Article Text

Abstract
These guidelines on the management of primary sclerosing cholangitis (PSC) were commissioned by the British Society of Gastroenterology liver section. The guideline writing committee included medical representatives from hepatology and gastroenterology groups as well as patient representatives from PSC Support. The guidelines aim to support general physicians, gastroenterologists and surgeons in managing adults with PSC or those presenting with similar cholangiopathies which may mimic PSC, such as IgG4 sclerosing cholangitis. It also acts as a reference for patients with PSC to help them understand their own management. Quality of evidence is presented using the AGREE II format. Guidance is meant to be used as a reference rather than for rigid protocol-based care as we understand that management of patients often requires individual patient-centred considerations.
- sclerosing cholangitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Executive summary and recommendations
There are many causes of cholangiopathy and these should be considered in the assessment of all patients presenting with biliary strictures. Primary sclerosing cholangitis (PSC) has a wide spectrum of symptomatology and stages of disease. Diagnosis is based on the cholangiographic (or histological) features of sclerosing cholangitis in the absence of identifiable causes of secondary sclerosing cholangitis. The diagnosis and management of PSC can be difficult and requires specialist referral for advanced disease or patients experiencing significant clinical events. Few randomised controlled trials have been carried out to define best management. Most recommendations derive from small case –control studies, retrospective series and expert opinion. There is little evidence for the use of medical therapy to prevent progression of disease. Ursodeoxycholic acid is not recommended for routine use in newly diagnosed PSC. Non-invasive investigations such as magnetic resonance cholangiopancreatography (MRCP), dynamic liver MRI and/or contrast CT should be performed in patients who have new or changing symptoms or evolving abnormalities in laboratory investigations. Worsening liver biochemistry and/or new high grade or evolving strictures should prompt further investigation for cholangiocarcinoma (CCA). Patients with PSC should ordinarily not undergo endoscopic retrograde cholangiopancreatography (ERCP) until there has been expert multidisciplinary assessment to justify endoscopic intervention. Colitis should be sought in all patients with PSC using colonoscopy and colonic biopsies. Patients with colitis should then have annual surveillance colonoscopy because of the increased risk of colorectal cancer. In these guidelines, we also review the management of PSC overlap syndromes and IgG4-related sclerosing cholangitis (IgG4-SC).
List of recommendations
There are multiple causes of cholangiopathy. We recommend that cholestatic liver biochemistry with typical cholangiographic features in the absence of other identifiable causes of secondary sclerosing cholangitis is usually sufficient for a diagnosis of PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that MRCP should be the principal imaging modality for the investigation of suspected PSC. ERCP should be reserved for patients with biliary strictures requiring tissue acquisition (eg, cytological brushings) or where therapeutic intervention is indicated (strength of recommendation: STRONG; quality of evidence: HIGH).
We recommend that liver biopsy is normally reserved for possible small duct PSC, assessment of suspected possible overlap variants or instances where the diagnosis is unclear (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend risk stratification based on non-invasive assessment. Clinical scores are an emerging theme but no single method can be recommended at present to predict individual patient prognosis. Given the unpredictable disease course and the serious nature of the complications of PSC, patients should receive lifelong follow-up (strength of recommendation: STRONG; quality of evidence: VERY LOW).
We recommend that ursodeoxycholic acid (UDCA) is not used for the routine treatment of newly diagnosed PSC (strength of recommendation: STRONG; quality of evidence: GOOD). For patients already established on UDCA therapy, there may be evidence of harm in patients taking high dose UDCA 28–30 mg/kg/day (strength of recommendation: WEAK; quality of evidence: LOW).
We recommend that UDCA is not used for the prevention of colorectal cancer or cholangiocarcinoma (strength of recommendation: STRONG; quality of evidence: HIGH).
We recommend that corticosteroids and immunosuppressants are not indicated for the treatment of classic PSC (strength of recommendation: STRONG; quality of evidence: HIGH). In those patients with additional features of autoimmune hepatitis (AIH) or IgG4-related sclerosing cholangitis (IgG4-SC), corticosteroids may be indicated (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that endoscopic screening for oesophageal varices should be done in line with international guidelines where there is evidence of cirrhosis and/or portal hypertension (strength of recommendation: STRONG; quality of evidence: HIGH).
We recommend that colitis should be sought in all patients with PSC using colonoscopy and colonic biopsies (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that patients with suspected PSC undergoing ERCP should receive prophylactic antibiotics (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that non-invasive investigations such as MRCP, dynamic liver MRI and/or contrast CT should be performed in patients who have new or changing symptoms or evolving abnormalities in laboratory investigations (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that patients with PSC should ordinarily not undergo ERCP until there has been expert multidisciplinary assessment to justify endoscopic intervention (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that in patients undergoing ERCP for dominant strictures, pathological sampling of suspicious strictures is mandatory (strength of recommendation: STRONG; quality of evidence: STRONG).
We recommend that in patients undergoing ERCP for dominant strictures, biliary dilatation is preferred to the insertion of biliary stents (strength of recommendation: STRONG; quality of evidence: MODERATE).
We suggest that provision of care should involve a partnership between patients, primary care and hospital-led specialty medicine with consideration made with regard to patient risk assessment, symptom burden and how local services are configured (strength of recommendation: WEAK; quality of evidence: LOW).
We recommend that patients with symptomatic, evolving or complex disease should be referred for expert multidisciplinary assessment. Patients with early, stable disease can be managed in general clinics (strength of recommendation: STRONG; quality of evidence: LOW).
We suggest that patients with PSC meeting inclusion criteria should be offered referral to a centre participating in clinical trials (strength of recommendation: WEAK; quality of evidence: LOW).
PSC is a well-recognised indication for liver transplantation. We recommend that eligibility and referral should be assessed in line with the national guidelines (strength of recommendation: STRONG; quality of evidence: HIGH).
We recommend that all patients with PSC should have a risk assessment for osteoporosis. Once osteoporosis is detected, treatment and follow-up should be in accordance with national guidelines (strength of recommendation: STRONG; quality of evidence: MODERATE).
Poor nutrition and fat-soluble vitamin deficiency are relatively common in advanced PSC and we suggest that clinicians should have a low threshold for empirical replacement (strength of recommendation: WEAK; quality of evidence: MODERATE).
We recommend that in patients with fatigue, alternative causes should be actively sought and treated (strength of recommendation: STRONG; quality of evidence: LOW).
We suggest that cholestyramine (or similar) is first-line medical treatment for pruritus. Rifampicin and naltrexone are second-line treatments (strength of recommendation: WEAK; quality of evidence: LOW).
We suggest that an elevated CA19.9 may support a diagnosis of suspected cholangiocarcinoma but has a low diagnostic accuracy. Routine measurement of serum CA19.9 is not recommended for surveillance for cholangiocarcinoma in PSC (strength of recommendation: WEAK; quality of evidence: MODERATE).
We recommend that when a diagnosis of cholangiocarcinoma is clinically suspected, referral for specialist multidisciplinary meeting (MDM) review is essential (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that where cholangiocarcinoma is suspected, contrast-enhanced, cross-sectional imaging remains the initial preferred investigation for diagnosis and staging (strength of recommendation: STRONG; quality of evidence: HIGH). Confirmatory diagnosis relies on histology with the approach to tissue sampling guided by MDM review. Options include ERCP-guided biliary brush cytology/fluorescence in situ hybridisation (FISH)/endobiliary biopsy/cholangioscopy/endoscopic ultrasound (EUS)-guided biopsy and/or percutaneous biopsy (strength of recommendation: STRONG; quality of evidence: HIGH).
We suggest that an annual ultrasound scan of the gallbladder should be performed in patients with PSC. If polyps are identified, treatment should be directed by specialist hepatopancreaticobiliary (HPB) MDM (strength of recommendation: WEAK; quality of evidence: LOW).
We recommend that patients with PSC who have coexistent colonic inflammatory bowel disease (IBD) should have annual colonoscopic surveillance from the time of diagnosis of colitis in line with the British Society of Gastroenterology (BSG) guidelines (strength of recommendation: STRONG; quality of evidence: HIGH). We suggest that those without IBD may benefit from less frequent 5-year colonoscopy or earlier in the advent of new symptoms (strength of recommendation: WEAK; quality of evidence: VERY LOW).
We suggest that in the presence of cirrhosis, hepatocellular carcinoma surveillance should be carried out in accordance with international guidelines (strength of recommendation: WEAK; quality of evidence: LOW).
We recommend that because pregnancy in cirrhotic patients carries a higher risk of maternal and fetal complications, patients should have preconception counselling and specialist monitoring (strength of recommendation: STRONG; quality of evidence: LOW).
We recommend that patients with PSC should be encouraged to participate in patient support groups (strength of recommendation: STRONG; quality of evidence: VERY LOW).
IgG4-related sclerosing cholangitis (IgG4-SC)
We recommend that elevated serum IgG4 levels support the diagnosis of clinically suspected IgG4-related disease (IgG4-RD) but cannot be relied on for making a definite diagnosis, or distinguishing IgG4-SC from PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that in patients with suspected IgG4-SC, attempts should be made to obtain a confirmatory histological diagnosis (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that other organ involvement (in particular, pancreatic manifestations of IgG4-RD) may provide important information to distinguish IgG4-SC from PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that IgG4-SC should be diagnosed according to the recommendations of the international consensus guidelines (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that patients with active IgG4–SC should be given corticosteroids as first-line treatment (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that all patients with IgG4-SC, including those with multiorgan involvement in IgG4-RD, should be considered for continued immunosuppressive therapy (strength of recommendation: STRONG; quality of evidence: MODERATE).
We recommend that patients with complex IgG4-SC and those with suspected malignancy should be referred to a specialist MDM for review (strength of recommendation: STRONG; quality of evidence: LOW).
Scope and purpose
These guidelines have been commissioned on behalf of the British Society of Gastroenterology (BSG) liver section and UK-PSC with the aim of assisting clinicians in the diagnosis and management of patients with PSC. Members of the writing committee included gastroenterologists, hepatologists, transplant physicians and patient representatives. Where possible, clear, clinically applicable recommendations are provided. The guidelines were reviewed by the BSG guideline commissioning group and council before circulation for international peer review. This document should be used in conjunction with other BSG guidelines and documents published by other international bodies in the USA, Europe and Japan.1–4 We recommend revision of the guidelines in 5 years. Where possible, we have tried to avoid duplicating advice published in related BSG guidelines.
Evidence base
These guidelines have been produced using systematic review of publications identified using PubMed, Medline and Cochrane database searches in line with the Appraisal of Guidelines for Research and Evaluation (AGREE) instrument II (www.agreetrust.org). The primary keywords for baseline searches were ‘primary sclerosing cholangitis’, ‘autoimmune pancreatitis’, ’IgG4,’ ’autoimmune overlap syndrome’ and ‘cholangiocarcinoma’. Additional keywords were included for specific searches such as therapy, ursodeoxycholic acid, ERCP, endotherapy, biliary dilatation, etc. The literature search was updated and completed in March 2018 before submission for peer review. Where possible, guidance is based on the highest levels of evidence available and cited. Where no high-quality studies or clear evidence exist, guidance is based on the majority consensus advice of expert opinion in the literature and the writing committee.
Grade of evidence is presented as ‘strong’ and ‘weak’ according to the international GRADE3 4 system:
High-quality evidence: The authors are very confident that the estimate presented lies very close to the true value. One could interpret it as: there is very low probability of further research completely changing the presented conclusions.
Moderate-quality evidence: The authors are confident that the presented estimate lies close to the true value, but it is also possible that it may be substantially different. One could also interpret it as: further research may completely change the conclusions.
