Article Text

Abstract
Objective Aldehyde dehydrogenase 2 (ALDH2), a key enzyme to detoxify acetaldehyde in the liver, exists in both active and inactive forms in humans. Individuals with inactive ALDH2 accumulate acetaldehyde after alcohol consumption. However, how acetaldehyde affects T-cell hepatitis remains unknown.
Design Wild-type (WT) and Aldh2 knockout (Aldh2 -/-) mice were subjected to chronic ethanol feeding and concanavalin A (ConA)-induced T-cell hepatitis. Effects of acetaldehyde on T-cell glucose metabolism were investigated in vitro. Human subjects were recruited for binge drinking and plasma cortisol and corticosterone measurement.
Results Ethanol feeding exacerbated ConA-induced hepatitis in WT mice but surprisingly attenuated it in Aldh2 -/- mice despite higher acetaldehyde levels in Aldh2 -/- mice. Elevation of serum cytokines and their downstream signals in the liver post-ConA injection was attenuated in ethanol-fed Aldh2 -/- mice compared to WT mice. In vitro exposure to acetaldehyde inhibited ConA-induced production of several cytokines without affecting their mRNAs in mouse splenocytes. Acetaldehyde also attenuated interferon-γ production in phytohaemagglutinin-stimulated human peripheral lymphocytes. Mechanistically, acetaldehyde interfered with glucose metabolism in T cells by inhibiting aerobic glycolysis-related signal pathways. Finally, compared to WT mice, ethanol-fed Aldh2 -/- mice had higher levels of serum corticosterone, a well-known factor that inhibits aerobic glycolysis. Blockade of corticosterone partially restored ConA-mediated hepatitis in ethanol-fed Aldh2 -/- mice. Acute alcohol drinking elevated plasma cortisol and corticosterone levels in human subjects with higher levels in those with inactive ALDH2 than those with active ALDH2.
Conclusions ALDH2 deficiency is associated with elevated acetaldehyde and glucocorticoids post-alcohol consumption, thereby inhibiting T-cell activation and hepatitis.
- glucose metabolism
- glycolysis
- T-cell hepatitis
- Concanavalin A
- binge drinking
- AKT
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Aldehyde dehydrogenase 2 (ALDH2) is a key enzyme to detoxify acetaldehyde in the liver and exists in both active and inactive forms in humans.
Approximately 35%–45% Asian people have inactive ALDH2 and develop ‘flushing syndrome’ after alcohol drinking due to acetaldehyde accumulation.
Acetaldehyde is considered one of the major culprits to promote liver disease progression by inducing adduct formation and inflammatory response.
Chronic ethanol feeding markedly exacerbates concanavalin A (ConA)-induced T cell hepatitis in mice.
What are the new findings?
Chronic ethanol feeding markedly attenuates ConA-induced hepatitis and cytokines in Aldh2-/- mice despite high levels of acetaldehyde in these mice.
In vitro treatment with acetaldehyde inhibits production of cytokine proteins without affecting their mRNAs in activated mouse and human T cells. Acetaldehyde treatment inhibits glucose metabolism in T cells by inhibiting aerobic glycolysis-related signal pathways.
Aldh2-/- mice had higher levels of serum corticosterone than wild-type mice post ethanol feeding. Blockade of corticosterone with the glucocorticoid receptor antagonist RU486 partially restores ConA-mediated hepatitis in ethanol-fed Aldh2-/- mice.
Acute alcohol consumption elevates plasma cortisol and corticosterone levels in human subjects with much higher levels in those with inactive ALDH2 than those with active ALDH2.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Physicians should pay attention to the potential immunosuppression in alcoholics with inactive ALDH2 gene due to elevated levels of acetaldehyde and cortisol, which may contribute to the pathogenesis of alcohol-associated diseases in these subjects.
Introduction
Viral hepatitis and alcohol abuse are the two major causes of chronic liver disease worldwide, and both factors frequently coexist and synergistically promote liver disease progression;1-4 however, the underlying mechanisms remain obscure. In viral hepatitis, activation of T cells plays an important role in eliminating viruses and promotes hepatocellular damage.5, 6 Injection of concanavalin A (ConA), a T-cell mitogen, has been widely used to study how T-cell activation induces liver injury in mice.7-9 In this model, administration of ConA rapidly activates T cells and natural killer T (NKT) cells and subsequently activates Kupffer cells/macrophages, followed by producing a variety of proinflammatory cytokines (eg, tumor necrosis factor (TNF)-α, interferon (IFN)-γ) that cause hepatocyte injury via the activation of their downstream signals (eg, nuclear factor (NF)-κB, signal transducer and activator of transcription 1 (STAT1)).10, 11 Activated immune cells also produce several hepatoprotective cytokines (eg, interleukin (IL)-6, IL-22) that promote liver repair via the activation of STAT3.10-12 We have previously demonstrated that chronic ethanol feeding exacerbated ConA-induced T-cell-mediated hepatitis via the upregulation of the TNF-α-NF-κB signalling pathway and subsequent upregulation of chemokines/adhesive molecules and recruitment of leucocytes into the liver.13 In contrast, chronic ethanol feeding attenuates the activation of the hepatocyte survival signal STAT3 in T-cell hepatitis.13 Thus, alcohol consumption dysregulates the activation of cytokines and their downstream signals, thereby exacerbating T-cell hepatitis, which likely contributes to the synergistic effect of alcohol and viral hepatitis on liver disease progression. However, how alcohol promotes T-cell-mediated hepatitis is not clear. For example, does ethanol or its metabolites enhance T-cell activation and accelerate liver injury?
