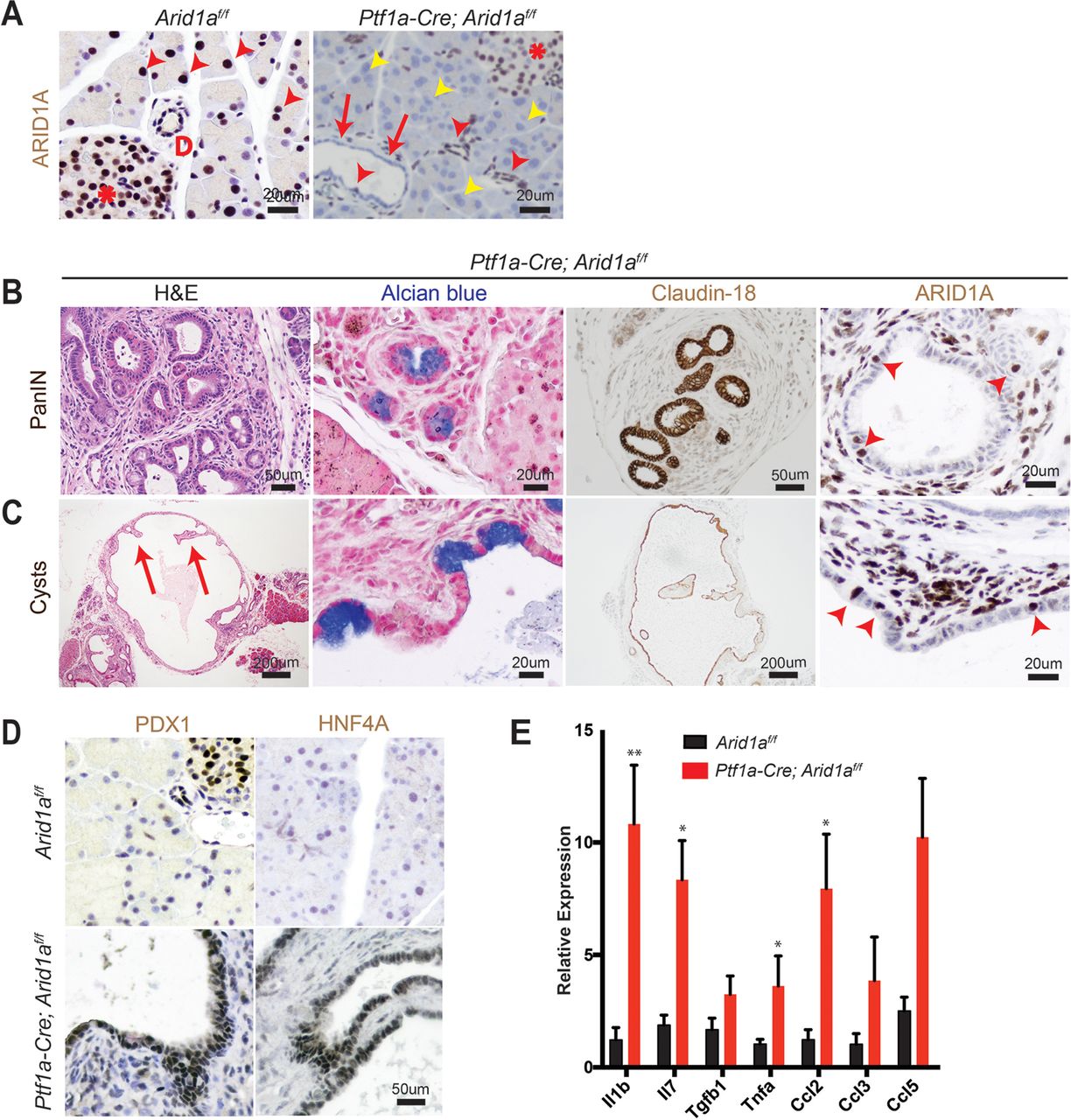
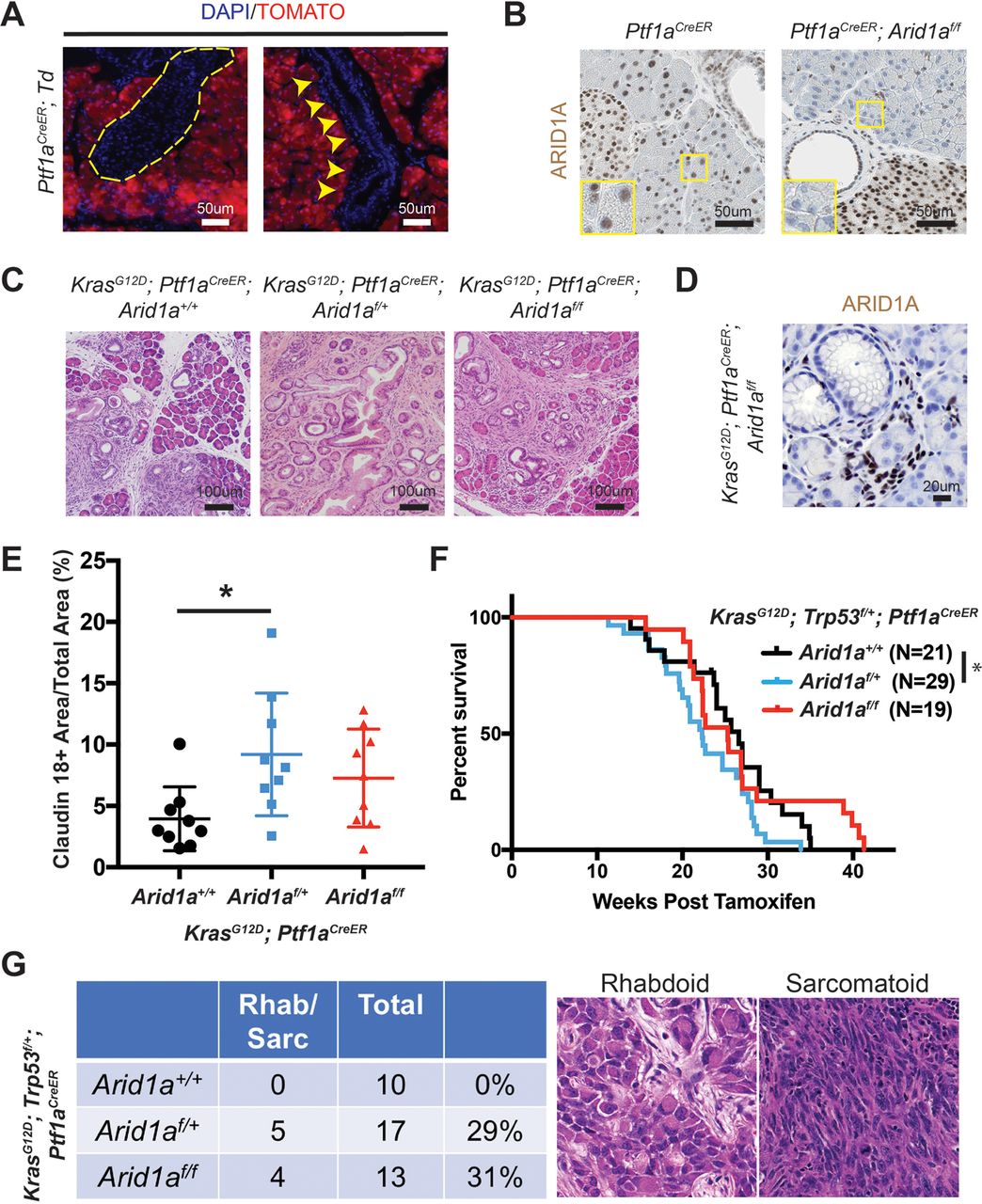
To determine the functional effects of Arid1a loss in pancreas, we generated Ptf1a-Cre; Arid1af/f (referred henceforth as CA) mice. Exon 8 of Arid1a is flanked by loxP sites and tissue specificity was achieved with Cre under regulatory control by Ptf1a, which is a transcription factor that is expressed in all pancreas cell types starting around E9.5 (online supplementary figure S1A).14 15 These mice were viable, fertile and appeared grossly normal. Arid1a was deleted without alternatively spliced gene products or truncated proteins when bulk pancreata was examined with western blotting (online supplementary figure S1B).16 However, on closer examination of individual cellular compartments, we found that Arid1a deletion was complete in acinar cells, partial in ductal cells and almost none in the endocrine cells (figure 1A). When we used the Rosa-LSL-tdTomato reporter to track Cre expression, we found that tomato was expressed in essentially all cells, consistent with previous reports (online supplementary figure S1C).15 17 This pattern of Arid1a deletion by Ptf1a-Cre is consistent with results shown by other groups.11

Conditional deletion of Arid1a in the pancreas induces inflammation, pancreatic intraepithelial neoplasia (PanIN) and mucinous cysts. (A) In wild-type mice, ARID1A was expressed in most acinar cells (red arrowheads), all duct cells (‘D’ adjacent) and all islet cells (asterisk). In Ptf1a-Cre; Arid1a f/f (CA) mice, ARID1A was lost in all acinar cells (yellow arrowheads), some duct cells (red arrows point to lost, red arrowheads point to retained) and none of the islet cells. Sixteen-week-old CA mice formed (B) PanIN and (C) large cysts with flat to cuboidal epithelium with papillary projections (arrows) that were Alcian blue and claudin-18 positive. Both PanIN and cysts showed focal ARID1A positivity (arrowheads point to ARID1A-positive cells). (D) The cysts expressed embryonic factors PDX1 and HNF4a. (E) The pancreata of CA mice showed significant upregulation of chemokines and cytokines, as measured by quantitative PCR, n≥5, *P<0.05, **P<0.01.