Low-quality evidence: The authors are not confident of the effect estimate and the true value may be substantially different. One could interpret it as: further research is likely to change the presented conclusions completely.
Very low-quality evidence: The authors do not have any confidence in the estimate and it is likely that the true value is substantially different from it. One could interpret it as: new research will most probably change the presented conclusions completely.
Background
Definitions
PSC is an immune-mediated chronic liver disease characterised by inflammation, fibrosis and destruction of intrahepatic and/or extrahepatic bile ducts leading to cholestasis, bile duct strictures and hepatic fibrosis, which in turn may progress to cirrhosis, portal hypertension and hepatic decompensation.5 6 A variant known as small duct PSC is characterised by typical cholestatic liver biochemistry and histological findings typical of PSC but with normal appearance of the bile ducts at cholangiography.7 PSC overlap/variant syndromes are conditions with diagnostic features of both PSC and other immune-mediated liver diseases, including autoimmune hepatitis (AIH). These guidelines refer specifically to PSC and its overlap syndromes, and include discussion of IgG4-related sclerosing cholangitis, which can mimic PSC. Causes of secondary sclerosing cholangitis related to other identifiable causes of biliary obstruction leading to injury of the bile ducts are listed in box 1 but are not considered further.
Causes of secondary sclerosing cholangitis and conditions with cholangiographic features that may mimic biliary strictures
Cholangiocarcinoma
IgG4-SC
Traumatic or ischaemic bile duct injury
Choledocholithiasis
Hilar lymphadenopathy
Ampullary or pancreatic cancer
Acute or chronic pancreatitis
Choledochal varices (portal biliopathy)
HIV cholangiopathy
Chronic biliary infestation (liver fluke, ascaris)
Congenital (choledochal cysts, biliary atresia)
Papillary stenosis
Critical illness ischaemic cholangiopathy
Recurrent pyogenic cholangitis
Hereditary haemorrhagic telangiectasia
Systemic mastocytosis
Langerhans' cell histiocytosis X
Drugs
Epidemiology
Population-based studies estimate the incidence of PSC to be 0.91 to 1.3 per 100 000 person-years and may be increasing.8–12 The incidence of small duct PSC is reported to be 0.15 per 100 000 person-years.9 These studies were undertaken in populations of northern European descent in whom the incidence is thought to be highest. The incidence in most other ethnic groups is less clear.
Aetiology
PSC is a progressive biliary disorder strongly associated with inflammatory bowel disease (IBD). The genetic associations with disease risk, presence of chronic inflammation in the portal tracts and the strong association with IBD suggest that PSC is an immune-mediated disease, in which the biliary epithelial cell is a key cell targeted. However, no reliable autoantibodies have been identified and there is no significant response to immunosuppression. To date, genome-wide studies have uncovered susceptibility loci for PSC-IBD, the majority of which have been previously reported as risk factors in other immune-mediated disorders. The strongest association resides within the human leucocyte antigen complex and suggests that disease-specific antigens drive pathogenic immune responses. Genetic determinants account for <10% of total disease liability in PSC-IBD, clearly emphasising the predominant role of environmental factors on ultimate disease susceptibility.
How is PSC diagnosed?
Consensus diagnostic criteria for PSC have been published as a workshop summary on behalf of the American College of Gastroenterology.1 Consensus guidelines relating to IgG4-SC, the biliary manifestation of IgG4-related disease (IgG4-RD),13 and on PSC/AIH variant syndrome, have also been published.14 A summary is outlined in figure 1.
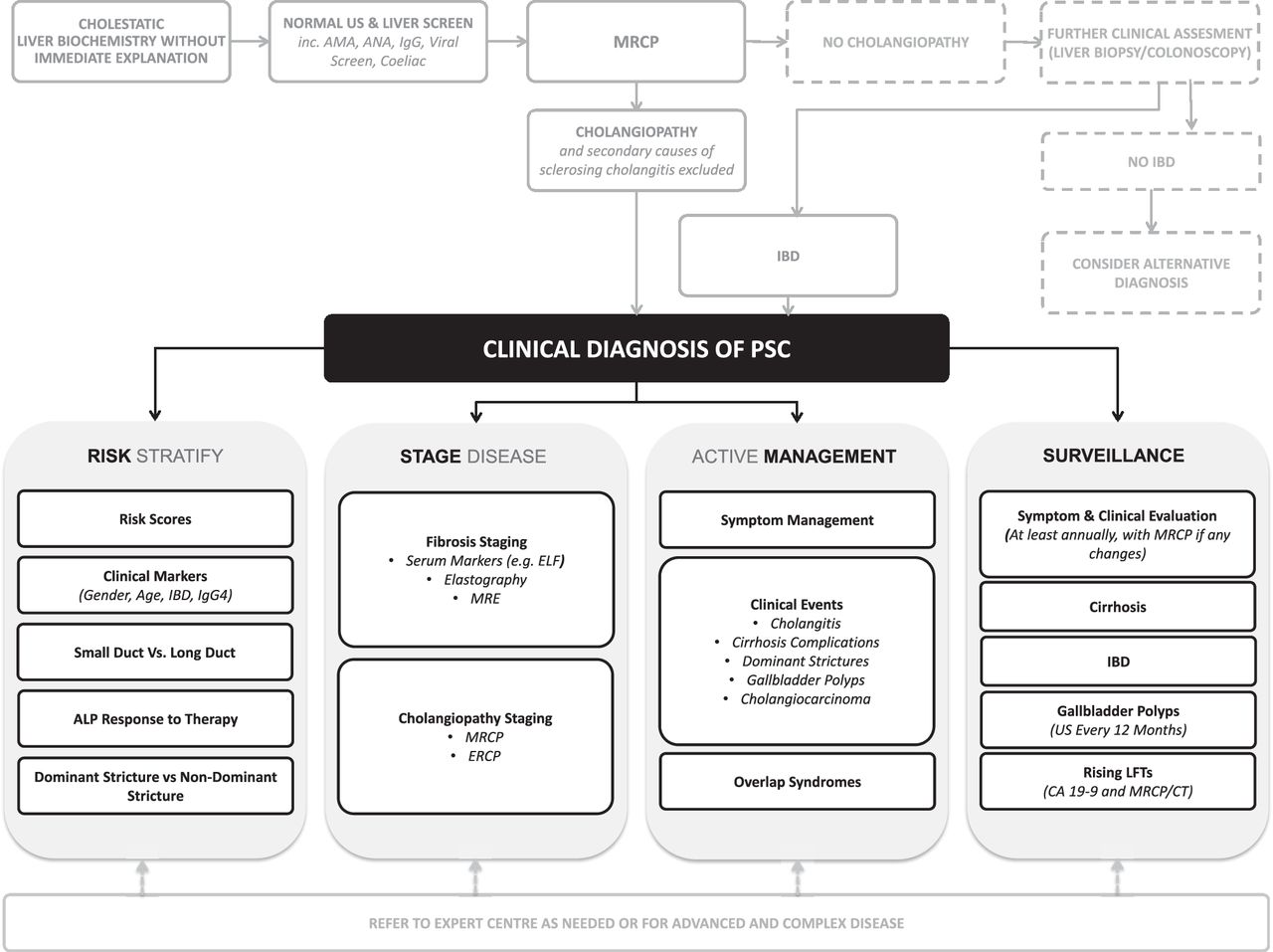
Algorithm for the management of suspected primary sclerosing cholangitis.
Modes of presentation
Symptoms are rare in early disease. In more established cases, symptoms such as right upper quadrant pain, pruritus, fatigue, jaundice, fever and weight loss are present in 47–56% of patients.11 15 Patients usually present in one of several ways: (i) no symptoms or signs but with an incidental finding of abnormal liver biochemistry, (ii) biochemical screening of patients with newly diagnosed or pre-existing IBD, (iii) jaundice and pruritus secondary to cholestasis, (iv) cholangitis, (v) jaundice secondary to liver failure, (vi) variceal bleeding and/or ascites from portal hypertension or (vii) cholangiocarcinoma (CCA).
Blood tests
Serum liver biochemistry tests are abnormal in approximately 75% of patients with PSC.1 The most common pattern is of a cholestatic picture with raised alkaline phosphatase (ALP) and γ-glutamyl transpeptidase. An elevated serum bilirubin is reported to be present in 28–40% and is a marker of poor prognosis,15–17 but this is likely to be an overestimate with more advanced cases reported by published series. An elevated ALP is a sensitive marker for diagnosis but is not specific. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are often mildly raised and do not necessarily suggest additional features of autoimmune hepatitis. As with other causes of liver disease, a raised AST>ALT may be an indicator of cirrhosis and poor prognosis.18 Other indicators of cirrhosis or portal hypertension include an elevated prothrombin time or international normalised ratio, low albumin and low platelets. There are no autoantibodies diagnostic of PSC. Serum perinuclear antinuclear cytoplasmic antibody is positive in 33–88% of those with PSC but is not specific and is not related to disease activity or prognosis.15 19–22
Similar to clinical outcomes in primary biliary cholangitis (PBC), recent data from retrospective studies support the use of falling ALP (normalisation or <1.5 x upper limit of normal (ULN)) as a stratifier for improved outcome in patients with PSC, independent of the therapeutic modality used23–25
There are contradictory data on whether a raised serum IgG4 in patients with PSC (IgG4 + PSC) correlates with the disease course of PSC. In the study by Mendes,26 IgG4 + PSC was associated with more aggressive disease and progression to transplantation, but this was not seen in a European cohort of 345 patients with PSC.27 A further study including histological assessment of IgG4 staining in 98 liver explants from patients with a diagnosis of PSC, reported raised serum IgG4 levels in 22%, and raised tissue IgG4 levels in 23%.28 Again, those patients with raised IgG4 had a more rapid progression and need for liver transplantation. It is uncertain whether these findings are explained by misdiagnosis in some cases (ie, cases of IgG4-SC incorrectly diagnosed as PSC), or whether they represent a more aggressive phenotype of PSC in those with elevated IgG4 levels. A further assessment of liver explants from patients with PSC undergoing transplantation reported at least moderate IgG4 immunostaining in 24.6% and was associated with higher rates of dominant strictures, although this did not appear to correlate with age or speed of progression of disease.29
Some causes of secondary sclerosing cholangitis may respond well to medical treatment and it is therefore important to exclude secondary causes before making a diagnosis of PSC. Measurement of other biochemical tests, including antinuclear antibodies (ANA), antimitochondrial antibodies (AMA), smooth muscle antibodies (SMA), HIV antibodies, serum angiotensin converting enzyme, total immunoglobulins and immunoglobulin subsets (including IgG4), should be performed and positive results should raise the suspicion of alternative diagnoses or overlap/variant syndromes.
Recommendation 1: There are multiple causes of cholangiopathy. We recommend that cholestatic liver biochemistry with typical cholangiographic features in the absence of other identifiable causes of secondary sclerosing cholangitis is usually sufficient for a diagnosis of PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
Imaging
Transabdominal ultrasound scanning is rarely useful in the diagnosis of PSC, but may be helpful in excluding other causes of biliary obstruction such as choledocholithiasis, which can complicate stricturing disease and cholestasis in PSC. Ultrasound is also useful in the detection and surveillance of gallbladder polyps and in identifying developing portal hypertension. Contrast-enhanced CT may demonstrate features of cholangiopathy but is used primarily for the diagnosis and staging of suspected CCA.