Ethanol is mainly metabolised in hepatocytes by cytosolic alcohol dehydrogenase to generate acetaldehyde, which is subsequently metabolised by mitochondrial acetaldehyde dehydrogenase 2 (ALDH2) to produce acetate.14 Many variants of human ALDH2 gene exist, but the most relevant ALDH2 variants are the ALDH2*1 and ALDH2*2 alleles. The ALDH2*1 allele encodes active acetaldehyde metabolising enzyme with G at nucleotide position 42 421 (its corresponding glutamate at amino acid position 487), whereas the ALDH2*2 allele encodes inactive enzyme with A at position 42 421 (its corresponding lysine at position 487). Individuals with homozygous ALDH2*1/1 have high levels of acetaldehyde metabolising activity in the liver, whereas those with homozygous ALDH2*2/2 have very low or undetectable enzyme activities. Individuals with heterozygous ALDH2*2/1 have approximately 80%–90% (not 50%) reduction in enzyme activities, which is because ALDH2 is the isotetramer enzyme and all four subunits of ALDH2 are required to be normal for keeping its full activity.15 The individuals with the inactive form of ALDH2, which is found in 35%–45% Asian population, develop ‘flushing syndrome’ after alcohol drinking, including facial flushing, palpitations and drowsiness.16 These unpleasant symptoms usually prevent these individuals from consuming large amounts of alcohol; however, many people with inactive ALDH2 still drink heavily and generate high levels of acetaldehyde, even after the consumption of only a moderate amount of alcohol.15-17 Acetaldehyde is considered one of the major culprits to promote liver disease progression by inducing adduct formation and inflammatory responses.18-20 Overexpression or activation of ALDH2 ameliorated alcohol-induced liver injury in mice.21-23 In contrast, deletion of the Aldh2 gene accelerated liver inflammation and fibrosis after carbon tetrachloride and/or alcohol treatment.24 Thus, we hypothesised that ethanol-fed Aldh2 -/- mice, which have high levels of acetaldehyde, are more susceptible to ConA-induced T-cell hepatitis. To our surprise, ethanol-fed Aldh2 -/- mice were less sensitive to ConA-induced hepatitis than wild-type (WT) mice. Further studies demonstrated that ALDH2 deficiency and alcohol consumption were associated with high levels of acetaldehyde, cortisol and corticosterone, leading to attenuated T-cell activation both in mice and humans.
Materials and methods
Human subjects
Six healthy subjects with active ALDH2*1/*1 (G/G) and six healthy subjects with heterozygous inactive ALDH2*1*/*2 (G/A) were recruited from the First Affiliated Hospital of Jilin University (Changchun, China) for isolation of peripheral blood mononuclear cells (PBMCs). Another 42 healthy subjects with active ALDH2*1/*1 (G/G) and 18 healthy subjects with inactive ALDH2*1*/*2 (G/A) were recruited from the same hospital for measurement of plasma cortisol and corticosterone post one-time acute alcohol drinking (0.4 g/kg) (see the detailed information in online supplementary tables 1–2). All participants provided written informed consent.
Supplementary file 1
Mice
Aldh2 knockout (KO) (-/-) and WT C57BL/6N mice were fed a liquid diet containing 4% ethanol for 4 weeks as described previously.24 All animal studies were approved by the NIAAA Animal Care and Use Committee.
Model of T-cell-induced hepatitis
T-cell-mediated hepatitis was induced by a single intravenous injection of 10 µg/g body weight ConA as described previously.10
Statistical analysis
Data are expressed as the means±SEM and were analysed using GraphPad Prism software (La Jolla, California, USA). To compare values obtained from three or more groups, a one-way analysis of variance (ANOVA) was used, followed by Tukey posthoc test. To compare values obtained from two groups, the Student’s t-test was performed. P<0.05 was considered significant.
Other methods are described in online supplementary document.