By 4 months of age, many CA mice developed acinar to ductal metaplasia (ADM, 12 of 14 mice, online supplementary figure S1D), which is a process where acinar cells respond to injury or stress by downregulating digestive enzymes, expressing duct-specific markers and taking on ductal phenotypes.18 ADM is reversible in wild-type mice on removal of the stress; however, ADM can progress to PanIN in the setting of oncogenic Kras.19–21 A significant proportion of CA mice also developed ductal lesions that were mucinous, as supported by Alcian blue staining, and appeared to be PanIN, based on morphology and claudin-18 positivity (11 of 14 mice, figure 1B). Four of 14 mice formed larger cysts with some lined by bland appearing epithelium, while others had papillary projections and mucinous components that were claudin-18 positive (figure 1C). ARID1A expression in PanIN and cysts were mosaic within the same lesion (figure 1B, C).
Further characterisation of the cysts showed reactivation of embryonic factors PDX1 and HNF4A, which are associated with reprogramming of heretofore terminally differentiated cells into a dedifferentiated state capable of progressing down a neoplastic pathway (figure 1D).21–23 Inflammation has also been previously shown to be a key component of pancreatic reprogramming.21 24 In CA mice, there were occasional robust inflammatory responses as demonstrated by infiltration of CD45-positive lymphoid cells and marked fibrosis (online supplementary figure S1E). This is consistent with quantitative PCR data on bulk pancreata, where multiple chemokines and cytokines, such as Il1b, Il7, Tnfa and Ccl2 were elevated (figure 1E). By 1 year of age, we did not observe any evidence of PDAC or significant progression of PanIN lesions. In addition, the cysts remained benign appearing, although reaching very large size in some instances, and there was also widespread adipocyte infiltration (online supplementary figure S1F). Thus, Arid1a loss in the pancreas was sufficient to initiate PanIN and simple mucinous cysts, but did not support further progression to higher grade dysplasia or frank carcinoma within 1 year.
Arid1a loss synergises with oncogenic Kras to accelerate IPMN formation
Activating KRAS mutations are found in more than 90% of human PanIN and PDAC samples and are thought to be the initiating driver event for PDAC.2 3 25 As a result, a large portion of ARID1A mutations in human PDAC are accompanied by KRAS mutations. IPMN have also been found to harbour KRAS mutations.1 To determine the effects of concomitant ARID1A deletion and KRAS activation, we generated KrasG12D; Ptf1a-Cre; Arid1af/f mice (henceforth called ‘KCA’ mice, online supplementary figure S2A). By 8 weeks of age, KrasG12D; Ptf1a-Cre (KC) mice had significant inflammation and developed numerous PanIN that were Alcian blue positive, as expected (figure 2A,B).25 While KCA mice also developed inflammation and PanIN (online supplementary figure S2B), oncogenic Kras significantly accelerated the cyst phenotype seen in CA mice. Whereas only about 30% of CA mice developed macroscopic cysts that were occasionally mucinous or complex, all KCA mice had large mucinous cysts with papillary projections (figure 2A,B). As was the case in CA mice, the KCA cysts were mosaic for ARID1A expression (figure 2B).

Kras activation synergises with Arid1a loss in the pancreas to induce intraductal papillary mucinous neoplasms (IPMNs). (A) KrasG12D; Ptf1a-Cre (KC) mice had oedema, while KrasG12D; Ptf1a-Cre; Arid1af/f (KCA) mice formed large pancreatic cysts. (B) KC mice showed significant inflammation and numerous pancreatic intraepithelial neoplasia. KCA mice formed large mucinous cysts with papillary projections that were mosaic for ARID1A (arrowhead points to ARID1A-positive cells). (C) KCA cyst fluid was amylase rich (normal serum amylase <10 U/L), suggesting a direct connection to the pancreatic ductal system, consistent with IPMN. n=4, error bars represent SEM. (D) KCA cysts with mucin expression patterns consistent with human gastric subtype IPMN. (E) Human gastric subtype (n=28) and pancreaticobiliary subtype (PB, n=10) IPMN showed areas with negative or low ARID1A staining, as compared with intestinal subtype (n=6). **P<0.01. (F) Kaplan-Meier curve estimating survival of KrasG12D ; Trp53f/+ ; Ptf1a-Cre mice that were Arid1a+/+, Arid1af/+ or Arid1af/f .
There are two types of mucinous pancreatic cysts, both of which can progress to PDAC. Mucinous cystic neoplasms (MCN) are cyst-forming epithelial neoplasms composed of mucinous epithelium associated with ovarian-type stroma (expressing oestrogen and progesterone receptors), which do not communicate with the pancreatic ductal system, while IPMN are intraductal, grossly visible epithelial neoplasm of mucin-producing cells, arising in the main pancreatic duct or its branches, and usually exhibiting a papillary growth pattern.26 Cysts in KCA mice demonstrated papillary projections and did not stain for oestrogen and progesterone receptors (online supplementary figure S2C). Analysis of the cyst fluid showed high amylase levels, suggesting a direct connection to the ductal system (figure 2C). Thus, histologic and biochemical analyses demonstrated that concurrent Kras activation and Arid1a deletion resulted in pancreatic mucinous cysts which resemble human IPMN, rather than MCN.