Endoscopic retrograde cholangiopancreatography (ERCP) has been conventionally regarded as the ’gold standard' for the diagnosis of PSC, where the presence of a typical beading appearance caused by short multifocal strictures of the bile ducts is considered the best supportive evidence for the diagnosis of PSC. However, the risks involved with ERCP and improvement in image acquisition led to magnetic resonance cholangiopancreatography (MRCP) becoming the preferred imaging modality for the diagnosis of PSC. A number of studies have reported that the diagnostic accuracy of MRCP is comparable to that of ERCP, with a sensitivity and specificity of 80–100% and 89–100%, respectively.30–36 A meta-analysis of the diagnostic utility of MRCP included six well-controlled prospective studies and reported a sensitivity and specificity of 86% and 94%, respectively, for the diagnosis of PSC.37 However, MRCP may be less sensitive than ERCP in detecting early changes of PSC and has less specificity in patients with cirrhosis.30 Contrast- enhanced MRI scanning may also provide additional information about liver parenchyma, presence of varices, CCA and lymphadenopathy.
Many of the studies describing and differentiating PSC from other diseases were done before the widespread recognition of IgG4-SC, which may be present in 20–88% of patients with IgG4-RD.38 Whereas some cholangiographic features, such as long biliary strictures with prestenotic dilatations, and low common bile duct strictures, are more suggestive of IgG4-SC, beading, peripheral duct pruning and pseudodiverticula point more towards PSC.39 Cholangiography alone is insufficient to distinguish IgG4-SC, PSC and CCA.40
Recommendation 2: We recommend that MRCP should be the principal imaging modality for the investigation of suspected PSC. ERCP should be reserved for patients with biliary strictures requiring tissue acquisition (eg, cytological brushings) or where therapeutic intervention is indicated (strength of recommendation: STRONG; quality of evidence: HIGH).
The role of liver biopsy
Modern imaging techniques have reduced the role of liver biopsy for diagnosis. A retrospective study of 138 patients with cholangiographic features of PSC concluded that liver biopsy rarely added diagnostic information in classic PSC.41 Liver biopsy should be considered when histopathology would help clarify diagnosis or alter management such as when there is a clinical suspicion of IgG4-SC, PSC overlap/variant syndromes and for diagnosis of small duct PSC. Liver biopsy may also help in otherwise unexplained cholestasis.
The hallmark of PSC on histological assessment is concentric ‘onion skin’ periductal fibrosis, but this is often not present on small liver biopsy specimens. Other features include bile duct proliferation, chronic periportal inflammatory change, cholangioectasia, ductopenia and varying degrees of fibrosis and cirrhosis.5 In practice, histological assessment is often non-specific, demonstrating general features of cholestasis. One recognised system describes four stages: (1) periportal inflammation, (2) periportal fibrosis, (3) ductopenia and bridging fibrosis and (4) cirrhosis.42
Recommendation 3: We recommend that liver biopsy is normally reserved for possible small duct PSC, assessment of suspected possible overlap variants or instances where the diagnosis is unclear (strength of recommendation: STRONG; quality of evidence: MODERATE).
Non-invasive assessment of liver fibrosis
While liver biopsy does provide information on the stage of liver fibrosis,43 there has been increasing interest in the value of non-invasive assessment in patients with PSC. One retrospective study highlighted the strong correlation between transient elastography and histological stage of liver fibrosis, as well as the prognostic significance.44 Serological assessment of liver fibrosis using the enhanced liver fibrosis test correlates with elastography and helps to stratify prognosis in patients with PSC.45 Both these modalities are undergoing further evaluation, and recent reports from a larger cohort suggest they may be effective markers of fibrosis and disease progression.46 Magnetic resonance elastography is also emerging as a possible non-invasive marker of cirrhosis in PSC.47 European Association for the Study of the Liver (EASL) clinical practice guidelines recommend the use of non-invasive markers for monitoring the degree of liver fibrosis, but evidence specifically related to patients with PSC is still evolving.
What other conditions should be considered in the differential diagnosis of PSC?
The main differential diagnoses for PSC include causes of secondary sclerosing cholangitis listed in box 1.
Overlap syndromes
PSC with additional features of AIH
There is a reported 1.4–17% overlap of AIH in adults diagnosed with PSC.48–50 Conversely, a prospective study of MRCP and liver biopsy in 59 patients with AIH demonstrated features of PSC in 1.7%.51 These patients typically have cholangiographic features of PSC in combination with findings suggestive of AIH, including younger age, higher transaminases, elevated immunoglobulins, positive ANA, SMA and/or liver/kidney microsomal antibodies and mixed histopathological changes with interface hepatitis as well as the typical biliary pathology of PSC.
Patients who fulfil the diagnostic criteria of AIH published by the International Autoimmune Hepatitis Group respond to treatment with steroids and have a better prognosis than classic PSC, but worse than non-overlap AIH.48 52 An AIH/PSC overlap syndrome is more common in children (where it may be labelled autoimmune sclerosing cholangitis (ASC)), with cholangiographic features of sclerosing cholangitis reported in up to 49% of children with antibody-positive AIH.53 We recommend management of PSC with additional features of AIH according to the EASL guidelines on the management of AIH.54 The importance of identifying an AIH overlap syndrome is due to the potential therapeutic benefit of immunosuppression, and hence liver biopsy is recommended for those with significantly raised transaminases, immunoglobulins or positive AIH autoantibodies (ALT >5 x upper limit of normal (ULN), IgG >x2 ULN, positive ANA, SMA and/or liver/kidney microsomal antibodies).54 Some patients with features of AIH overlap syndrome progress to a more typical PSC phenotype.55 In this situation, ongoing treatment with immune suppression may not be effective and patients may require repeat assessment with cholangiography and consideration of repeat biopsy.
Other overlap syndromes
A PSC/PBC overlap syndrome has been reported in only small case series.56 57 This may reflect the diagnostic difficulty in those with small duct PSC where the classic cholangiographic features are absent and liver biopsy is often not diagnostic. AMAs are positive in <5% of cases of PSC.58
It is not clear whether IgG4-SC can be an overlap syndrome or if it represents a separate condition with similar clinical features. However, a subset of patients with a diagnosis of PSC do have elevated serum levels of IgG4 as discussed elsewhere in the guidelines26 59
What other conditions may be associated with PSC
PSC in inflammatory bowel disease
Abnormal liver biochemistry is common in patients with IBD. In a cohort of 544 patients with IBD, 29% had at least one abnormal liver biochemical test, but only 5.8% had a clinical diagnosis of chronic liver disease (biopsy was not required for diagnosis in this series). When performed in a subset with a suspicion of biliary disease, cholangiographic features of PSC were present in 4.6% of all patients.60 In a recent study, 7.5% of patients with longstanding IBD (over 20 years' duration) with normal liver biochemistry, had evidence of cholangiopathy (9% of Crohn’s disease, 6.8% ulcerative colitis), indicating that PSC may be underdiagnosed within cohorts of patients with IBD.61
IBD is present in 62–83% of patients with PSC of Northern European descent, but rates are as low as 21% elsewhere in the world.8 11 16 62–65 Patients often have extensive colitis, which may be of ulcerative colitis or Crohn’s colitis type. Rectal sparing and backwash ileitis are more common in IBD associated with PSC.66 PSC may be diagnosed before IBD but generally IBD is diagnosed some years before the identification of PSC. Despite potential mechanisms linking active colonic inflammation with the aetiology and activity of PSC, this has never been properly demonstrated. Clinically, the activity of IBD can follow an unpredictable course. Patients with PSC and IBD often describe minimal symptoms even in the presence of endoscopically and biopsy proven active IBD. Treatment of active colitis appears to have no impact on the progression of PSC.67 Case series also show that patients can develop changes of PSC years after colectomy for ulcerative colitis. A number of small case series have described the pattern of IBD in PSC with and without liver transplantation and/or immunosuppression, demonstrating some cases of de novo presentation of IBD after transplantation or a paradoxical worsening of disease activity after liver transplantation despite immunosuppression.68–70 Conversely, other transplant series report a milder course of IBD in those with more progressive PSC and/or improved IBD activity with immunosuppression after liver transplantation.71 72 The reasons for these variable reports and patterns of disease are unknown but suggest that there is at most a weak correlation between activity of PSC and IBD. IBD appears to be rare in IgG4-SC, providing additional means to help distinguish this from IgG4 + PSC.
Other associated conditions
PSC may rarely be associated with some other immune-mediated diseases, including coeliac disease, thyroid disease, Sjögren’s syndrome, type 1 diabetes mellitus, systemic sclerosis, retroperitoneal fibrosis, autoimmune haemolytic anaemia, sarcoidosis and rheumatoid arthritis. An association with these conditions is uncommon and some may relate to IgG4-RD misdiagnosed as PSC (eg, when associated with retroperitoneal fibrosis).
What is the natural history of PSC?
The natural history of PSC is variable and often unpredictable. Most patients are diagnosed in the fourth or fifth decades of life. The mean age of diagnosis is between 32 and 41 years.11 15 73 PSC is uncommon in childhood. Men are affected more commonly, with a male to female ratio of 2:1.
The mean time from diagnosis to death or liver transplantation is 10–22 years.8 12 15 73 74 It should be noted that most published data come from tertiary referral units and probably overestimate the risk of complications and death. A population-based assessment of natural history in Holland demonstrated improved prognosis in the overall PSC population compared with those in liver transplant centres, with a median time from diagnosis to death or transplantation of 21 years and 13 years, respectively.12 Asymptomatic patients are reported to have a better prognosis, but this is probably due in part to lead time bias with diagnosis at an early disease stage. Historically and before liver transplantation, most patients died of complications of cirrhosis. In more recent series, most deaths are due to CCA (58%), liver failure (30%) and variceal bleeding (9%).15 Patients with small duct PSC appear to have a better prognosis and a very low risk of developing CCA, but a significant minority (23%) will develop cholangiographic features of classic PSC over time.75 76 Large retrospective series suggest that patients with PSC and Crohn’s disease have a better prognosis than those with ulcerative colitis.77
These studies have been reinforced by the recent International PSC Study Group (IPSCSG) cohort study of 7121 patients, of whom 2616 progressed to liver transplantation or death (median 14.5 years); and 721 developed hepato-pancreato-biliary malignancy, mainly CCA (n=594) (incidence rate: 5.4 and 1.4 per 100 patient-years, respectively). Of these patients, 65.5% were men, 89.8% had classic/large duct disease and 70.0% IBD.78
Prognostic scoring systems
It is difficult to predict the rate of progression or outcome for individual patients with PSC. Asymptomatic patients are likely to have a better prognosis than those with symptoms. Multivariate analyses in a number of series demonstrate clinically predictable parameters as being markers of poor prognosis. Some groups have devised prognostic models (summarised in table 1) using a variety of parameters, including age, blood results, liver biopsy staging, cholangiographic findings and complications such as a history of variceal bleeding.15–17 74 The most widely used is the revised Mayo natural history model for PSC. As with other models it has a complex formula reflecting the variability and complexity of the natural history of PSC. These models probably have little role for ordinary patient care and are rarely used in clinical practice in the UK. Their main roles are to assist in the timing of liver transplantation and for research studies. Model for End Stage Liver Disease (MELD) and UK Model for End Stage Liver Disease (UKELD) scores may be applied to patients with PSC as for patients with other causes of liver disease, but both may fluctuate highly and overestimate the stage of liver disease in view of the impact of biliary obstruction on the bilirubin component of the scores. The Child-Pugh score has been applied specifically to PSC, with 7-year survival rates of 90%, 68% and 25% for scores A, B and C, respectively.79 Prognostic models using clinical and laboratory parameters for established PSC do not vary widely from data using the simple Child-Pugh score.