Results
ALDH2 deficiency attenuates ConA-induced hepatitis in response to chronic ethanol feeding in mice
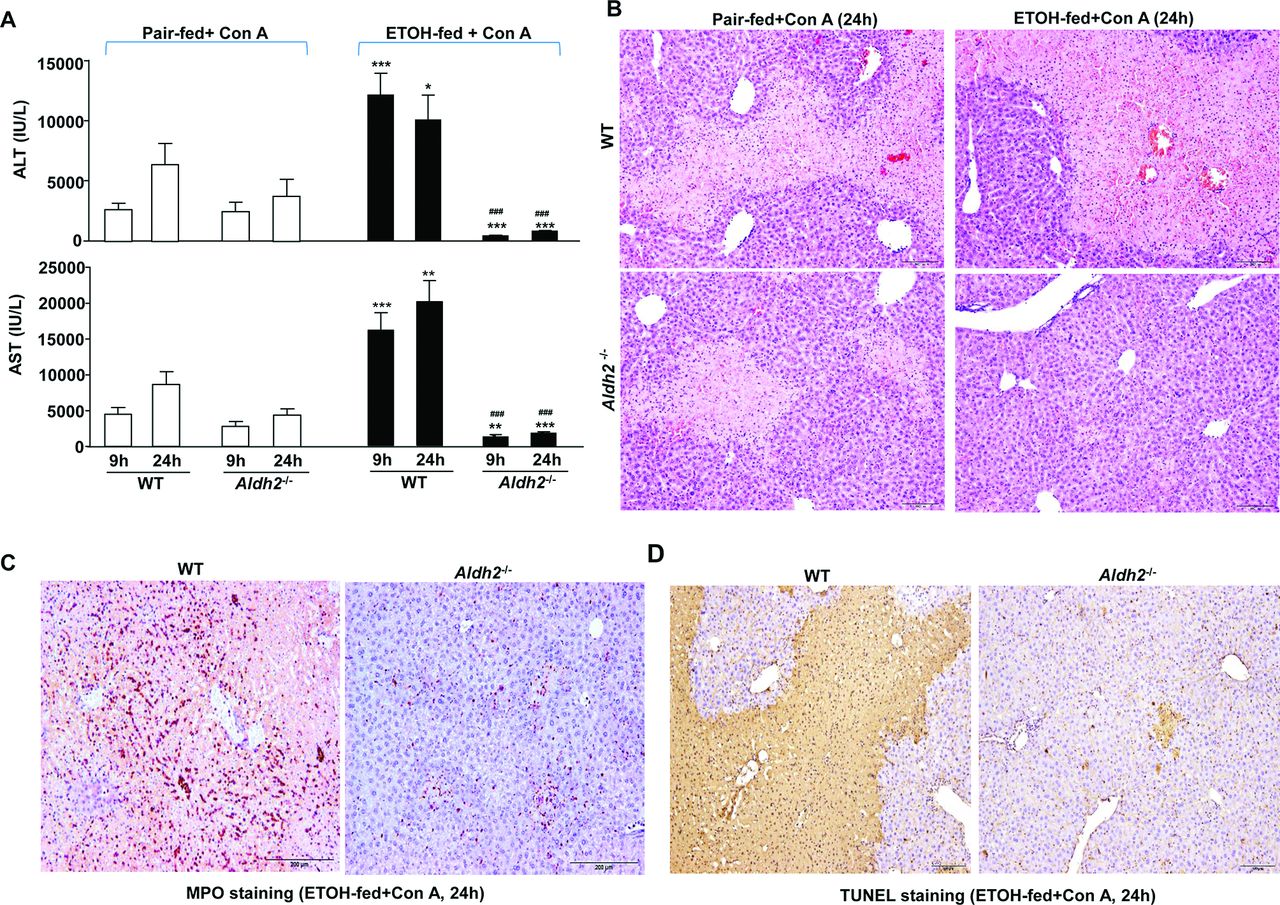
We previously demonstrated that chronic ethanol feeding aggravated ConA-induced hepatitis in mice.13 Here we also confirmed that chronic ethanol feeding exacerbated ConA-induced liver injury in WT mice, as demonstrated by greater levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), liver necrosis, hepatic neutrophil infiltration and hepatocyte death (as shown by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining) in ethanol-fed WT+ConA vs pair-fed WT+ConA (figure1A–D). Surprisingly, ethanol feeding markedly reduced ConA-induced liver injury in Aldh2-/- mice, as demonstrated by lower levels of serum ALT and AST, liver necrosis, hepatic neutrophils and TUNEL+ hepatocytes in ethanol-fed Aldh2-/- mice vs pair-fed Aldh2-/- mice post ConA injection (figure 1A–D). Compared with ethanol-fed WT mice, ethanol-fed Aldh2-/- mice had much lower (approximately 100 times) levels of ALT and AST post-ConA injection (figure 1A), whereas without alcohol feeding, pair-fed WT and Aldh2-/- mice had comparable levels of serum ALT and AST (figure 1A). Taken together, without alcohol feeding, WT and Aldh2-/- mice have the same susceptibility to ConA-induced liver injury, whereas after alcohol feeding, Aldh2-/- mice have much less ConA-induced hepatitis than WT mice.

Chronic ethanol exacerbates ConA-induced liver injury in WT mice but attenuates it in Aldh2 -/- mice. WT and Aldh2 -/- mice were fed an ethanol diet or control diet for 4 weeks, followed by ConA injection. The mice were euthanised 9 hours and 24 hours after ConA injection. (A) Serum ALT and AST levels. (B,C,D) Representative H&E staining, MPO staining and TUNEL staining of liver tissues from the mice treated with ConA for 24 hours. The values represent means±SEM (n=8 in pair-fed WT or KO group, n=15 in ethanol-fed WT of KO group). *P<0.05; **P<0.01; ***P<0.001 in comparison with the corresponding pair-fed mice. ###P<0.001 in comparison with the corresponding ETOH-fed WT mice. ALT, alanine aminotransferase; AST, aspartate aminotransferase; ConA, concanavalin A; ETOH, ethanol; MPO, myeloperoxidase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; WT, wild type.
ConA-induced elevation of serum cytokines and their downstream signals in the liver is attenuated in ethanol-fed Aldh2-/- mice vs WT mice
Elevation of serum cytokines and activation of their downstream signals in the liver play a critical role in the control of ConA-induced hepatitis.10, 11 To understand the mechanisms by which ethanol feeding modulates ConA-induced hepatitis in WT and Aldh2-/- mice, we measured serum levels of cytokines and their downstream signals in the liver. As illustrated in figure 2A, injection of ConA rapidly elevated serum levels of several cytokines in ethanol-fed WT mice. Such elevation was markedly attenuated in ethanol-fed Aldh2-/- mice. In agreement with serum cytokine levels, activation of their downstream signals including STAT1 and STAT3 in the liver was lower in ethanol-fed Aldh2-/- mice than in ethanol-fed WT mice (figure 2B). For example, hepatic expression of pSTAT1 (activated STAT1) and pSTAT3 (activated STAT3) was still highly activated in ethanol-fed WT mice but not detected in ethanol-fed Aldh2-/- mice 24 hours post ConA injection (figure 2B). In addition, hepatic expression of BCL-2 was higher in ethanol-fed Aldh2-/- mice than in ethanol-fed WT mice, whereas hepatic expression of BCL-xL was comparable between these two groups (figure 2B).