IPMN may be further categorised into four epithelial subtypes based on histomorphology and mucin expression patterns.27 The intestinal and oncocytic subtypes progress to colloid and oncocytic adenocarcinomas, respectively, and have a more favourable outcome than PDAC not arising in these precursor lesions. Gastric and pancreaticobiliary subtypes tend to form tubular adenocarcinomas, which have significantly worse outcomes than colloid and oncocytic carcinomas, but still better than PDAC not arising in IPMN.28 IPMN from KCA mice appeared to be predominantly of the gastric subtype, as most areas had short papillae and epithelium morphologically resembling gastric foveolar epithelium and did not express MUC1 or MUC2 (figure 2B,D). However, there were some occasional areas that resembled the pancreaticobiliary subtype, which is characterised by more interconnected and arborising papillae composed of cuboidal cells with little mucin (online supplementary figure S2D). We did not observe any lesions resembling the intestinal subtype and further confirmed by negative CDX2 immunohistochemistry (online supplementary figure S2E).
To determine if KCA cysts represent a faithful recapitulation of human disease, we asked if ARID1A expression was lost in human IPMN. We performed a retrospective review of our institutional surgical database and identified 35 patients who underwent pancreatectomies from 2006 to 2016 and had IPMN identified on final pathology. In these patients, there were 28 gastric, 6 intestinal, 1 oncocytic and 10 pancreaticobiliary components (some patients had mixed IPMN containing more than one subtype). We performed immunohistochemistry for ARID1A and graded the expression as negative, low or high (online supplementary figure S2F). We found that 61% of gastric and 10% of pancreaticobiliary subtype IPMN had ARID1A null or low expression, whereas no intestinal type had ARID1A loss (figure 2E). Even when IPMN contained cells with no or low ARID1A staining, there were pockets of strong staining consistent with the mosaic expression pattern seen in KCA mice cysts (online supplementary figure S2G). These data show that ARID1A loss is present in human IPMN and highlights the clinical relevance of the KCA model.
While ARID1A has been considered a tumour suppressor, we recently showed that it has both context-dependent oncogenic and tumour-suppressive effects in liver cancer.29 To determine the effect of Arid1a loss on PDAC formation, we generated KrasG12D; Trp53f/+; Ptf1a-Cre (KPC) mice that were Arid1a wild-type, heterozygous or null. Surprisingly, we found no difference in survival between the three cohorts (figure 2F). While we noted cancer in all three KPC groups, KPC Arid1a-null mice often had massive cysts replacing almost all of the pancreas (online supplementary figure S2H). Thus, it was unclear if the cause of death for KPC Arid1a-null mice was cancer or mass effect from the large cysts.
Arid1a loss in pancreatic duct cells promotes cystic duct dilation and PDAC
Since it was difficult to discern cancer-specific deaths in the KPC system, we hypothesised that we could isolate the IPMN phenotype using the inducible duct-compartment specific Sox9-CreER line (online supplementary figure S3A).30 Mice were given tamoxifen (TAM) after weaning at 3.5 to 4.5 weeks of age. We found that 3 to 4 months after induction KrasG12D ; Sox9-CreER; Arid1a+/+ mice remained histologically bland, while KrasG12D ; Sox9-CreER; Arid1af/f mice had mildly dilated pancreatic ducts (figure 3A,B). We observed a progressive increase in the calibre of pancreatic ducts at 26 weeks and then 52 weeks after TAM (figure 3B). At 52 weeks, we found that one of five KrasG12D ; Sox9-CreER; Arid1af/f mice had a mucinous cystic lesion that exhibited focal mild epithelial papillary tufting, resembling the earliest signs of IPMN formation (figure 3A,C). KrasG12D ; Sox9-CreER; Arid1af/f mice also occasionally had significant localised inflammation and PanIN (online supplementary figure S3B), but there was no evidence of frank carcinoma or dysplasia by 1 year after TAM. Three of 15 KrasG12D ; Sox9-CreER; Arid1a+/+ mice had only very small foci of inflammation or ADM. There was no difference in proliferation in ductal cells (online supplementary figure S3C). To understand the phenotypic variation seen in the Sox9-CreER cohort, we performed lineage tracing using a Rosa-LSL-tdTomato (Td) reporter. We examined Sox9-CreER; Td mice 12 weeks after TAM and identified mosaic ARID1A expression that corresponded to tomato signal (figure 3D). In addition, we found that Sox9-CreER mice had 36% to 98% of the CK-19 positive ductal cells showing tomato positivity (online supplementary figure S3D).
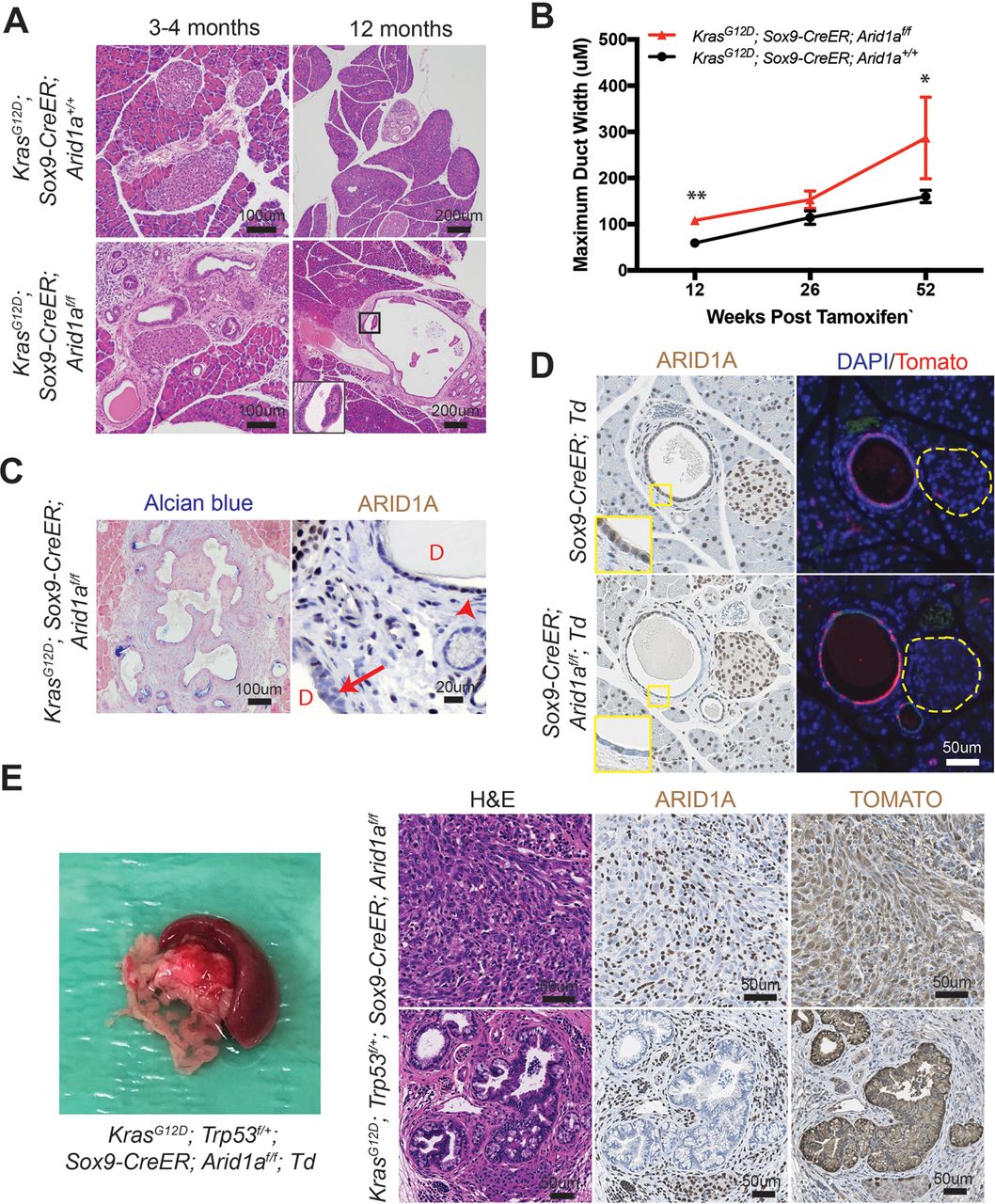
Phenotype of Arid1a deletion in pancreatic duct cells. (A) Three to 4 months after tamoxifen (TAM) induction, both KrasG12D; Sox9-CreER; Arid1a+/ + and KrasG12D; Sox9-CreER; Arid1af/f mice formed small ductal lesions. Twelve months after TAM, KrasG12D; Sox9-CreER; Arid1af/f mice occasionally formed large cystic lesions, while KrasG12D; Sox9-CreER; Arid1a+/+ mice had only smaller lesions that most resembled pancreatic intraepithelial neoplasia. (B) KrasG12D; Sox9-CreER; Arid1af/f mice showed progressively enlarging ducts over time. At 12 weeks, n=12 and 9, at 26 weeks, n=6 and 10 and at 52 weeks, n=15 and 5 for KrasG12D; Sox9-CreER; Arid1a+/+ and Arid1af/f mice, respectively. *P<0.05, **P<0.01. (C) Lesions in KrasG12D; Sox9-CreER; Arid1af/f mice were Alcian blue positive and mosaic for ARID1A (arrow to ARID1A-negative cells and arrowhead to ARID1A-positive cells, ‘D’ denotes duct/cyst lumens). (D) Serial section showed that ARID1A loss in Sox9-CreER; Arid1af/f; ROSA26-LSL-tdTomato (Td) corresponded to where tomato was expressed. Yellow dashed lines denote islets. (E) KrasG12D; Trp53f/+; Sox9-CreER; Arid1af/f mice formed occasional pancreatic ductal adenocarcinoma that had sarcomatoid features (top panels) and incipient intraductal papillary mucinous neoplasm (bottom panels). Both lesions were ARID1A negative and tomato positive. ARID1A positivity were seen in stromal cells only.