Comparison of published primary sclerosing cholangitis prognostic scoring systems
Recommendation 4: We recommend risk stratification based on non-invasive assessment. Clinical scores are an emerging theme but no single method can be recommended at present to predict individual patient prognosis. Given the unpredictable disease course and the serious nature of the complications of PSC, patients should receive lifelong follow-up (strength of recommendation: STRONG; quality of evidence: VERY LOW)
Sclerosing cholangitis in children
Raised ALP is normal in growing children and adolescents, and is unreliable for screening for PSC. Abnormal liver biochemistry in children with IBD is common but most is thought not to be related to PSC. In a cohort of 300 children with IBD, sclerosing cholangitis was reported in 6%, with a persistently raised γ-glutamyl transpeptidase being the most predictive marker.80 The proportion of children with abnormal liver biochemistry who develop features suggestive of PSC is not well reported. A prospective study of 55 consecutive children presenting with abnormal liver biochemistry and positive autoantibodies suggests that the distinction between classical PSC and AIH is much less clear in the paediatric population.53 Half of these children with AIH also had changes of sclerosing cholangitis at cholangiography. Most had features of an overlap syndrome with positive autoantibodies, elevated transaminases, elevated immunoglobulins and mixed histological findings of interface hepatitis and portal inflammation. The disease tends to progress to a more classic PSC pattern and become resistant to immunosuppressive treatment in the adult years. Classic PSC has also been described in children but appears to be rare. Some have therefore suggested that PSC is a ‘sequential syndrome’ or long-term consequence of damage from childhood AIH.81 The term autoimmune sclerosing cholangitis (ASC) has been used in children, but whether this is an early phase and/or the same condition as adult PSC remains unclear. MRCP is recommended in children with AIH that responds poorly to medical treatment in order to screen for changes of sclerosing cholangitis.
Children often require liver transplantation at a young age and have a high rate of disease recurrence in the graft.82 Children with classic PSC have a disease pattern mirroring that of adults with a poor response to treatment and a median survival before developing significant events or transplantation of 10–12 years.83 84 The outcome of classic PSC in children is worse than for children with steroid-responsive AIH or ASC, resulting in shorter transplant-free survival (78% at 5 years compared with 87–90% for AIH/ASC).85
Adolescents with PSC should, where possible, be managed in transition clinics before long-term management in adult clinics.
How should patients with PSC be managed?
Drug therapies
Ursodeoxycholic acid (UDCA)
UDCA is a hydrophilic bile salt used to treat cholestatic liver diseases, including PBC. Retrospective observational studies from centres with high UDCA use demonstrated worse actuarial survival in comparison with predicted survival using PSC prognostic models, suggesting a lack of therapeutic benefit from UDCA.15 A number of randomised controlled trials have been performed, but these have been generally small (n=6–110 patients) with a short follow-up of usually 12–24 months (range 12–60 months) and hence underpowered for identification of clinical events.86–92 Overall, the early studies using low doses of 10–15 mg/kg demonstrated improvement in liver biochemistry but not of liver histology and none have shown improvement in outcome measured by death or transplantation. Three small pilot trials of higher doses (20–30 mg/kg) have been published.88 93 94 All resulted in improvement of liver biochemistry and two included liver biopsy in the final clinical evaluation. One of these showed a non-significant improvement in histological score in the high dose (30 mg/kg) group (n=9) and the other (n=21) demonstrated improvement in Ishak staging in 3 out of 11 patients (p=0.05) and cholangiographic findings (p=0.015) in two patients at 2 years. None showed improved outcome, but these pilot studies were not sufficiently powered to demonstrate a survival benefit.
In a large, but still underpowered study of 219 patients randomised to moderate dose UDCA (17–23 mg/kg) or placebo for 5 years,95 there was no significant difference in outcomes between the two groups, including symptoms, liver biochemistry, CCA, death or transplantation, although there was a trend towards reduction in mortality or transplantation in the UDCA group (7.2% vs 10.9%, p=0.4). Liver biopsy was not included in the protocol. A further, large, multicentre study of high-dose UDCA (28–30 mg/kg) in 150 patients with PSC was terminated early after results showed higher rates of serious adverse events and primary endpoints of death, liver transplantation and development of varices in the UDCA-treated group.96 Meta-analyses of published data report no benefit from UDCA in patients with PSC.97 98 One uncontrolled study examined the effect of stopping UDCA in patients already established on treatment and demonstrated worsening of liver biochemistry and pruritus after stopping treatment, but the study was not able to assess the effect on longer-term outcomes.99
In small duct PSC, small case series suggest that UDCA improves liver biochemistry but has no effect on development of complications, progression to classic large duct PSC or risk of death or transplantation.76 100
Overall it appears that UDCA improves liver biochemistry, but there is no evidence that it improves outcome and may be harmful in high doses.
Recommendation 5: We recommend that UDCA is not used for the routine treatment of newly diagnosed PSC (strength of recommendation: STRONG; quality of evidence: GOOD). For patients already established on UDCA therapy, there may be evidence of harm in patients taking high dose UDCA 28–30 mg/kg/day (strength of recommendation: WEAK; quality of evidence: LOW).
Does ursodeoxycholic acid reduce cancer risk in PSC?
Early evidence suggested that patients with PSC treated with UDCA had a lower incidence of colorectal cancer than untreated patients. A retrospective study of 52 patients treated with UDCA for PSC followed up for >10 years showed a significant reduction in the incidence of colonic dysplasia or colorectal cancer (10% vs 35%).101 A second, cross-sectional study, reported the prevalence of colonic dysplasia or malignancy in 59 patients with PSC undergoing surveillance colonoscopy. A comparison of those treated or not treated with UDCA suggested a significant protective effect of UDCA on risk of colonic dysplasia or colorectal cancer (OR=0.14, 95% CI 0.03 to 0.64).102 However, a further retrospective study reported no difference in the rate of colorectal cancer or dysplasia in those treated with UDCA (n=28) compared with PSC controls not using UDCA (n=92).103 A randomised controlled trial of 1285 patients (without PSC) undergoing surveillance colonoscopy following polypectomy showed a significant reduction in the risk of high-grade dysplasia in recurrent adenomas in those patients treated with UDCA (OR=0.61, p=0.03). However, the overall incidence of new adenomas was not statistically different (p=0.31) between UDCA treated (41%) or untreated groups (44%).104 A randomised controlled trial (n=98) of UDCA (17–23 mg/kg) for the treatment of PSC reviewed the incidence of colorectal neoplasia as a secondary endpoint at almost 5 and 15 years. The rates of neoplasia were high but no difference was seen between the UDCA treated and untreated groups at either 5 years (13% and 16%) or 15 years (27% and 30%).105 One study reported a higher rate of colorectal cancer associated with the use of UDCA106 Two meta-analyses report no significant effect of UDCA on rates of colorectal neoplasia in patients with PSC, although there was a trend towards lower rates of neoplasia in patients taking low-dose UDCA.107 108
There is little evidence for a beneficial effect of UDCA in reducing the risk of CCA, with no placebo controlled trials specifically examining this question. Two observational studies reported a lower incidence of CCA in patients taking UDCA in comparison with previously reported incidence rates.109 110 The largest of these studies followed up 150 patients for a median of 6.4 years, with CCA developing in five patients (3.3%), which represents about half the expected incidence of CCA in PSC. The large US randomised control trial of UDCA versus placebo was terminated early, but also failed to show a significant difference in the rate of either CCA (2.6% vs 2.7%) or colonic dysplasia in either the UDCA or placebo arms, respectively, at 5 years.96
Recommendation 6: We recommend that UDCA is not used for the prevention of colorectal cancer or cholangiocarcinoma (strength of recommendation: STRONG; quality of evidence: HIGH).
Immunosuppression and other treatments
Despite the presumed immune-mediated disease process in PSC, clinical experience of treating those with active colitis using steroids and other immunosuppressant agents has not demonstrated improvement in PSC disease activity or outcome. Small randomised trials have investigated the role of prednisolone, budesonide, colchicine, penicillamine, azathioprine, ciclosporin, methotrexate, mycophenolate and anti-tumour necrosis factor monoclonal antibodies. There is no evidence that any of these drugs are effective and therefore none can be recommended for the treatment of classic PSC.111 112 Nevertheless, some of these drugs may have a role in an overlap syndrome, since paediatric patients and those with additional features of AIH are more likely to respond to immunosuppressive treatments.50 A retrospective study in adults suggested a beneficial role of steroids in a subgroup with additional features of AIH.113 Those with good evidence of PSC and additional features of AIH should be treated similarly to those with classic AIH.114 The choice of the most appropriate systemic steroid therapy is not clear.
Steroids have been given to the subset of patients with PSC and a raised serum IgG4 (after exclusion of IgG4-SC). In a small study of 18 patients, steroids led to a fall in bilirubin in 9/10 patients with raised bilirubin, and a significant fall in ALP, but steroid-related side effects and post-steroid relapse were common.59
A review of small case series with limited evidence suggests modest improvement in liver biochemistry in patients treated with vancomycin.115 These data may justify a larger clinical trial but currently do not support the use of vancomycin (or other antibiotics) for treatment of PSC liver disease in the absence of cholangitis.
Recommendation 7: We recommend that corticosteroids and immunosuppressants are not indicated for the treatment of classic PSC (strength of recommendation: STRONG; quality of evidence: HIGH). In those patients with additional features of AIH or IgG4-SC, corticosteroids may be indicated (strength of recommendation: STRONG; quality of evidence: MODERATE).
Role of endoscopy, ERCP and endotherapy
Joint European Society of Gastrointestinal Endoscopy (ESGE) and EASL guidelines on the role of endoscopy in patients with PSC have recently been published and should be reviewed along with these guidelines4
Oesophageal and gastric varices have been reported in 7–36% of patients with PSC.15 116 Screening and appropriate treatment of varices should be considered in those with evidence of cirrhosis and/or portal hypertension, such as those with thrombocytopenia, jaundice and an elevated AST/ALT ratio or those with evidence of cirrhosis on elastography or imaging.116 117
Colitis is common in patients with PSC and patients may have few or no symptoms. A full colonoscopy with colonic biopsies is therefore strongly recommended after a diagnosis of PSC in order to identify occult IBD, and to determine the need for colonoscopic surveillance of colorectal neoplasia.
ERCP has historically been the preferred investigation for suspected PSC, but carries significant risks. One retrospective study of almost 9000 ERCPs, including 141 patients with PSC, reported higher rates of pancreatitis (7.8%), cholangitis (7.1%) and overall complications (18%) in patients with PSC compared with other indications.118 However, other large series reported a relatively low complication rate of 4.3% from ERCP in patients with PSC (pancreatitis 1.2%, cholangitis 2.4%, bleeding 0.7%).119 Patients with PSC should ordinarily not undergo ERCP until there has been expert clinico-radiological assessment to justify endoscopic intervention.
In PSC, after ERCP, cholangitis rates of up to 36% are reported in case series.120 121 National Institute for Health and Care Excellence (NICE) and BSG guidelines advise that prophylactic antibiotics are required if complete biliary drainage at ERCP is unlikely to be, or is not, achieved. PSC with intrahepatic and/or extrahepatic stricturing is considered such a situation and so prophylactic antibiotics should be used for ERCP in patients with PSC.122 The recommended antibiotic regimens vary according to local policies but commonly include co-amoxiclav, quinolones, gentamicin or cephalosporins for 3–5 days. There is no role for the addition of antibiotics to contrast agents used during ERCP.123
Recommendation 8: We recommend that endoscopic screening for oesophageal varices should be done in line with international guidelines where there is evidence of cirrhosis and/or portal hypertension (strength of recommendation: STRONG; quality of evidence: HIGH).