ConA-induced elevation of serum cytokines and their downstream signals in the liver are attenuated in ethanol-fed Aldh2 -/- mice compared with those in ethanol-fed WT mice. WT and Aldh2 -/- mice were fed an ethanol diet for 4 weeks, followed by ConA injection. (A) Serum samples were collected at different time points post-ConA injection and were subjected to measurement of cytokines. (B) Liver tissues were collected for western blot analyses. The values represent means±SEM (n=5–7 at each time point). *P<0.05; **P<0.01; ***P<0.001 in comparison with the corresponding WT groups. ConA, concanavalin A; ETOH, ethanol; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; WT, wild type.
Acetaldehyde inhibits production of cytokine proteins but not their mRNAs from ConA-stimulated mouse splenocytes in vitro
T and NKT cells are known to play an important role in ConA-induced hepatitis;25, 26 we wondered whether chronic ethanol feeding differentially affects these cells in WT and Aldh2 -/- mice. Our data revealed that the number of total T and NKT cells were not reduced and some of them were even higher in ethanol-fed Aldh2-/- mice compared with ethanol-fed WT mice. In vitro incubation with acetaldehyde did not alter T-cell proliferation in ConA-stimulated WT mouse splenocytes (see details in online supplementary materials and online supplementary figures 1–4). Collectively, our data suggest that the reduced susceptibility of ethanol-fed Aldh2-/- mice to ConA-induced T-cell hepatitis is not due to the reduced T and NKT-cell number.
To further understand the mechanisms by which ConA-induced T-cell hepatitis is attenuated in ethanol-fed Aldh2 -/- mice, we measured acetaldehyde levels from ethanol-fed mice and examined the effects of acetaldehyde on ConA-induced cytokine production from splenocytes in vitro. As illustrated in figure 3A, serum acetaldehyde levels were higher in ethanol-fed Aldh2 -/- mice than in ethanol-fed WT mice, whereas serum ethanol levels were comparable between these two groups. Because serum acetaldehyde levels were approximately 150 µM in chronic ethanol-fed Aldh2-/- mice (figure 3A) and were even much higher in Aldh2-/- mice after ethanol gavage, reaching to more than 200 µM27, 28 and acetaldehyde is very volatile at room temperature (the boiling point is 20.2°C), therefore, we used 200 µM acetaldehyde in mouse cell culture experiments. Figure 3B shows that treatment with acetaldehyde decreased protein levels of IL-2, IL-4 and TNF-α in the supernatant from splenocytes at early time points (24 hours) of ConA stimulation, whereas acetaldehyde reduced IFN-γ protein levels at all time points. Surprisingly, acetaldehyde treatment did not alter mRNA expression levels of these cytokines (figure 3C). These data suggest that acetaldehyde inhibits production of cytokine proteins but not their mRNAs from ConA-stimulated mouse splenocytes in vitro.

Acetaldehyde (ACH) attenuates ConA-induced T-cell response in mouse spleen cells in vitro. (A) WT and Aldh2 -/- mice were fed an ethanol diet for 4 weeks. Serum acetaldehyde and ethanol concentrations were measured. (B,C) Spleen cells from C57BL/6N mice were isolated and seeded on a 96-well plate with a same cell number/well. Acetaldehyde (200 µM) was added 5 hours prior to ConA was added. Levels of cytokines in the cell supernatant were measured (panel B). Cells were also harvested and subjected to real-time PCR analysis of cytokine mRNAs (panel C). The values represent means±SEM from three independent experiments. *P<0.05. ACH, acetaldehyde; ConA, concanavalin A; IL, interleukin; PBS, phosphate buffered saline; WT, wild type.
Acetaldehyde inhibits aerobic glycolysis in mouse T cells in vitro
Glucose metabolism plays a critical role in the regulation of T-cell activation and cytokine secretion.29-31 Thus, we asked whether acetaldehyde inhibits T-cell cytokine production via the inhibition of T-cell metabolism. During activation, T cells use glycolysis to metabolise glucose despite the presence of oxygen, a procedure called aerobic glycolysis, which is critical for the efficient secretion of cytokines such as IFN-γ and IL-2.29-31 One marker for aerobic glycolysis is the rapid consumption of glucose in the culture medium because aerobic glycolysis is less efficient in producing adenosine triphosphate (ATP) compared with oxidative glycolysis. Therefore, we analysed the glucose concentration in T-cell culture medium. As illustrated in figure 4A, acetaldehyde-treated T cells had higher levels of glucose in the medium than PBS-treated T cells 48 hours postculture, indicating that acetaldehyde inhibits aerobic glycolysis. In agreement with these findings, acetaldehyde treatment downregulated the expression of glucose transporter 1 (Glut1) and hypoxia-inducible factor-1α (Hif-1a) (figure 4B), both are critical for aerobic glycolysis.29-31 Furthermore, we analysed the glucose uptake by T cells by staining 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-d-glucose (a fluorescent glucose analogue) and found that the fluorescence intensities in total T, CD4+ and CD8+ T cells were lower in acetaldehyde-treated cells than those in PBS-treated cells (figure 4C). Finally, we examined several signal pathways that are involved in aerobic glycolysis, including mammalian target of rapamycin (mTOR), protein kinase B (PKB)/AKT, eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1).29-31 As shown in figure 4D and online supplementary figure 5, acetaldehyde markedly reduced p-mTOR, p-AKT and p-4E-BP1 in ConA-activated T cells with the strongest inhibition of pAKT.