To determine if tumour suppressor loss could accelerate cancer formation, we generated KrasG12D; Trp53f/+; Sox9-CreER; Td mice that were Arid1a wild-type, heterozygous or null. A similar proportion of mice from all three genotypes developed soft tissue masses or ocular lesions that necessitated euthanasia, but no pancreas-related deaths were seen 1 year after TAM (online supplementary figure 3E). We found no pancreatic phenotypes in 14 KrasG12D; Trp53f/+; Sox9-CreER; Arid1a+/+ mice and only small areas of inflammatory infiltrates in 2 of 16 KrasG12D; Trp53f/+; Sox9-CreER; Arid1af/+ mice. Of the 12 KrasG12D; Trp53f/+; Sox9-CreER; Arid1af/f mice, 2 had ADM or early PanIN and another 2 had cystic lesions (online supplementary figure 3F). Two other mice had firm, large pancreatic tail masses that were red on gross examination, suggesting tomato expression (figure 3E). On histologic examination, the masses were PDAC containing undifferentiated components with sarcomatoid features. In addition, they were ARID1A negative and tomato positive, confirming ductal origin (figure 3E). One of the two mice with PDAC had areas of duct dilation with some epithelial tufting suggestive of incipient IPMN (figure 3E), while the other had only PanIN-associated PDAC. In sum, these data show that Arid1a restrains tumourigenesis, either in the form of pre-IPMN lesions or PDAC, in ductal cells.
Heterozygous Arid1a loss accelerates PDAC formation
To determine the effects of Arid1a loss in the acinar compartment, we used the Ptf1aCreER line to induce Kras and Arid1a mutations (online supplementary figure S4A).31 Ptf1aCreER mice were given TAM between 3.5 and 4.5 weeks of age and we observed complete recombination in the acinar compartment without leakage into the duct cells or endocrine cells, consistent with previous reports (figure 4A,B and online supplementary figure S4B).31 32 We generated KrasG12D ; Ptf1aCreER mice that were wild-type, heterozygous or null for Arid1a and observed inflammation, fibrosis and mucin-positive lesions consistent with PanIN in all three lines (figure 4C). In the Arid1a null mice, the PanINs lacked Arid1a expression, consistent with acinar origin (figure 4D). When we quantitated the amount of PanIN from each line by measuring the amount of claudin-18 positive lesions 16 weeks after TAM, we found Arid1a heterozygous mice had more PanIN than Arid1a wild-type mice (figure 4E). Similarly, when we made KrasG12D; Trp53f/+; Ptf1aCreER mice with all three Arid1a states, we found that Arid1a heterozygous mice had the worst survival (figure 4F). When we measured proliferation with Ki-67, we found no difference between the three groups (online supplementary figure S4C). While all three genotypes had moderately to poorly differentiated ductal adenocarcinomas, about 30% of mice in both Arid1a heterozygous and null groups had tumours with undifferentiated rhabdoid or sarcomatoid features (figure 4G). Arid1a null mice had an intermediate phenotype with respect to both PanIN quantity and overall survival.

Heterozygous Arid1a loss in acinar cells accelerates pancreatic intraepithelial neoplasia (PanIN) formation and shortens survival. (A) After tamoxifen, tomato expression in Ptf1aCreER; ROSA26-LSL-tdTomato (Td) mice was restricted to acinar cells. None was seen in islets (yellow dashed outline) or ducts (yellow arrowheads). (B) After tamoxifen, ARID1A expression was lost in acinar cells but retained in duct and islet cells in Ptf1aCreER; Arid1af/f mice. (C) After tamoxifen, KrasG12D; Ptf1aCreER; Arid1a+/+ , Arid1af/+ and Arid1af/f mice all developed PanIN. (D) PanIN in KrasG12D; Ptf1aCreER; Arid1af/f mice did not express ARID1A. (E) KrasG12D; Ptf1aCreER; Arid1af/+ formed more PanIN, n=9 for each group, *P<0.05. (F) KrasG12D; Trp53f/+; Ptf1aCreER; Arid1af/+ mice had worse survival than KrasG12D; Trp53f/+; Ptf1aCreER; Arid1a+/+ mice. *P<0.05. (G) KrasG12D; Trp53f/+; Ptf1aCreER mice with heterozygous and homozygous Arid1a loss developed components of undifferentiated pancreatic ductal adenocarcinoma with rhabdoid or sarcomatoid features.