Recommendation 9: We recommend that colitis should be sought in all patients with PSC using colonoscopy and colonic biopsies (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 10: We recommend that patients with suspected PSC undergoing ERCP should receive prophylactic antibiotics (strength of recommendation: STRONG; quality of evidence: MODERATE).
Dominant bile duct strictures
It is important that in patients presenting with signs of biliary obstruction and/or those who develop changing symptoms or evolving abnormalities in laboratory investigations, non-invasive investigations such as MRCP, dynamic liver MRI and/or contrast CT should be performed and reviewed by a hepatopancreaticobiliary (HPB) MDT before any high-risk invasive endoscopic interventions.
A dominant stricture is often not simple to define in clinical practice but a pragmatic definition is of functional stenoses of the major bile ducts with signs of biliary obstruction shown by worsening liver biochemistry and/or proximal biliary dilatation or symptoms of cholestasis. The prevalence of dominant bile duct strictures in PSC is 36–50%.15 121 124 125 Patients with dominant strictures have significantly worse outcomes than those without, even when regular endoscopic treatment of stenoses is applied and CCA is excluded.125
Decision-making about intervention for dominant strictures is important but complex. A common consensus has been that patients with PSC who do not have significant jaundice and/or have not had episodes of cholangitis in the presence of assumed functionally significant extrahepatic strictures should avoid ERCP unless a clinical suspicion of CCA based on non-invasive imaging is high.
Differentiating benign from malignant causes of dominant strictures is crucial but difficult. Biliary brush cytology is the standard investigation for suspicious biliary strictures but despite excellent specificity, its sensitivity is poor. A systematic review of the published literature (n=747) on the use of biliary brushings in the diagnosis of CCA in PSC reported a sensitivity of 43% and specificity of 97%.126 The sensitivity of cytology from bile aspirates is lower. A single-centre prospective study of systematic biliary brushings at index ERCP in 261 patients with PSC reported malignancy or dysplasia suspicious for malignancy in 7%.127 Additional cases of biliary dysplasia were identified in explants of patients who underwent transplantation. Some international centres use dysplasia in brushings as a marker of in situ carcinoma and refer these patients for consideration for liver transplantation.127 128 However, CCA remains a contraindication to liver transplantation in the UK. Other markers using fluorescence in situ hybridisation (FISH) analysis in biliary brushings or KRAS and p53 in bile have been evaluated, but are not sufficiently sensitive or specific to be useful as screening or diagnostic tests.129–132 A meta-analysis of 828 patients with PSC undergoing assessment by FISH demonstrated sensitivity and specificity of 68% and 70%, respectively.133 Other approaches such as using a panel of biomarkers may show more promise in the future.
Another approach to tissue diagnosis at ERCP is fluoroscopically guided intraductal biopsy without direct cholangioscopy. Selected studies (not PSC specific) demonstrate relatively high rates of tissue confirmation of malignancy (70%) using larger biopsy forceps.134 Rates for confirmatory tissue diagnosis can be improved by multimodal sampling using brushings, biopsy and EUS.135 Similarly, the use of multiple crushed biopsy specimens analysed immediately by a pathologist during the ERCP procedure may improve diagnosis rates.136
Intraductal cholangioscopy can aid the diagnosis of indeterminate biliary strictures. Early case series, including patients with PSC, suggested that very high sensitivities (92–100%) could be achieved for the diagnosis of malignant biliary strictures using direct visualisation without biopsy, although with a decline in specificity to 87–93%.137 138 Larger, more recent studies using video cholangioscopy and multicentre registries have reported high sensitivities (62–99%) and specificities (64–100%) for the diagnosis of biliary strictures.139–143 A UK multicentre experience of cholangioscopy for the diagnosis of CCA in PSC and IgG4- related cholangitis suggests that in comparison with investigation of possible CCA in patients without cholangiopathy, it is similarly efficacious (sensitivity 50%).144 Technological developments and the wider availability of cholangioscopy will probably lead to an increasing role for this procedure in the assessment of strictures in PSC.145 Early studies on the addition of adjunctive techniques to EUS and cholangioscopy, such as intraductal chromoendoscopy, narrow band imaging and confocal laser microscopy, are emerging.139 146–148 These techniques are limited to specialist centres, but availability is expanding.
Endotherapy of dominant bile duct strictures
The exact role of endoscopic therapy in the management of dominant strictures in PSC remains incompletely understood. Investigations in animals and humans suggest that decompression of biliary obstruction prevents further damage and can reverse fibrotic liver disease.149 This is supported by data demonstrating that patients with PSC who achieve normalisation or near normalisation of ALP have improved outcomes compared with those who do not.24 25 It is clear that endoscopic treatment of biliary strictures often improves liver biochemistry and pruritus, and may reduce the risk of recurrent cholangitis. Consensus has been for repeated endoscopic intervention (usually stricture dilatation ± stenting) of dominant biliary strictures in those with symptomatic disease.150–152 Evidence from studies comparing jaundice, cholangitis, transplantation and actuarial survival rates with figures from prognostic models, tend to suggest a benefit of endoscopic intervention for dominant biliary strictures.124 153–155 In contrast, a Swedish study comparing liver biochemistry in those with and without dominant strictures suggested that variations in cholestasis and jaundice are a feature of PSC liver disease and are not a direct consequence of endotherapy of dominant strictures.121
The optimum method and frequency of dilatation of dominant strictures is unclear. Plastic stent insertion with or without prior stricture dilatation has been commonly used. The difficulty with this approach is that further ERCPs are required to remove or replace the stent and there is a high rate of stent occlusion and/or cholangitis within 3 months of insertion. One uncontrolled study of short-term stenting (mean 9 days) reported improved outcome, particularly for resolution of jaundice and symptoms of cholestasis (81% compared with 57% in historical controls undergoing 2–3 monthly elective stent changes).155 Other studies have compared the role of stenting with balloon dilatation, with similar efficacy and lower rates of complications such as cholangitis associated with balloon dilatation alone (18% vs 50%).156 Multiple dilatations are usually required over months or years in order to maintain patency once dominant strictures are identified. A large (n=171) uncontrolled prospective study of patients with PSC included 96 patients with dominant strictures undergoing regular balloon dilatations over a median follow-up of 7 years.157 Over 500 dilatations were performed (only five stents inserted) with low complication rates of 2.2% for pancreatitis, 1.4% for cholangitis and 0.2% for bile duct perforation, and 5- and 10-year transplant free survival rates of 81% and 52%, respectively.
Balloon dilatation in preference to stenting has been advised in European and American guidelines on the management of patients with PSC.1 2 4 Some strictures do not open satisfactorily with balloon dilatation alone and stent insertion is usually recommended in these cases. A meta-analysis in other benign causes of biliary stricture and/or obstruction, shows that insertion of multiple plastic stents offers higher rates of relief of cholestasis (94% vs 60%) and lower complication (mostly cholangitis) rates (20% vs 36%) than single stent insertion.158 In a multicentre randomised trial of patients with PSC and a dominant stricture (n=65), short-term stents were not superior to balloon dilatation and were associated with significantly higher complications of pancreatitis and cholangitis in the stent group (45%) than in the balloon dilatation group (7%)159
Fully covered self-expandable metal stents are now well established in the management of benign biliary strictures of different aetiologies.160 161 Case series including small numbers of patients with PSC also suggest a role for these stents in dominant strictures below the liver hilum due to PSC.161–163
Some strictures are not amenable to, or do not require, endoscopic intervention. In these patients, careful consideration should be made about a conservative, radiological or surgical (including liver transplantation) approach to treatment before ERCP is performed. If ERCP is performed in the presence of dominant stricture, it is important that consideration is made of possible CCA and that appropriate sampling is undertaken if there is any clinical suspicion of malignancy.
Recommendation 11: We recommend that non-invasive investigations such as MRCP, dynamic liver MRI and/or contrast CT should be performed in patients who have new or changing symptoms or evolving abnormalities in laboratory investigations (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 12: We recommend that patients with PSC should ordinarily not undergo ERCP until there has been expert multidisciplinary assessment to justify endoscopic intervention (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 13: We recommend that in patients undergoing ERCP for dominant strictures, pathological sampling of suspicious strictures is mandatory (strength of recommendation: STRONG; quality of evidence: STRONG).
Recommendation 14: We recommend that in patients undergoing ERCP for dominant strictures, biliary dilatation is preferred to the insertion of biliary stents (strength of recommendation: STRONG; quality of evidence: MODERATE).
Specialist referral
We suggest that care provision should involve a partnership between patients, primary care and hospital-led specialty medicine. Care delivery for an individual patient should encompass patient risk assessment, symptom burden and how local services are configured. All patients should have at least an annual review, which should become more frequent as required if symptoms or complications develop. The timing of referral to specialist regional HPB units will vary and depend on physicians’ experience in caring for patients with PSC, and in biliary intervention. In practice, referral will be at the point where a patient’s clinical management is beyond both the local expertise and knowledge of their responsible physician and team. As a general rule, all symptomatic patients should be under the care of a specialist clinician or HPB centre with an interest and experience in managing PSC. In the absence of effective medical treatment and with the unpredictable natural history of PSC, it is important that patients are referred early for consideration of liver transplantation. Patients with jaundice from suspected parenchymal disease, rising liver disease scores (MELD >11, UKELD >46) or complicated biliary strictures, require discussion with specialist units for consideration of endoscopic, radiological and/or surgical biliary intervention or liver transplantation. Other reasons to consider referral include persistently raised ALP levels,23 transient elastography of >9.9 kPa44 or an enhanced liver fibrosis test result of >10.6.45 Patients with early, asymptomatic, stable disease can usually be managed by non-specialist gastroenterologists or other clinicians with adherence to management guidelines. Centres with a particular interest in PSC may be undertaking clinical trials, and patients should be offered the chance to enter into such trials. All patients with suspected CCA or other malignancies should be referred to the appropriate regional multidisciplinary cancer meeting for review.
Recommendation 15: We suggest that provision of care should involve a partnership between patients, primary care and hospital-led specialty medicine with consideration made with regard to patient risk assessment, symptom burden and how local services are configured (strength of recommendation: WEAK; quality of evidence: LOW).
Recommendation 16: We recommend that patients with symptomatic, evolving or complex disease should be referred for expert multidisciplinary assessment. Patients with early, stable disease can be managed in general clinics (strength of recommendation: STRONG; quality of evidence: LOW).
Recommendation 17: We suggest that patients with PSC meeting inclusion criteria should be offered referral to a centre participating in clinical trials (strength of recommendation: WEAK; quality of evidence: LOW).