Acetaldehyde (ACH) reduces aerobic glycolysis in mouse T cells in vitro. C57BL/6N mouse splenocytes were treated with acetaldehyde (200 µM) or PBS for 5 hours, followed by stimulation with ConA. (A) The supernatant glucose concentrations were measured 24 and 48 hours post-ConA stimulation. (B) Expression of Glut1 and Hif-1a mRNA was measured by using real-time PCR analysis. (C) The glucose uptake in total T cells, CD4+ T cells and CD8+ T cells were measured with 2-NBDG incorporation assay 24 and 48 hours after ConA stimulation. (D) Western blot analyses of phosphorylated mTOR, AKT and 4E-BP1 24 and 48 hours post-ConA stimulation. Left panel shows representative western blot images (all original blots are shown in online supplementary figure 5) and right panel shows the density quantification. The values represent means±SEM (n=4–6 independent experiments), *P<0.05; **P<0.01. ACH, acetaldehyde; AKT, protein kinase B; 4E-BP1, 4E-binding protein 1; ConA, concanavalin A; mTOR, mammalian target of rapamycin; 2-NBDG:2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-d-glucose; PBS, phosphate buffered saline.
Acetaldehyde inhibits IFN-γ production and aerobic glycolysis from phytohaemagglutinin (PHA)-stimulated human T cells in vitro
ALDH2-inactive individuals are known to have much higher levels of acetaldehyde after alcohol consumption compared with those with normal ALDH2, reaching to approximately 100 µM.15, 32 Thus, next we examined whether acetaldehyde also inhibits human T-cell cytokine production such as IFN-γ in vitro. As illustrated in figure 5A, in vitro treatment with acetaldehyde markedly inhibited T-cell mitogen PHA-stimulated IFN-γ production in T cells from individuals with normal ALDH2 (ALDH2*1*/1*) or with inactive ALDH2 (ALDH2*1*/2*) in a dose-dependent manner. Such inhibition was comparable between those T cells of normal ALDH2 and inactive ALDH2 individuals (figure 5A and online supplementary figure 6). Lack of difference in inhibition of IFN-γ production between normal ALDH2 and ALDH2 deficient T cells is probably because lymphocytes such as T cells express low levels of ALDH2 and do not efficiently metabolise acetaldehyde.33

Acetaldehyde (ACH) attenuates PHA-induced T-cell response and aerobic glycolysis in human PBMCs and T cells in vitro. (A) PBMCs were isolated from six healthy volunteers with active ALDH2 and six healthy volunteers with inactive ALDH2 and seeded on a 96-well plate with a same cell number/well. Acetaldehyde (50–400 µM) was added 5 hours before PHA was added. Levels of cytokines in the cell supernatant were measured 48 hours post-PHA stimulation. *P<0.05; **P<0.01; ***P<0.001 in comparison with the corresponding group without acetaldehyde treatment (0 µM). (B–D) T cells were isolated from human PBMCs and treated with acetaldehyde (100 µM) or PBS for 5 hours, followed by stimulation with PHA for 24 and 48 hours. The supernatant glucose concentrations were measured 24 and 48 hours post-PHA stimulation (panel B); expressions of GLUT1 and HIF-1A mRNA were measured by using real-time PCR analysis (panel C). (D) Western blot analyses of phosphorylated mTOR, AKT and 4E-BP1 24 and 48 hours post-PHA stimulation. Left panel shows representative western blot images (all original blots are shown in online supplementary figure 7), and right panel shows the density quantification. In panels B–D, the values represent means±SEM (n=6 independent experiments), *P<0.05, **P<0.01. ACH, acetaldehyde; AKT, protein kinase B; ALDH2, aldehyde dehydrogenase 2; 4E-BP1, 4E-binding protein 1; IFN, interferon; mTOR, mammalian target of rapamycin; PBMC, peripheral blood mononuclear cell; PBS, phosphate buffered saline; PHA, phytohaemagglutinin.
Figure 5B–C shows that treatment with acetaldehyde significantly increased glucose levels in the supernatant but downregulated expression of GLUT1 and HIF-1A mRNAs in PHA-stimulated human T cells, suggesting that acetaldehyde also inhibits aerobic glycolysis in human T cells. In agreement with the data from figure 5B–C, activation of aerobic glycolysis pathways especially pAKT was markedly suppressed by acetaldehyde in PHA-stimulated human T cells 24 and 48 hours post-treatment (figure 5D, online supplementary figure 7). Moreover, online supplementary figure 8 shows that acetaldehyde did not affect the expression of phosphatase and tensin homolog (PTEN) and protein phosphatase 2 (PP2A) (both inhibit AKT activation34, 35) in PHA-stimulated human T cells.
The above data show that acetaldehyde inhibits T-cell cytokine secretion by inhibiting the AKT pathway 24 and 48 hours post-treatment. A recent study reported that activation of the pyruvate dehydrogenase (PDH) kinase 1 (PDHK1)-PDH pathway contributes to early T-cell activation, lactate and cytokine production.36 To examine whether acetaldehyde also regulates early T-cell cytokine production, we measured PHA-induced cytokine, lactate production and activation of several signalling pathways 1 and 6 hours post-PHA stimulation. As illustrated in online supplementary figure 8 and online supplementary figure 9A–B, acetaldehyde treatment neither affected cytokine and lactate production nor altered the phosphorylation of PDH, an enzymatic target of PDHK1,36 at 1 and 6 hours post-PHA treatment, suggesting acetaldehyde does not affect activation of the PDHK1-PDH-lactate pathway. In addition, acetaldehyde treatment did not affect the m-TOR-AKT activation 1 and 6 hours post-PHA treatment (online supplementary figure 9C). Collectively, acetaldehyde treatment seems not affecting early T-cell cytokine production.