Arid1a loss increases protein synthesis through the induction of MYC
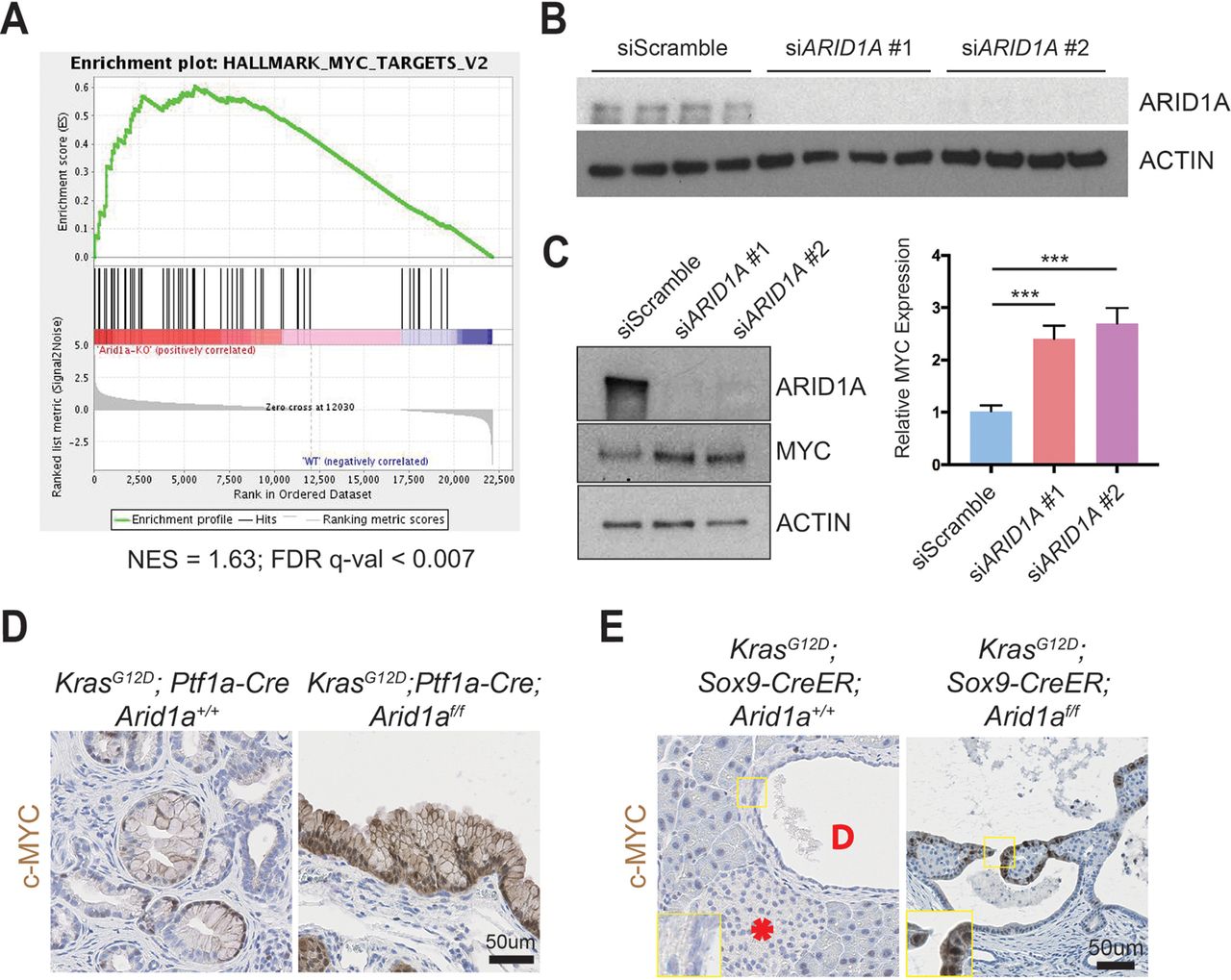
To understand the downstream transcriptional effects of Arid1a deletion in the pancreas, we performed RNA sequencing of pancreata collected from CA mice and their wild-type littermates. We used gene set enrichment analysis and found that the Myc target gene network was the highest ranked among the ‘Hallmark’ gene sets, suggesting that Arid1a loss in CA mice pancreas induced increased MYC activity (figure 5A and online supplementary table 1).33 We chose to focus on the ductal compartment to identify possible therapeutic targets for IPMN, as surgery is currently the only treatment option. We performed small-interfering RNA (siRNA) ARID1A knockdown in immortalised HPDE cells (figure 5B)34 and found an increase in MYC protein expression (figure 5C). When we performed immunohistochemistry for c-MYC, we found only weak and patchy signals in PanIN of KC mice but intense signals in the cysts of KCA mice (figure 5D). Similarly, the cystic lesions in KrasG12D ; Sox9-CreER; Arid1af/f mice expressed c-MYC, while the duct cells in KrasG12D ; Sox9-CreER; Arid1a+/+ mice did not have any expression (figure 5E). In both cohorts, there was very little to no signal in the acinar and islet compartments (online supplementary figures S5A,B).
Supplementary file 3

ARID1A loss induces gene signatures associated with MYC activity. (A) Gene set enrichment analysis of RNA-seq of Ptf1a-Cre; Arid1af/f (CA) pancreata using the ‘Hallmark’ gene sets demonstrated that the MYC target gene signature was the most upregulated signature in CA mice. NES, normalised enrichment score; FDR, false discovery rate. (B) Western blot confirmed that siARID1A 1 and 2 were effective in knocking down ARID1A in human pancreatic ductal epithelial (HPDE) cells. (C) Western blot and quantification demonstrating that ARID1A knockdown induced MYC expression in HPDE cells. n=8, ***P<0.001. (D) PanIN in KrasG12D; Ptf1a-Cre; Arid1a+/+ mice had patchy c-MYC expression, while cysts in KrasG12D; Ptf1a-Cre; Arid1af/f mice stained intensely for c-MYC. (E) In KrasG12D; Sox9-CreER; Arid1a+/+ mice, acinar, duct (‘D’ in duct lumen) and islet (asterisk) cells had very little c-MYC staining, while cysts from KrasG12D; Sox9-CreER; Arid1af/f mice expressed some c-MYC.
To determine what pathways might be regulated by MYC, we analysed the RNA-seq data using the ‘Molecular Signatures Database’ and the top four highest ranked signatures were protein synthesis related (figure 6A and online supplementary table 1).33 Because MYC directly regulates ribosomal protein transcription as one way to mediate increased translation, we measured ribosomal protein expression in CA pancreata using real-time quantitative PCR and found significant elevation in Arid1a null tissues (online supplementary figure S6A). To more directly determine if this was a result of acute Arid1a loss, we measured ribosomal protein expression in Ptf1a-CreER; Arid1af/f pancreata 1 week after TAM and also found increased expression (online supplementary figure S6B).