Liver transplantation
Advanced liver disease secondary to PSC is a well-established indication for liver transplantation.164 165 Patients receiving a transplant owing to PSC have excellent outcomes compared with many other indications. The European Liver Transplant Registry (which includes the UK data) has recorded 1-, 3- and 5-year survival rates of 86%, 80% and 77%, respectively, in patients transplanted between 1988 and 2005. Data from the US registries of more recent cases indicate even better survival, although the results for PSC are poorer than for PBC, even though patients with PSC were younger.166 The optimal timing of referral for transplant assessment is difficult because jaundice can be caused by both liver failure and/or biliary obstruction, which may respond to endoscopic therapy. Owing to the difficulties in predicting outcome and the particular risks of severe recurrent cholangitis and CCA, in some national allocations schemes, patients with PSC are given exemption points to balance their risk compared with other causes of cirrhosis when using scoring systems such as MELD. Some have advocated early transplantation in PSC because of the risk of CCA, but the risk of recurrent PSC in the graft and long-term survival data being poorer than a conservative approach in early disease do not favour this opinion.166
A large study analysing the American transplant United Network for Organ Sharing (UNOS) database reported a lower death rate for patients with PSC on the waiting list (13.6%) than for other indications (20.5%).167 A variable potentially skewing these data is the higher rate of living donor transplants in the PSC population, resulting in dropout from the standard waiting list.167 The most common cause of death for patients with PSC on a transplant waiting list is development of cholangiocarcinoma. Complications of portal hypertension are much lower than for other listing indications, which has been proposed as the reason that patients with PSC on the waiting list have a more favourable outlook than patients with other indications.167 168 Furthermore, bacterial cholangitis does not appear to be associated with an increased risk of waiting list removal for death or clinical deterioration, calling into question the rationale for granting additional exemption points for this indication.169
In general, patients with PSC should be referred early for consideration of transplantation if there is cirrhosis and/or portal hypertension associated with any complications or when the UKELD or MELD scores rise towards minimal listing criteria (currently 49 and 15, respectively).170 171 The presence of intractable pruritus (uncommon in PSC) and recurrent cholangitis are also accepted indications for orthotopic liver transplant within the UK and should justify earlier referral for consideration of liver transplantation (http://odt.nhs.uk/pdf/liver_selection_policy.pdf).
Recurrence of PSC in transplanted livers is seen in 10–40% of cases.165 172–177 The main identifiable risk factors for recurrent disease are male sex, the presence of an intact colon and/or active colitis after transplantation.174–176 178 There is no evidence that post-transplant immunosuppression with single or multiple agents reduces the risk of recurrent disease, although most units favour a triple immunosuppression regimen. Diagnosis of recurrence is based primarily on clinical findings of typical cholangiopathy (either by radiographic or liver biopsy assessment) after 90 days in the absence of other causes, including hepatic artery ischaemia, ABO incompatibility and established ductopenic rejection. Recurrent disease can be difficult to treat and necessitates retransplantation in approximately 50% of cases. Duct to duct biliary anastomosis should be undertaken whenever possible as it is associated with a reduced risk of cholangitis.179 Anastomotic or de novo dominant strictures are usually managed with balloon dilatation and/or biliary stent insertion (plastic or possibly removable fully covered metal stents) but occasionally require surgical repair.
As for other immune-mediated liver diseases including AIH and PBC, there is a higher frequency of acute and chronic rejection, with reported rates of early acute rejection between 39% and 71%.165 176 In a retrospective series of over 3000 patients included in the American UNOS database and a smaller UK series166 174 graft dysfunction in PSC, from whichever cause resulted in a retransplantation rate of 12.4– 13.5%.
Other subjects relevant to transplantation include complications of coexisting IBD which can make surgery more complex, the need for annual surveillance for colorectal cancer (predicted colorectal cancer incidence of 1% a year associated with PSC and long-term immunosuppression) and the higher rate of recurrent disease in those with IBD.176 180 For these reasons, some centres have advocated colectomy at the time of liver transplantation, but this remains contentious in the absence of colonic dysplasia or difficult to control colitis before transplantation. Patients with IBD being considered for transplantation should stop smoking and their IBD should be in remission by the time of transplantation as both these measures improve the outcome from liver transplantation.181
Recommendation 18: PSC is a well-recognised indication for liver transplantation. We recommend that eligibility and referral should be assessed in line with the national guidelines (strength of recommendation: STRONG; quality of evidence: HIGH).
How do I manage complications of PSC?
Cholangitis
Cholangitis is a common complication of PSC. Bacterobilia is reported in 55% of patients at the time of liver transplantation, increasing to at least 77% in those who have predisposing factors such as biliary strictures or previous biliary instrumentation.182 Cholangitis can present without significant change in baseline liver biochemistry as infections can be limited to small liver segments. A clear risk factor for cholangitis or positive bile cultures is previous ERCP with or without therapeutic intervention, with the highest risk seen when stents are left in situ. A study (not in PSC) reported a positive bile culture rate of 98% in those with a stent in situ and 55% in those without.183 The number and variety of bacterial isolates were inversely proportional to the time since the last ERCP.182 Another potential source of cholangitis is portal bacteraemia, which has been described in patients with active colitis.184 ERCP is a risk major factor for cholangitis in PSC and antibiotics should be routinely used as recommended above.
Biliary infections are often polymicrobial, but the most common organisms are Eschericia coli, Klebsiella, Enterococcus, Clostridium, Steptococcus, Pseudomonas and Bacteroides species.185 The choice of antibiotic agent should be directed by local practice after taking into consideration the history, severity of liver or renal disease and bacterial sensitivities. A common first-line agent for mild episodes is a fluoroquinolone such as ciprofloxacin. More severe cases are usually treated with intravenous cephalosporins or extended spectrum penicillins with the addition of anaerobic cover.185 186 Candida species have been isolated from the bile of 8/67 (12%) patients with PSC undergoing ERCP.187 However, the clinical relevance of fungal contamination of bile is unknown. Antifungal therapy should be considered in those with cholangitis not responding to antibiotic therapy.
Patients with severe acute cholangitis and dominant bile duct strictures require urgent biliary decompression, as the mortality in those untreated is high.186 In patients with recurrent cholangitis secondary to complex intrahepatic cholangiopathy, rotation of antibiotics is occasionally used. This can lead to multiple antibiotic resistances and should be avoided where possible. Where this option is considered, expert multidisciplinary assessment, including formal microbiology advice, should be sought.
Cirrhosis, portal hypertension and liver failure
In an observational series of 174 patients with PSC who underwent a liver biopsy, advanced fibrosis or cirrhosis was found in 43% of patients with asymptomatic disease, and in 69% of those who were symptomatic64; 25% died of liver failure. Other studies have shown similar results.15 188 It is likely that these series are subject to referral bias with patients at a more advanced stage than many patients routinely followed up in local centres, but they indicate a high prevalence of advanced parenchymal liver disease in PSC. The true prevalence of portal hypertension is not known, but extrapolating data from clinical findings, such as the presence of splenomegaly and oesophageal varices, suggests that clinically significant portal hypertension is present in 30%.15 188
Metabolic bone disease
As with other cholestatic liver diseases, osteopenia and osteoporosis are common in PSC.189 190 In a study of 237 patients who underwent annual measurement of bone mineral density, 15% had evidence of osteoporosis, equating to a 24-fold risk of osteoporosis compared with an age-matched population.191 In this study, the presence of older age (>54 years), low body mass index (<24 kg/m2) and presence of IBD were strong risk factors of low bone density (prevalence of 75% with all three risk factors and 3% with none), but interestingly, cumulative dose of corticosteroids was not. Patients may also have coexistent vitamin D deficiency, but overt osteomalacia is uncommon. UK guidelines on the management of osteoporosis associated with chronic liver disease advise that all patients should receive lifestyle advice and those with cirrhosis or advanced cholestasis should have bone densitometry performed every 2 years.192 In practice, young patients with early disease are at low risk of low bone density and will not usually require formal testing. Patients with a high risk of bone disease and those requiring steroid treatment for IBD or liver transplantation should be treated with daily vitamin D 400 IU (10 μg) and calcium supplements if calculated dietary calcium intake is insufficient. Those with confirmed osteoporosis should be treated according to BSG and NICE guidelines and fracture risk scores (http://www.nice.org.uk).192
Recommendation 19: We recommend that all patients with PSC should have a risk assessment for osteoporosis. Once osteoporosis is detected, treatment and follow-up should be in accordance with national guidelines (strength of recommendation: STRONG; quality of evidence: MODERATE).
Poor nutrition and fat soluble vitamin deficiency
Poor nutrition is common in chronic liver disease and should be considered and treated appropriately in patients with PSC. Advanced cholestasis can result in malabsorption of fat-soluble vitamins. In advanced disease before transplantation, deficiency of vitamin A, D and E in 82%, 57% and 43%, respectively, are reported, but much lower levels of deficiency are seen earlier in the disease process.193 Evidence of deficiency of any measurable vitamin should lead to consideration of empirical replacement with multivitamins.
Recommendation 20: Poor nutrition and fat-soluble vitamin deficiency are relatively common in advanced PSC and we suggest that clinicians should have a low threshold for empirical replacement (strength of recommendation: WEAK; quality of evidence: MODERATE).
Fatigue and depression
Fatigue is a common symptom of patients with chronic liver disease, but no treatments have been proved to be beneficial.194 Depression is also common in people with chronic illnesses, and there are mixed reports of the association between depression and fatigue in PSC.194 195 One study directly assessing quality of life and fatigue scores in PSC, reported a lower incidence of fatigue than in the general population and when present, symptoms were associated with depression rather than severity of liver disease.194 There does not appear to be a role for the treatment of fatigue using antidepressants without clear symptoms of depression.196–198
Recommendation 21: We recommend that in patients with fatigue, alternative causes should be actively sought and treated (strength of recommendation: STRONG; quality of evidence: LOW).
Pruritus
Pruritus has a significant detrimental effect on quality of life for patients with PSC.199 The mechanism of pruritus in cholestasis remains unclear, which makes targeted treatment difficult. Antihistamines are not effective for the pruritus of cholestasis. There are few data for treatment in PSC and most recommendations are extrapolated from trials in PBC.200 Pruritus associated with advanced disease is difficult to treat medically, and treatable biliary obstructions should be sought and relieved as above. The first line of medical treatment is usually cholestyramine, colesevelam or colestipol. Care is needed to avoid administering soon before or after other medications. Further possible treatments include rifampicin and opiate antagonists such as naltrexone. The efficacy of these drugs is variable and they tend to have significant side effects. Patients with intractable pruritus not responsive to standard medical treatment should be offered referral to a specialist and/or transplant unit for further management.
Recommendation 22: We suggest that cholestyramine (or similar) is first-line medical treatment for pruritus. Rifampicin and naltrexone are second-line therapies (strength of recommendation: WEAK; quality of evidence: LOW).
Cancer
Cholangiocarcinoma (CCA)
Malignancy, particularly CCA, is now the the most common cause of death in patients with PSC who have not undergone transplantation.201 The incidence of CCA in PSC is between 0.6% and 1.5% a year, with a prevalence of 6–13% and a lifetime risk of up to 20%.15 62 64 125 188 202 203 Approximately half of cases of PSC-associated CCA are identified within a year of presentation of PSC, and may be the reason for presentation of previously unrecognised PSC.125 203–205 There is no clear evidence that the risk of developing cancer is related to the duration of PSC disease. The incidence of CCA is highest in those with dominant strictures, with up to 76% located within the perihilar region.125 CCA is rare in small duct PSC.75 76 203 206 A summary for management of suspected cholangiocarcinoma is outlined in figure 2.

{kind=link}
{kind=link}
Algorithm for the investigation of possible cholangiocarcinoma in patients with primary sclerosing cholangitis.
The usual modes of presentation are with upper abdominal pain, worsening liver biochemistry, jaundice and a raised serum CA19.9. CCA tends to spread by local invasion of the bile ducts and much less often with mass formation, so that cross-sectional imaging may fail to identify the tumour. It is difficult to distinguish benign from malignant biliary strictures by MRCP or ERCP. Endoscopic assessment of suspicious biliary strictures is discussed above. Physicians should refer patients to units with experience in ERCP and EUS if there is continuing concern about biliary malignancy in patients with PSC.