Finally, we also examined the effect of acetaldehyde on PKA activation that is involved in metabolic metabolism and T-cell activation.37 As illustrated in online supplementary figure 10, PKA activity was comparable in PHA-treated cells with or without acetaldehyde treatment, suggesting that acetaldehyde does not affect PKA activation in PHA-treated human T cells.
Corticosterone contributes to the resistance of ethanol-fed Aldh2-/- mice to ConA-induced hepatitis
The above data show acetaldehyde attenuates T-cell cytokine production by inhibiting aerobic glycolysis, contributing to the resistance of Aldh2-/- mice to ConA-induced hepatitis. Corticosteroids also inhibit aerobic glycolysis in immune cells.38, 39 Chronic ethanol consumption is associated with elevation of corticosteroids in the blood40 and treatment with corticosteroids attenuated ConA-induced hepatitis.41 Thus, we asked whether the reduced susceptibly to ConA-induced hepatitis in ethanol-fed Aldh2 -/- mice was also due to elevation of corticosteroids. As illustrated in figure 6A, serum corticosterone levels were higher in ethanol-fed Aldh2 -/- mice vs WT mice. To examine whether acetaldehyde, which is higher in ethanol-fed Aldh2 -/- mice, contributes to corticosterone release, we injected mice with acetaldehyde. As illustrated in figure 6B, acetaldehyde injection elevated serum levels of corticosterone and adrenocorticotrophic hormone (ACTH, which stimulates corticosterone release) in both WT and Aldh2 -/- mice with higher levels in the latter group. In addition, it is well known that glucocorticoids can exert negative feedback on both the hypothalamus and pituitary gland by targeting glucocorticoid receptor (GR) to reduce corticosterone levels. Ethanol can disrupt this feedback loop by interfering with glucocorticoid response element-DNA binding and downregulating GR expression in various brain regions,42 thereby elevating corticosterone levels. Then we asked whether acetaldehyde also targets GR to elevate corticosterone levels. To answer this question, mice were treated with RU486, which blocked GR, followed by injection of acetaldehyde. As illustrated in figure 6C, injection of RU486 markedly elevated serum corticosterone levels, which was further increased after acetaldehyde injection, suggesting acetaldehyde elevates corticosterone independent of GR.

Chronic ethanol feeding induces higher serum corticosterone levels in Aldh2 -/- mice than in WT mice, and treatment with the steroid antagonist partially restores ConA-induced liver injury in Aldh2 -/- mice. (A) WT and Aldh2 -/- mice were fed an ethanol diet for 4 weeks. Serum corticosterone levels were measured. (B) WT and Aldh2 -/- mice were injected intraperitoneally with 200 mg/kg body weight acetaldehyde. Serum corticosterone and ACTH levels were measured 30 and 60 min post injection. The values in panels A and B represent means±SEM (n=5 in WT and KO). *P<0.05 as indicated. #P<0.05 in comparison with WT groups without acetaldehyde treatment. (C) WT mice was injected RU486 (35 mg/kg body weight, a single injection), 1 hour later, PBS or acetaldehyde (200 mg/kg body weight) was injected. Serum corticosterone levels were measured 30 min post-PBS or acetaldehyde injection. The values represent means±SEM (n=6). *P<0.05, **P<0.01 as indicated. (D,E) WT and Aldh2 -/- mice were fed an ethanol or control diet for 4 weeks. At the end of 4 weeks, mice were treated with vehicle or the steroid antagonist RU486 (10 mg/kg/day) for 3 days, followed by ConA injection. Serum ALT levels were measured (panel D) and representative H&E staining of liver tissues are shown (panel E). The values represent means±SEM (n=4 in pair-fed group, n=8 in ethanol-fed groups with vehicle or RU486 treatment). *P<0.05. ACTH, adrenocorticotrophic hormone; ALT, alanine aminotransferase; ConA, concanavalin A; ETOH, ethanol; WT, wild type.
Figure 6D shows that in WT mice, chronic ethanol feeding increased ConA-induced elevation of serum ALT compared with the pair-fed group 9 and 24 hours post-ConA injection. This elevation was not affected by the treatment with the steroid antagonist RU486. In Aldh2 -/- mice, chronic ethanol feeding decreased serum ALT levels compared with the pair-fed group at both 9 and 24 hours after ConA injection. Treatment with RU486 restored elevation of serum ALT levels 24 hours post-ConA injection. In addition, RU486 treatment did not affect liver necrosis in ethanol-fed WT mice, but partially restored it in ethanol-fed Aldh2 -/- mice post-ConA injection (figure 6E).