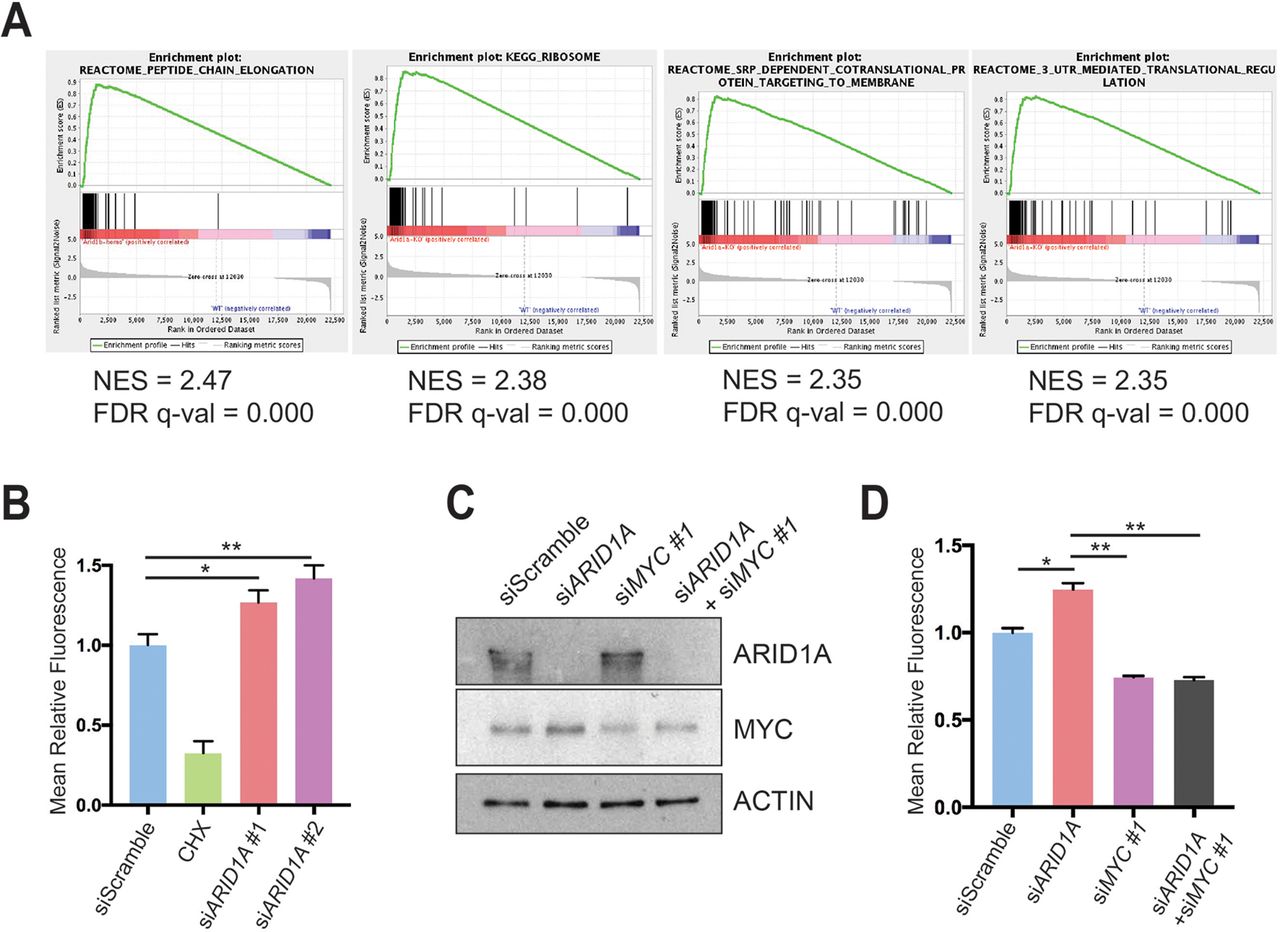
ARID1A loss induces MYC-mediated increase in protein synthesis. (A) Gene set enrichment analysis of RNA-seq of Ptf1a-Cre; Arid1af/f (CA) mice pancreata using the ‘Molecular Signatures Database’ demonstrated that the top four upregulated signatures in CA mice were associated with protein translation. NES, normalised enrichment score; FDR, false discovery rate. (B) Human pancreatic ductal epithelial (HPDE) cells were treated with O-propargyl-puromycin (OPP), which were incorporated into nascent peptides, labelled with fluorescence and quantified with fluorescence activated cell sorting (FACS). Representative FACS plot (left) and quantification (right) are shown. HPDE cells were treated siScramble (n=7), siARID1A 1 (n=6) and siARID1A 2 (n=6). Cycloheximide (CHX) is a translation inhibitor and served as a negative control for the assay in cells treated with siScramble (n=2). *P<0.05, **P<0.01. (C) Western blot demonstrating partial knockdown of MYC with siMYC in HPDE cells. (D) HPDE cells were treated with OPP and treated with siScramble (n=8), siARID1A (n=3), siMYC 1 (n=7) and siARID1A and siMYC 1 (n=3). Quantification of relative fluorescence is shown. *P<0.05, **P<0.01.
We next investigated whether increased expression of ribosomal proteins resulted in more or less protein synthesis and quantitatively assessed protein synthesis in the context of Arid1a loss. We used a fluorescent-labelling assay based on the incorporation of O-propargyl-puromycin (OPP), which is taken up by ribosomes and incorporated into new peptides. We found that ARID1A knockdown in HPDE cells significantly increased OPP incorporation, signifying increased translation (figure 6B and online supplementary figure S6C). Because MYC and mTOR drive essential signalling networks that integrate growth and proliferative signals to activate protein translation, we examined mTOR activity.35 After ARID1A knockdown, there was no difference in mTOR activity, thus favouring MYC as the causative mechanism of protein synthesis activation (online supplementary figure S6D). To test if MYC was responsible for the increase in translation due to ARID1A loss, we used siRNA to partially knock down MYC in the setting of siARID1A (figure 6C). When we measured protein synthesis, we found that returning MYC to baseline levels in the setting of ARID1A knockdown led to a reduction of OPP levels comparable to the levels seen in siMYC-treated cells (figure 6D and online supplementary figure S6E). This suggests that MYC, in part, mediates the control of protein translation by ARID1A.
ARID1A loss leads to increased activated enhancer/promoter marks
We next sought to understand the effects of ARID1A loss on chromatin activation. Using HPDE cells, we generated an ARID1A-null line using CRISPR/Cas9 (online supplementary figure S7). ChIP-seq was then performed against H3K27ac, which marks active enhancers and promoters. We found that ARID1A null cells had more peaks than cells with intact ARID1A (figure 7A). When we examined genes associated with MYC and protein translation signatures, which were the most upregulated gene networks identified from RNA-seq of CA pancreata (figure 5A and figure 6A), both also had significantly more peaks in ARID1A null cells (figure 7B,C). This shows that ARID1A loss led to increased activation of these target genes.
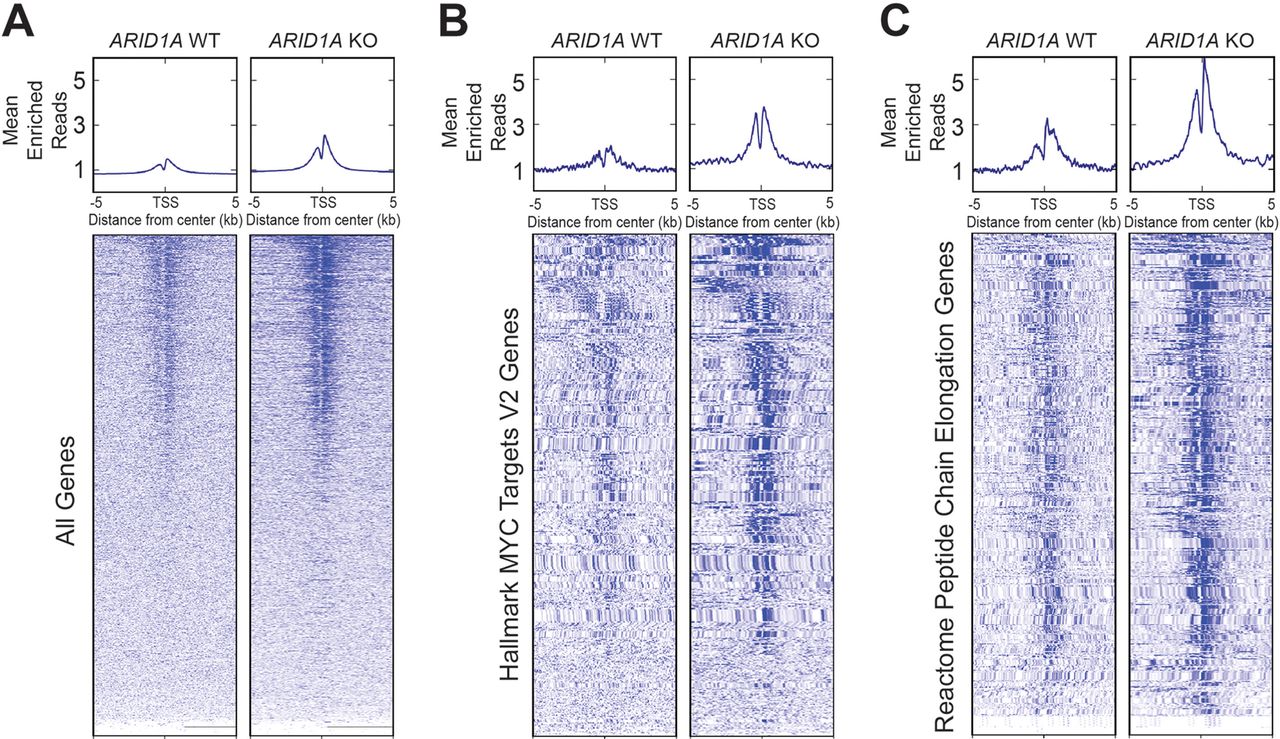
ARID1A loss leads to increased H3K27ac marks. Metagene plot and heatmap of H3K27ac sites in human pancreatic ductal epithelial cells that were wild type (WT) or null (KO) for ARID1A for the given gene sets. TSS, transcription start site. (A) All genes. (B) ‘Hallmark MYC Targets V2’ signature from Gene Set Enrichment Analysis (GSEA). (C) ‘Reactome Peptide Chain Elongation’ signature from GSEA.