Tumour markers used in clinical practice are CA19.9 and CEA but neither is sufficiently sensitive or specific for the diagnosis of CCA either alone or in combination.207–210 These studies used primarily CA19.9 with a cut-off point of 37–186 kU/L. The sensitivity and specificity ranged between 50% and 89% and 54% and 98%, respectively. The positive predictive value is low. The main problem with this marker is that it is frequently elevated in cholestasis and cholangitis, both of which are common in PSC. Another finding was that those with high levels of CA19.9 had unresectable disease, suggesting that it is not useful in surveillance.211 The utility of CA19.9 and various imaging modalities was reported in a large series of patients with PSC; no level of CA19.9 demonstrated reliable detection of CCA.212 Early retrospective data suggest that a rising trend in CA19.9 for individual patients may be more predictive than the actual level for the presence of early cholangiocarcinoma.213 Therefore, CA19.9 may be useful for supportive evidence of CCA but is not reliable for screening or for confirmation of the diagnosis of CCA.
Early reports suggested positron emission tomography (PET) scanning may be useful for surveillance or investigation of suspected CCA.214 However, a further study of 36 patients (without PSC) with suspected CCA demonstrated a sensitivity of 85% for mass-forming tumours, 65% for metastases but only 18% for infiltrating tumours.215 One small prospective study on the use of PET in transplant assessment (n=10, four with CCA) reported a sensitivity of 75% and false-positive rates of 20% in the presence of cholangitis.216 Studies of PET scanning in sporadic CCA, demonstrate sensitivities of 61–92% and specificities of 75–93%.215 217 The detection rate falls to as low as 36–59% in cases of extrahepatic disease with infiltrating rather than mass-forming disease.218 219 Therefore, PET is not routinely recommended for surveillance or diagnosis of CCA in PSC.
Cases of suspected CCA should be referred for review in the regional specialist hepatobiliary and pancreatic MDM or liver transplant centre. Decisions on treatment depend on the stage of disease. Chemotherapy remains the main palliative treatment for patients with CCA and will be directed by the specialist MDM. Although resectional surgery may be curative, this is usually not possible in patients with PSC (particularly with hilar/intrahepatic malignancy) because of either advanced stage malignancy at diagnosis, complex biliary structuring or coexistent parenchymal liver disease precluding a major liver resection. A retrospective multicentre study of 47 patients with liver transplantation for hilar CCA (not PSC specific) reported a high rate of recurrence (34%) and poor 5-year survival (22%).220 A Canadian study of patients who received a transplant for PSC reported an 80% recurrence rate, and a median time to recurrence of 26 months in 10 patients with an incidental finding of CCA in the explant.221 UK data demonstrate similar outcomes.222
In the UK, CCA remains a contraindication to liver transplantation after poor outcomes, with high rates of recurrence reported, even in cases of incidental CCA found in liver explants.171 However, in selected cases, 3-year survival rates of 35–50% have been achieved.223 A series from the USA suggests that highly selected cases with early-stage hilar CCA have good outcomes when treated with preoperative chemotherapy and radiotherapy followed by transplantation.224 225 A more recent review of 287 patients in 12 US centres undergoing neoadjuvant chemo-radiotherapy followed by transplantation for early CCA reported a 65% 5-year recurrence-free survival.226 Decisions about transplantation when biliary dysplasia is identified at tissue sampling are complex and require in-depth discussions in a formal liver transplant MDM.
Recommendation 23: We suggest that an elevated CA19.9 may support a diagnosis of suspected cholangiocarcinoma but has a low diagnostic accuracy. Routine measurement of serum CA19.9 is not recommended for surveillance for cholangiocarcinoma in PSC (strength of recommendation: WEAK; quality of evidence: MODERATE).
Recommendation 24: We recommend that when a diagnosis of cholangiocarcinoma is clinically suspected, referral for specialist MDM review is essential (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 25: We recommend that where cholangiocarcinoma is suspected, contrast-enhanced, cross-sectional imaging remains the initial preferred investigation for diagnosis and staging (strength of recommendation: STRONG; quality of evidence: HIGH). Confirmatory diagnosis relies on histology with the approach to tissue sampling guided by MDM review. Options include ERCP-guided biliary brush cytology/FISH/ endobiliary biopsy/cholangioscopy/EUS-guided biopsy and/or percutaneous biopsy (strength of recommendation: STRONG; quality of evidence: HIGH).
Other hepatopancreaticobiliary cancers
Gallbladder polyps are more often malignant in patients with PSC than in those without PSC, and malignancy may occur even in polyps <1 cm.227 228 A study of explanted livers from patients with PSC reported a high prevalence of dysplasia or cancer of the gallbladder, with 14% of resected gallbladders having foci of adenocarcinoma.229 No studies have been carried out examining prospective ultrasound surveillance of gallbladder polyps in patients with PSC. The US guidelines recommend cholecystectomy when polyps are identified, but this should be balanced against the increased risk of surgery in patients with more advanced cirrhosis and portal hypertension.
Early reports suggested that hepatocellular carcinoma (HCC) has a prevalence of 2–4% in PSC.62 230 However, other reports suggest that the incidence of HCC in patients with PSC is extremely low, even in the presence of cirrhosis.231 Pancreatic cancer is reported to have a 14-fold standard incidence ratio compared with expected rates in the general population.62
Recommendation 26: We suggest that an annual ultrasound scan of the gallbladder should be performed in patients with PSC. If polyps are identified, treatment should be directed by specialist HPB MDM (strength of recommendation: WEAK; quality of evidence: LOW).
How should one screen for cancer in PSC
There is a high incidence of colorectal cancer in IBD associated with PSC. A Swedish population-based study reported a prevalence of 7.4%, and other observational studies suggest the cumulative risks are as high as 14%, 31% and 50% at 10, 20 and 25 years, respectively.204 232 In a meta-analysis of 116 studies, the prevalence of colorectal cancer was 3.7% with cumulative risks of 2%, 8% and 18% at 10, 20 and 30 years, respectively.62 233 A second meta-analysis confirmed an increased risk of colorectal cancer in ulcerative colitis associated with PSC and calculated a relative risk of 4.79 (95% CI 3.58 to 6.41) in comparison with ulcerative colitis without PSC.234 The risk of colorectal cancer in patients with ulcerative colitis and concomitant PSC rises further (1% a year) for those who have undergone liver transplantation.180 A case–control study compared 27 patients with PSC and IBD-related colorectal cancer with 127 cases of IBD-related colorectal cancer without PSC and reported a higher prevalence of right-sided cancers (67% vs 36%, p=0.006) in patients with PSC.235 A population-based study suggested that PSC-related colitis has a 10-fold increased risk of colorectal cancer compared with ulcerative colitis without PSC and that screening improves outcomes.12 The BSG guidelines for screening and surveillance of colorectal cancer in patients with IBD advise that patients with colitis and PSC should be screened annually from the time of diagnosis, ideally using adjunctive techniques such as dye spray to highlight dysplasia.236 Management of dysplastic polyps and foci of colonic dysplasia within segments of colitis should follow guidance laid out in other guidelines237
There is a lack of data for patients with PSC without colitis, but one observational study of 211 patients reported a 10-year risk of 2% for the development of colorectal cancer.204 A second observational study of 200 patients with PSC also reported three cases of colorectal cancer in those without evidence of IBD.201 Some clinicians undertake 1, 2, 3 or 5 yearly surveillance for all patients with PSC, but there is no evidence that outcomes are altered using any of these strategies.
There is little evidence to support the use of ERCP to screen for CCA. Surveillance with non-invasive imaging, such as ultrasound and MRCP, has not been shown to be effective and is not routinely recommended. The use of serum tumour markers, particularly CA19.9, is widely used, but there is little evidence to justify its use for reliable surveillance (see CCA above). Data presented in a large cohort of patients with PSC describe the limited utility of single and multiple biochemical and imaging modalities for the screening of CCA.212
HCC is thought to be uncommon in PSC. A retrospective cohort of 509 patients with PSC identified a high risk of colorectal cancer, CCA and gallbladder cancer, but no reported cases of HCC were identified in 119 patients with cirrhosis.231 One large retrospective study of 830 patients with PSC suggests that those underdoing surveillance imaging had earlier diagnosis and better 5-year survival rates.238
Even though there are no clear data to support particular surveillance methods for each of the main HPB cancer risks in patients with PSC, annual transabdominal ultrasonography, in addition to other clinically indicated imaging for new symptoms, is weakly recommended.
Recommendation 27: We recommend that patients with PSC who have coexistent colonic IBD should have annual colonoscopic surveillance from the time of diagnosis of colitis in line with the BSG guidelines (strength of recommendation: STRONG; quality of evidence: HIGH). We suggest that those without IBD may benefit from less frequent 5-year colonoscopy or earlier in the advent of new symptoms (strength of recommendation: WEAK; quality of evidence: VERY LOW).
Recommendation 28: We suggest that in the presence of cirrhosis, HCC surveillance should be carried out in accordance with international guidelines (strength of recommendation: WEAK; quality of evidence: LOW).
Pregnancy in women with PSC
Data on pregnancy in PSC are lacking. A German case–control series of 229 people with PSC assessed 25 pregnancies in 17 women and reported no difference in fertility rates or outcomes of pregnancy in PSC for either mother or baby compared with matched controls.239 However, in this study, 20–32% of women with PSC had a rise in liver biochemical tests during and after delivery compared with pre-pregnancy levels. A second retrospective series of 13 pregnancies in 10 women with PSC also reported no significant complications to mother or baby but did suggest a higher rate of pruritus during pregnancy, which in two cases led to early delivery of the baby.240 In neither series were there reports of gastrointestinal bleeding or other complications of portal hypertension. Data from the Swedish National Patient Register, including 229 babies born to women with PSC, reported a higher rate of caesarean section and preterm delivery but no adverse effect on outcome as measured by size, Apgar score or neonatal death.241
As discussed above, long-term UDCA is not routinely indicated for patients with PSC. However, in those women developing pruritus and worsening cholestasis in pregnancy, there is a role for short-term UDCA (10–15 mg/kg) use for symptomatic relief owing to the possibility of coexistent intrahepatic cholestasis of pregnancy, which has a much higher prevalence (1.5% according to a large prospective Swedish study) than PSC in pregnant women.242 However, there is no strong evidence to support this recommendation and clinicians should judge each case individually.
Recommendation 29: We recommend that because pregnancy in cirrhotic patients carries a higher risk of maternal and fetal complications, patients should have preconception counselling and specialist monitoring (strength of recommendation: STRONG; quality of evidence: LOW).
Patient perspectives and support groups
Patients with PSC are reported to have lower measured physical and psychological health-related quality of life scores than controls—related in part to liver and IBD symptoms as well as anxiety, depression and social isolation.198 Dealing with uncertainties about progression of disease and risk of cancer can generate considerable anxiety for people diagnosed with PSC. The main UK support groups for people with PSC are PSC Support (www.pscsupport.org.uk) and UK-PSC (www.uk-psc.com). The websites provide patient friendly information, support forums, lists of liver units and meetings, new developments and other items. Patients with IBD should also be encouraged to contact related support groups, such as Crohn’s & Colitis UK (www.crohnsandcolitis.org.uk) and Guts UK (www.gutscharity.org.uk)
Recommendation 30: We recommend that patients with PSC should be encouraged to participate in patient support groups (strength of recommendation: STRONG; quality of evidence: VERY LOW).
Service standards and audit
PSC can be difficult to manage. Key service standards may assist in developing good clinical practice. Clinicians who are uncertain about managing patients with, or complications of, PSC should refer them to a specialist clinician or centre for review. Key markers of appropriate service standards include:
Patients with PSC should have at least an annual clinical assessment. Those with more advanced disease require more frequent evaluation and follow-up.