Acute alcohol consumption elevates higher plasma levels of cortisol and corticosterone in human subjects with ALDH2 deficiency than those with normal ALDH2
While in rodents, corticosterone is the main glucocorticoid produced by the adrenal glands cortex, in human, cortisol is the primary glucocorticoid. Thus, we measured both cortisol and corticosterone in human subjects with inactive or active ALDH2 post-alcohol consumption. As illustrated in figure 7, serum cortisol and corticosterone levels were rapidly elevated with 10–20 min after acute alcohol drinking in both groups with no differences. Serum cortisol and corticosterone levels returned to normal levels after 60 min in individuals with normal ALDH2. Interestingly, in individuals with ALDH2 deficiency, serum cortisol and corticosterone levels also declined to normal levels 60 min post drinking, but further elevated again at later time points with a peak occurring at 180 min post drinking. Serum cortisol and corticosterone levels at later time points in ALDH2-deficient individuals were much higher than those at earlier time points.

Acute alcohol consumption induces much higher plasma levels of cortisol and corticosterone in ALDH2-inactive human subjects than in those with normal ALDH2. Individuals with active ALDH2 or inactive ALDH2 were recruited and subjected to acute alcohol drinking (0.4 g/kg). Plasma samples were collected and subjected to cortisol (panel A) and corticosterone (panel B) measurement. Individual data from each subject are presented in the left panels. Summarised data are presented in the right panel. Values represent means±SEM. *P<0.05; **P<0.01 and ***P<0.001 in comparison with corresponding time points in individuals with normal ALDH2. ALDH2, aldehyde dehydrogenase 2.
Discussion
In the current study, we revealed some unexpected interesting findings that chronic ethanol exposure reduced rather than enhanced T-cell hepatitis in Aldh2 -/- mice, which is in contrast to ethanol exacerbation of T-cell hepatitis in WT mice.13 Mechanistically, we found that acetaldehyde directly inhibited T-cell glucose metabolism and cytokine release, which contributed to the suppressed T-cell hepatitis in Aldh2 -/- mice after alcohol consumption. In addition, mice and human subjects with ALDH2-defiency have higher levels of corticosterone and cortisol after alcohol consumption than those with normal ALDH2, which may also contribute to the resistance of ethanol-fed Aldh2 -/- mice to ConA-induced T-cell hepatitis. We have integrated our findings into a model depicting the mechanisms by which ALDH2 deficiency is associated with attenuated T-cell response post alcohol consumption (figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A model illustrating the mechanisms by which ALDH2 deficiency is associated with T-cell suppression post-alcohol consumption. ALDH2-deficient individuals post-alcohol consumption is associated with high levels of acetaldehyde, which can directly block T-cell cytokine production via the inhibition of AKT activation and glucose metabolism or indirectly attenuate T-cell response via the elevation of corticosteroids. ‘+’ means promotion, ‘–’ means suppression. ALDH2, aldehyde dehydrogenase 2; AKT, protein kinase B; GLUT1, glucose transporter 1; HPA, hypothalamic-pituitary-adrenal; PKB, protein kinase B.
Acetaldehyde is believed to be very toxic,18-20 thus we originally hypothesised that ethanol-fed Aldh2 -/- mice, which had higher acetaldehyde levels compared with WT mice after alcohol feeding, are more sensitive to ConA-induced hepatitis. To our surprise, we found that ethanol-fed Aldh2 -/- mice were resistant to ConA-induced hepatitis and elevation of serum cytokines, but equally susceptible to lipopolysaccharide (LPS)-induced elevation of serum cytokines as WT mice (online supplementary figure 11), suggesting that the reduced susceptibility of ethanol-fed Aldh2 -/- mice to ConA-induced hepatitis is specific to this model, which is due to suppression of ConA-induced T-cell activation. Our mechanistical studies suggest that one of the mechanisms by which ConA-induced hepatitis is suppressed in ethanol-fed Aldh2 -/- mice is acetaldehyde-mediated direct inhibition of T-cell cytokine production without affecting T-cell proliferation. Furthermore, we identified a novel mechanism by which acetaldehyde inhibits T-cell cytokine production via the suppression of T-cell glucose metabolism. During activation, T cells uptake glucose via the upregulation of Glut1 expression, followed by generation of energy to promote cytokine secretion.29, 30 Here, we provided several lines of evidence suggesting that acetaldehyde inhibits glucose metabolism in T cells via the downregulation of Glut1 expression and inhibition of several metabolic signalling pathways. First, in vitro exposure to acetaldehyde reduced glucose uptake in cultured T cells, indicating that acetaldehyde reduces glucose metabolism in T cells. Second, incubation with acetaldehyde downregulated expression of two important genes involved in aerobic glycolysis, Glut1 and Hif-1a in T cells.29-31 Third, several metabolic signal pathways including mTOR, AKT, 4E-BP1, which are involved in aerobic glycolysis in T cells,29-31 were suppressed after exposure to acetaldehyde in vitro with the strongest inhibition of pAKT activation. Interestingly, although acetaldehyde treatment induced strong inhibition of AKT, such treatment did not affect the expression of PTEN and PP2A proteins (both inhibit AKT activation),34, 35 suggesting that acetaldehyde-mediated inhibition of AKT is independent of PTEN and PP2A. It is known that many other factors also affect AKT activation, including a wide variety of cytokines, growth factors and hormones as well as cross-talk with other signalling pathways.43 Whether acetaldehyde inhibits AKT activation by affecting these pathways requires further extensive studies. In addition to AKT activation, activation of the PDHK1-PDH pathway contributes to early T-cell activation and cytokine production.36 Interestingly, our data revealed that activation of this pathway and early T-cell cytokine production was not altered by acetaldehyde. Collectively, inhibition of AKT is probably the major mechanism contributing to acetaldehyde-mediated inhibition of cytokine production and glucose metabolism in T cells. Finally, epigenetic regulation has been suggested to play a complex role in controlling T-cell activation, cytokine production and glucose metabolism, which still remains poorly understood.44 Acetaldehyde and its metabolite acetate are known to affect gene expression at the epigenetic level (histone acetylation for instance).45 However, whether acetaldehyde inhibits T-cell cytokine production and glucose metabolism via the epigenetic regulation is not clear and requires extensive future studies to answer this question.