EZH2 inhibition does not abrogate Arid1a mutant IPMN
Since IPMN growth is associated with PDAC risk, therapies that slow IPMN progression would be highly desirable. Previous studies have suggested that SWI/SNF deficient tumours, including those with ARID1A mutations, are sensitive to inhibition of EZH2, which is the catalytic subunit of the Polycomb repressive complex 2.36 Thus, we asked whether Arid1a mutant IPMN were susceptible to EZH2 inhibition. We first confirmed that trimethylated histone H3 lysine 27 (H3K27me3), a marker of EZH2 activity, was highly expressed in KCA cysts (figure 8A). Next, we treated Capan-1 pancreatic cancer cells with the potent and specific EZH2 inhibitor EPZ011989 and confirmed that it was highly effective in reducing H3K27me3 levels (figure 8B).37 We then performed abdominal MRI on 20 KCA mice at 3 to 4 weeks of age, when significant cyst burden was already present. Mice were randomised to vehicle or EPZ011989 and stratified based on gender and cyst volume. After 4 weeks of therapy, repeat MRIs were performed (figure 8C). We found that mice treated with EPZ011989 gained less weight than vehicle-treated mice (figure 8D) and that the compound effectively reduced H3K27me3 levels in the pancreata of the treatment group (figure 8E). However, we found no difference in cyst volumes between the two groups (figure 8F). We also did not find any differences in proliferation and apoptosis in the cysts (online supplementary figure S8). Thus, Arid1a mutant IPMN epithelial cells did not respond to EZH2 inhibition in vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EZH2 inhibition does not block intraductal papillary mucinous neoplasm growth in KrasG12D; Ptf1a-Cre; Arid1af/f (KCA) mice. (A) Immunohistochemistry showing KCA murine cysts expressed high levels of H3K27me3. (B) Western blot showing that Capan-1 cells treated with EPZ011989 had decreased levels of H3K27me3. (C) Treatment plan, n=10 for each arm (vehicle and EPZ011989). Day 1 was defined by when the first dose was given. (D) Mice treated with EPZ011989 had a lower rate of weight gain than mice treated with vehicle. (E) Pancreata from the treatment group had decreased H3K27me3 levels on western blot. (F) Representative coronal section of MRI from vehicle and EPZ011989 treated KCA mice before and after treatment (arrowheads pointing to cysts, which were fluid filled, thus showing up as white signal on T2 weighted MRI). There was no difference in the change in volume after EPZ011989 treatment. AU, arbitrary unit. BID, two times daily.
Discussion
SWI/SNF components are frequently mutated in a variety of cancers, including PDAC.38 Although ARID1A is the most commonly mutated SWI/SNF subunit in PDAC, the functional effects of ARID1A alterations are unclear.2 3 To clarify the effects of ARID1A mutations in pancreatic neoplasms, we generated pancreas-specific Arid1a deficient mice using the Ptf1a-Cre system that resulted in complete Arid1a loss in acini, mosaic Arid1a deletion in duct cells and complete Arid1a retention in the islets. This suggests that in the ductal and endocrine compartments Arid1a was either required during development or its locus is inaccessible to Cre recombinase. We found that Arid1a loss alone was sufficient to generate PanIN and mucinous cysts that re-expressed progenitor markers such as PDX1 and HNF4A in a manner similar to that seen in pancreatitis and Kras activation.21 22 32 39 Our work shows that ARID1A activity was necessary to maintain terminal differentiation of the pancreatic epithelium, and its loss resulted in a more dedifferentiated state that allowed neoplastic transformation, consistent with results recently reported by Kimura et al. and Livshits et al.11 40 The concept that ARID1A is permissive for the process of tissue differentiation is also consistent with our previous work in the liver, where Arid1a loss resulted in downregulation of liver gene expression programmes involving coagulation, bile acid metabolism and xenobiotic metabolism.16 In this way, ARID1A joins a group of other transcription factors and epigenetic modifiers, such PTF1A, SOX9 and BRG1, as important players in maintaining pancreatic identity.32 39 41 42
We found that concurrent Kras activation and Arid1a loss led to IPMN formation, consistent with the findings of Kimura et al.11 These lesions were predominantly of gastric subtype, but there were areas of pancreaticobiliary epithelium. Because we also observed ARID1A loss in human gastric and pancreaticobiliary IPMN resection samples, the cysts that formed from murine Arid1a deletion appeared to recapitulate human disease. Both our findings and those of Kimura et al. showed that only a small subset of mice with duct-specific Kras activation and Arid1a deletion developed dilated cyst with dysplasia. We extended this work by also incorporating the loss of one Trp53 allele and found that a subset of mice developed PDAC. It is likely that additional tumour suppressor loss or activation of oncogenes such as GNAS and RNF43, the most commonly mutated genes in IPMN, play a more catalytic role in driving duct cells into IPMN.43 In addition, other non-ductal cell autonomous effects may be required. For example, in the embryonic Ptf1a-Cre model, there was a significant inflammatory response induced by the activated Kras mutation in the acinar cells. Whether inflammation can accelerate IPMN formation is a topic that requires further study.