In the current paper, we have also identified an additional important mechanism underlying immunosuppression of T cells in ethanol-fed Aldh2-/- mice that is due to high levels of glucocorticoid, a well-known factor that inhibits aerobic glycolysis in immune cells by downregulating HIF-α and GLUT1 and subsequently suppresses immune cell functions.38, 39 First, ethanol-fed Aldh2 -/- mice had higher levels of corticosterone than WT mice. Second, injection of the synthetic glucocorticoid prednisolone blunted ConA-induced hepatitis in mice.41 Last, in vivo treatment with the GR antagonist RU486 partially restored ConA-induced hepatitis in ethanol-fed Aldh2 -/- mice. Higher levels of corticosterone in ethanol-fed Aldh2 -/- mice were probably due to higher levels of acetaldehyde in these mice versus ethanol-fed WT mice because acetaldehyde can directly stimulate corticosterone release with 10 times more potency than ethanol.46-48 Importantly, we also demonstrated that acute alcohol drinking rapidly elevated serum levels of both cortisol and corticosterone in human subjects with much higher levels in those with ALDH2 deficiency. Serum cortisol and corticosterone levels were rapidly elevated within 10–20 min in human subjects with normal ALDH2 or ALDH2 deficiency. Interestingly, serum corticosteroid levels were further elevated at later time points (between 120 and 210 min) in the individuals with inactive ALDH2 but not in those with normal ALDH2. It was reported that after binge drinking, blood ethanol levels were rapidly elevated in both active and inactive ALDH2 individuals, whereas blood acetaldehyde elevation was delayed with much higher levels in ALDH2-deficient individuals than in those with normal ALDH2.15 Given acetaldehyde is 10 times more potent than ethanol to stimulate corticosterone release in vivo,48 we speculate that the delayed elevation and high levels of serum corticosteroids in ALDH2-deficient individuals after alcohol drinking was due to accumulated acetaldehyde in these subjects. 15, 16
The mechanisms by which acetaldehyde elevates serum corticosteroid levels have been explored previously by several studies. Treatment of ethanol-fed rats with cyanamide (ALDH2 inhibitor) elevated plasma levels of acetaldehyde and corticosterone concentrations along with upregulation of corticotropin-releasing hormone (CRH) in the paraventricular nucleus and pro-opiomelanocortin in the anterior pituitary, suggesting a role of acetaldehyde in activating the hypothalamic-pituitary-adrenal (HPA) axis.49 Furthermore, ex vivo studies revealed that incubation with acetaldehyde directly stimulated CRH release from hypothalamic explants in a dose-dependent manner.50 In the current study, we demonstrated that injection of acetaldehyde directly elevated serum corticosterone and ACTH levels in both WT and Aldh2 -/- mice with much higher levels in the latter group. The contribution of corticosterone to the suppressed T-cell hepatitis in ethanol-fed Aldh2 -/- mice was demonstrated by the fact that blockade of corticosterone with the steroid antagonist RU486 restored T-cell hepatitis in these mice. Interestingly, such restoration was only partial, suggesting additional mechanisms (such as acetaldehyde directly inhibits T-cell function) are also involved. Collectively, ALDH2-deficency-associated high levels of acetaldehyde post-alcohol consumption can directly stimulate HPA axis and corticosteroid release, resulting in suppression of T cells. Although the immunosuppressive effects of corticosteroids have been well documented in a variety of immune cells including T cells,51 the exact mechanisms underlying corticosterone-mediated inhibition of ConA-induced hepatitis in ethanol-fed Aldh2 -/- mice remain obscure. For example, it is not clear whether corticosterone inhibition of ConA-induced hepatitis in these mice is mediated by directly targeting GR on T cells and/or other types of immune cells. This question can be answered by using immune cell-specific GR and global Aldh2 double knockout mice in future studies.
In summary, ALDH2 deficiency is associated with suppression of T-cell functions post-alcohol consumption, which is due to elevation of corticosteroid and acetaldehyde levels. Such T-cell suppression may attribute to the increased risk of alcohol-related cancers and diseases in ALDH2-deficient individuals.52 Future studies on this area may help identify novel mechanisms and therapeutic strategies for those alcohol-associated maladies in ALDH2-inactive individuals.
Appendix
Supporting Materials.pdf
References
Footnotes
YG, ZZ and TR contributed equally.
Contributors YG, ZZ and TR planned and conducted the animal and cell culture study and clinical study, analysed the data and wrote the paper. S-JK, YH, WS, AG, WW, DF, MX and EH conducted some experiments related to mice and cell culture. YD, RW, SS, XW and HZ performed some human PBMC experiments and measurement of human cortisol and corticosterone. WZ and ZXZ measured serum ethanol and acetaldehyde levels. PP analysed the data and edited the paper. JN planned and conducted clinical study, analysed the data and edited the paper. BG designed the whole project, supervised the study and wrote the paper.
Funding This work was supported by the intramural program of NIAAA, NIH (BG) and Natural Science and Technology Major Project (2014ZX10002002) (JN). YG was supported by Chinese National Science Foundation Scholarship.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The First Affiliated Hospital of Jilin University Institutional Review Board and the Research and Development Committee.
Provenance and peer review Not commissioned; externally peer reviewed.