We also showed that Arid1a has tumour-suppressive effects in acinar cells. We observed that heterozygous Arid1a loss in conjunction with activated Kras in acini resulted in increased PanIN formation and reduced survival. Previous work by Hosada et al. that identified ARID1A mutations in high-grade PanIN hinted at ARID1A’s tumour-suppressive effects in the PanIN–PDAC axis.44 We also found that Arid1a loss led to tumours that contained components of undifferentiated PDAC with rhabdoid and sarcomatoid features. These results are in line with several recent studies linking loss of Smarcb1, another SWI/SNF component, to a more mesenchymal or dedifferentiated PDAC phenotype.45 46 Interestingly, the Arid1a null mice had a phenotype more similar to Arid1a wild-type mice in terms of PanIN formation and long-term survival. This likely speaks to the complicated context-dependent role of Arid1a in cancer biology consistent with what we had previously explored in the liver. There, we found that Arid1a was required in normal tissues to initiate cancer (ie, Arid1a was pro-tumorigenic) but in established cancers, Arid1a loss led to more aggressive cancer phenotypes (ie, Arid1a was tumour suppressive).29
While the PanIN to PDAC pathway is well established, the true cell of origin for human PDAC remains unclear. Using the transgenic mouse model, multiple groups have demonstrated that the acinar compartment can give rise to PanIN and PDAC in the setting of activating Kras mutations.19–21 47 Gidekel Friedlander et al. showed that, in the setting of chronic injury, endocrine cells were also capable of giving rise to PanIN.48 Our data showing that PanIN-associated PDAC arose from KrasG12D; Trp53f/+; Sox9-CreER; Arid1af/f mice supports recent work by Lee et al., who demonstrated that duct cells were capable of giving rise to PanIN and PDAC, using the Sox9-CreER line, and Bailey et al., who generated PDAC using the Hnf1b-CreERT line.49 50 Similarly, while Sox9-CreER-induced Arid1a loss led to cyst dilation, likely originating from the ductal compartment, this does not exclude the possibility that these lesions formed via a progenitor/ADM-like state, since a small percentage of acinar cells were labelled by Sox9-CreER.32
While ARID1A loss of function mutations have been identified in ~10% of PDAC samples in multiple large sequencing studies,2 3 the mutational profiles of IPMN are currently being delineated with next-generation sequencing.51–53 While some studies found ARID1A mutations in only a small number of resected IPMN,54 Tan et al. showed that ARID1A was mutated in approximately 10% of all IPMN samples and was one of the more commonly mutated genes.4 In addition, a recent study by Suenaga et al. found that about 15% of patients with IPMN containing high-risk features had ARID1A mutations detectable in pancreatic juice.5 Other mechanisms for ARID1A downregulation are likely be involved as we found a significant proportion of resected IPMN with areas expressing little or no ARID1A. Similarly, Kimura et al. found that in their resected IPMN cohort, 22% of the samples lacked ARID1A on the protein level.11
Mechanistically, we found that genes involved in protein synthesis were overexpressed in Arid1a-deficient pancreata and that ARID1A knockdown in immortalised human pancreatic duct cells resulted in increased protein translation. In addition, we observed that ARID1A knockdown induced MYC expression, and concurrent knockdown of ARID1A and MYC in HPDE cells abrogated the increased translation, indicating that ARID1A loss operates, at least in part, through MYC activation. Intriguingly, both we and Kimura et al. observed that there was no increased proliferation in the Arid1a-null duct cells to explain the increase in duct size.11 They had speculated that dilated cysts must thus occur due to mucin overproduction. Our finding that Arid1a loss led to increased protein translation supports that theory of IPMN formation. Based on the observation that mucinous cysts developed in mice with only Arid1a loss, perhaps Arid1a loss can be an initiating event that results in increased protein/mucin synthesis that mechanically increases duct size based on mass effect. A future acquisition of a driver mutation in genes such as KRAS, GNAS or RNF43 then leads to transformation. This hypothesis would be consistent with the fact that many IPMN do not transform and progress to cancer.
The functional connection we identified between Arid1a loss and Myc expression is consistent with previous reports. First, Nagl et al. showed that the Myc promoter was a direct target of the SWI/SNF complex.55 Second, Genovese et al. reported that the loss of Smarcb1, another component of the SWI/SNF complex, in PDAC cell lines activated Myc programmes that led to elevated protein synthesis and increased the aggressiveness of cancer cells.45 Significantly, the increased protein synthesis rendered Smarcb1 deficient cancer cell lines particularly sensitive to drugs that targeted the proteostatic stress response machinery, suggesting that this strategy may also be effective in ARID1A null IPMN and warrants further study. Furthermore, we found that the increase in protein production induced by ARID1A loss was not accompanied by activated mTOR signalling, which is an important activator of translation. This suggests that ARID1A loss may activate a translation programme specific to MYC activity. In addition, we also found that ARID1A loss led to increased H3K27ac marks at the loci of MYC targets and involved in protein translation, signifying increased transcriptional activity at these sites.
While increased protein synthesis has been considered a general hallmark of rapidly proliferating malignant cells, recent reports have demonstrated that elevated translation is necessary in only certain oncogenic contexts. Barna et al. showed that while elevated protein synthesis is required for Myc-driven lymphoma formation, moderating translation did not affect lymphomagenesis in the Trp53-/- background. These data suggest that protein synthesis is a specific requirement for Myc oncogenesis and not a function of carcinogenesis as a whole.13 However, since ARID1A and SWI/SNF potentially influence loci across the entire genome, it is likely that ARID1A mediates pancreatic neoplasm formation and progression through a variety of other pathways in addition to ones involving protein synthesis. Further studies will be required to confirm that aberrant translation is a specific necessity for cyst formation resulting from Arid1a deletion and to elucidate additional pathways that are preferentially activated by SWI/SNF loss to drive cyst formation.
The survival rate for patients with PDAC remains dismal, and more than 90% of patients will die of the disease.1 One strategy to improve outcomes is to intervene in patients with premalignant lesions, such as IPMN. Conversion from premalignancy to carcinoma significantly worsens survival as demonstrated by the fact that 50% of patients who undergo resection of localised disease still die within 2 years from recurrent disease.56 However, there are no medical treatments for IPMN, and the only intervention available is prophylactic pancreatectomy, which carries morbidity and risk of death. Previous studies have described an antagonistic relationship between the SWI/SNF and PRC2 complexes, prompting preclinical and early phase clinical tests.57 However, we found no difference in cyst size, or apoptosis or proliferation in cyst epithelium after treating a cohort of KCA mice with a potent EZH2 inhibitor. One possible explanation for this negative finding comes from the work of Kim et al., who found that in KRAS gain-of-function mutants EZH2 structural inhibition, rather than functional inhibition of the catalytic site, was essential to induce apoptosis.58
In conclusion, we have identified ARID1A as a pancreatic tumour suppressor that acts by repressing KRAS-induced PanIN and IPMN formation. ARID1A loss leads to increased MYC expression, elevated protein synthesis and a globally activated chromatin state. Continued insights into the mechanisms that drive premalignant PDAC lesions may lead to novel therapeutic strategies against this lethal disease.
Supplementary file 1
Supplementary file 2
Acknowledgments
We thank Dr. Rolf Brekken for sharing various reagents and mouse lines and critical reading of the manuscript, John Shelton and the UTSW Histology/Pathology Core for histology assistance, and the Children’s Research Institute Sequencing Core for sequencing.
References
Footnotes
SCW and IN contributed equally.
Contributors SCW, IN and HZ conceived of and designed the experiments. SCW, IN, SX, SZ, LHN, J-CC performed experiments. LP and JS performed histological evaluations. XL and JL performed bioinformatics analyses. SCW and IN performed statistical calculations. SD provided key reagent and helped design the experiment. All authors contributed to data interpretation. SCW, IN and HZ wrote the manuscript.
Funding SCW was supported by the UT Southwestern Disease Oriented Clinical Scholarship, American College of Surgeon Faculty Research Fellowship, American Cancer Society Institutional Research Grant, and National Institutes of Health (NIH) 1K08CA222611 grant. IN was supported by the Alpha Omega Alpha Honor Medical Society, ChiRhoClin and the National Center for Advancing Translational Sciences of the NIH UL1TR001105. XS was supported by the Hamon Center for Regenerative Science and Medicine at UT Southwestern Medical Center. JL was supported by the Cancer Prevention Research Institute (CPRIT) (RP150596). HZ was supported by a Burroughs Wellcome Career Medical Award, CPRIT New Investigator Grant (R1209), CPRIT Early Translation Grant (DP150077), NIH/NIDDK R01DK111588, and a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG 10-16). Stand Up To Cancer is a program of the Entertainment Industry Foundation and its research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Competing interests SD is an employee and stockholder of Epizyme.
Patient consent Not required.
Ethics approval Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.