Annual blood tests are the minimum baseline investigation, with additional tests done as clinically indicated.
All patients are screened for the presence of IBD at diagnosis.
Clear documentation of the need for surveillance colonoscopy (depending on presence of IBD) and screening for gallbladder pathology and cancer with ultrasound.
All patients should have multidisciplinary review before they undergo ERCP to minimise unnecessary intervention and risk.
All patients with suspicion of malignancy should be referred to the appropriate MDM. This includes patients with new, suspicious or evolving dominant biliary strictures being reviewed at the regional specialist HPB MDM to direct appropriate investigations and interventions.
Patients with evidence of advanced cirrhosis or complications from complex strictures should be referred to the local expert/liver centre.
Future studies
Research into PSC has been mostly through small case series and uncontrolled trials in specialist units. Most centres will have an insufficient number of patients to affect their clinical service, for audit purposes or to undertake independent clinical trials. A recent consensus on endpoints for future studies was reported by an international PSC working group.243
Studies in progress include investigations into the role of norUDCA, farnesoid X receptor agonists, bile salt transporter protein inhibitors, antibiotics and monoclonal antibody blockade of receptors thought to be activated in the pathogenesis of PSC.244
Gastroenterology units should be encouraged to link patient databases in order to improve knowledge of the natural history of PSC and response to treatments, and to improve future clinical trials. These efforts should be coordinated by UK-PSC, BSG and/or the British Association for the Study of the Liver (BASL) and the National Institute for Health Research (NIHR) rare diseases initiative. Studies should include a prospective national repository of clinical samples. Future clinical trials should be registered with the NIHR.
Research and clinical audit questions that may require more clarity include:
What constitutes a diagnosis of PSC in UK practice?
How should patients with unexplained abnormal liver biochemistry and IBD be evaluated, labelled and diagnosed, particularly when MRCP is normal?
How can we improve stratification for risk of (a) liver disease, (b) disease progression and (c) cancer?
What is the optimal timing and type of intervention for patients with dominant strictures?
Are there any other existing and/or new drugs which may be useful in the medical management of PSC?
Can we develop a robust database to map the epidemiology, progression and management of PSC in the UK as well as to assess causes of death?
What is the optimal approach to surgical management in patients with PSC requiring colectomy and/or liver transplantation?
Can we report patient perceptions of problems and risks of living with PSC with the aim of improving wellbeing for patients and carers.
Conclusions
PSC is a complex disease with a wide variation in prognosis. No drug treatments alter the outcome of classic PSC, but patients with evidence of overlap syndromes, including AIH and IgG4-SC, may respond to treatment with corticosteroids. Complications include development of dominant strictures, which may respond to endoscopic therapy, and a high risk (up to 1% a year) of developing CCA. Those with coexistent colitis should undergo annual surveillance colonoscopy. Patients with troublesome symptoms, evidence of advanced liver disease (or deemed to be at risk of this), jaundice with dominant stenoses, evidence of CCA or who express an interest in participating in clinical trials should be referred to specialist centres.
IgG4-related sclerosing cholangitis
Background
Definitions
IgG4-RD is a recently described multisystem disorder characterised by the presence of an IgG4-positive lymphoplasmacytic infiltrate in affected organs. It can affect almost any organ (the term IgG4-RD is used when referring to general aspects or multisystem phenotype of the disease) but most commonly the pancreas (type 1 autoimmune pancreatitis/IgG4-related pancreatitis (IgG4-RP)) and biliary tract (IgG4-SC). IgG4-SC has been classified into four types, with type 1 referring to biliary disease confined to the intrapancreatic bile duct (often in association with IgG4-RP), and types 2–4 being manifested by various degrees of hilar and intrahepatic biliary disease.245
Presentation and disease course
IgG4-SC may present with symptomatic biliary obstruction (often manifested as obstructive jaundice), due either to isolated biliary disease, but frequently in association with a pancreatic mass/diffuse enlargement (IgG4-RP). In a series,246 77% of the 53 patients with IgG4-SC presented with jaundice, which was associated with IgG4-RP in 91%. IgG4-SC may develop in 24–39% of patients previously diagnosed with IgG4-RP.247 248 Symptomatic biliary disease in IgG4-SC does occur in the absence of pancreatic disease, and IgG4-SC may also be an incidental finding in patients presenting with clinical manifestations of IgG4-RD in other organs (eg, kidneys, retroperitoneum, lungs and salivary glands). In a UK study of 115 patients with IgG4-RD, 59% had IgG4-SC.249 In patients undergoing surgery for presumed hilar CCA, pathological features of IgG4-SC, rather than malignancy, have been reported in up to 8% of cases.250 The disease course of IgG4-SC is poorly defined. Although a recent retrospective study of 527 patients followed up for a median of 4 years suggested that the disease ran an indolent course,251 other studies have reported progression to cirrhosis in 7.7–9% of patients,246 249 and a need for liver transplantation has been reported.249 A higher incidence of morbidity, malignancy and mortality in patients with systemic IgG4-RD than in age-matched controls has been reported.249
Diagnostic investigations
In clinical practice PSC is one of the most important differential diagnoses for IgG4-SC, with others including CCA, and alternative causes of secondary sclerosing cholangitis (box 1). There are no definitive diagnostic tests for IgG4-SC. Serum IgG4 may be elevated in 50–80% of patients, and while elevated levels may be supportive of the diagnosis they are insufficient in isolation to make the diagnosis.252 253 Raised serum IgG4 levels are found in only 1% of patients with PBC,26 but elevated serum IgG4 levels are found in 9–15% of patients with PSC,26 254 making distinction between the two diagnoses challenging. An IgG4/IgG1 ratio of >0.24 may improve the positive predictive value and specificity of serum IgG4 measurement in distinguishing IgG4-SC from PSC,254 and a serum IgG4 >x4 ULN appears to be highly specific for IgG4-SC, compared with IgG4 + PSC.255 More recently, a blood IgG4/IgG RNA ratio of >5% obtained by quantitative polymerase chain reaction on a cohort of 95 patients with IgG4-RD, CCA or PSC has been shown to have excellent sensitivity (94%) and specificity (99%) for IgG4-RD, although this test is not widely available.256
Recommendation 1: We recommend that elevated serum IgG4 levels support the diagnosis of clinically suspected IgG4-RD but cannot be relied on for making a definite diagnosis, or distinguishing IgG4-SC from PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
As with PSC, non-invasive imaging should be the cornerstone of imaging in IgG4-SC. Cross-sectional imaging (CT, MRI/MRCP) carries the advantage of allowing definition of the pancreaticobiliary ductal system, and also of other organs or inflammatory ‘pseudotumours’ which may be involved in IgG4-RD. PET scanning may identify fluorodeoxyglucose uptake at sites distant from the biliary tree characteristic of multisystem IgG4-RD (eg, salivary and lachrymal glands), reinforcing the diagnostic suspicion of IgG4-SC. However, PET positivity localised only to the biliary tree, and appearances of a ‘pseudotumour’ rarely allows definitive distinction from other pathologies, such as malignancy. Cholangiography is a central requirement in the investigation of all patients with suspected IgG4-SC. As with PSC, this should preferentially be by non-invasive means, using MRCP. Any part of the biliary tree can be involved in IgG4-SC, and four characteristic patterns have been defined39: type 1, stenosis in the lower common bile duct (often in association with IgG4-related pancreatitis); type 2a, intrahepatic stenosis with prestenotic dilatation; type 2b, intrahepatic stenosis and peripheral bile duct pruning; type 3, hilar and lower common bile duct stenosis; type 4, hilar stenosis only. MRCP may also demonstrate associated pancreatic abnormalities of IgG4-RP, including a long pancreatic duct stricture (more than one-third of the length of the main duct), multifocal stricturing and lack of upstream pancreatic duct dilatation.257
A pathological diagnosis should be pursued in cases of suspected IgG4-SC, as this may allow distinction from disease mimics (including PSC and CCA). All affected tissues are characterised by similar pathological findings, including an IgG4-positive lymphoplasmacytic infiltrate, often in association with storiform fibrosis and obliterative phlebitis.258 The finding of >10 IgG4-positive plasma cells per high power field in endoscopic biopsy specimens from the biliary tree (or other affected organs) may support a diagnosis of IgG4-SC, with an IgG4+/IgG + plasma cell ratio >40% providing additional histological evidence. Brush cytology does not allow a definitive diagnosis of IgG4-SC to be made, but a diagnosis on histology may be obtained via fluoroscopically guided endobiliary biopsy, or via visually directed cholangioscopic biopsies.259 Biopsies from the major papilla are a safe and easy method to obtain tissue; infiltration of IgG4-positive plasma cells has been reported in 53–80% of IgG4-related pancreatitis.260 261 In patients with suspected pancreatic and biliary disease associated with IgG4-RD, endoscopic ultrasound-guided, fine-needle aspiration cytology is effective in excluding malignancy, but rarely allows a definitive diagnosis of IgG4-RD.262 A core biopsy may provide more definitive pathological evidence.
Recommendation 2: We recommend that in patients with suspected IgG4-SC, attempts should be made to obtain a confirmatory histological diagnosis (strength of recommendation: STRONG; quality of evidence: MODERATE).
Differentiating IgG4-related sclerosing cholangitis from PSC
In clinical practice the most common challenge is differentiating IgG4-SC from either PSC or CCA. The presence on cross-sectional imaging of other organ manifestations of IgG4-RD, including the pancreas, kidneys, and retroperitoneum, may point towards a diagnosis of IgG4-SC, as opposed to PSC or CCA.13 263–265 IgG4-SC can occur in isolation, but has been reported in association with type 1 autoimmune pancreatitis (IgG4-RP) in >80% of cases.246 266
As outlined, MRCP is a useful non-invasive test in the investigation of possible IgG4-SC. Features such as long strictures with prestenotic dilatations, the absence of peripheral duct pruning and a lack of biliary pseudodiverticulae are more suggestive of IgG4-SC than PSC.39 However, a multicentre study of cholangiograms in patients with PSC, CCA or IgG4-SC, demonstrated that, even among specialists, a correct diagnosis by interpretation of cholangiography alone is difficult and unreliable, with a high interobserver variation.40 Certain features appear to help differentiate IgG4-SC from PSC, including the much higher prevalence of IBD in PSC (approximately 70%) compared with IgG4-SC (5.6% in 71 patients with autoimmune pancreatitis/IgG4-SC),267 and predominance of pancreatic disease and extra-gastrointestinal involvement in IgG4-SC (see table 2).
Clinical parameters in differentiating IgG4-related sclerosing cholangitis (IgG4-SC) from primary sclerosing cholangitis (PSC)
Although an IgG4-positive lymphoplasmacytic infiltrate may be found in liver biopsy specimens from patients with PSC, it rarely reaches the concentration of >10 IgG4-positive plasma cells per high power film often seen in IgG4-SC.268 Where there is a possibility that strictures are related to IgG4-SC, endoscopic ampullary biopsy sampling should be considered with immunostaining for IgG4, which may be present in 52–72% of cases of IgG4-RD,269 270 and may facilitate discrimination between IgG4-SC and PSC.271
A characteristic feature of IgG4-RD and IgG4-SC is that of a prompt clinical and radiographic response to steroid treatment (although subsequent relapse may occur in >40% of cases).246 266 This is in stark contrast to PSC in which routine steroid use ordinarily provides no therapeutic benefit. Nevertheless, in one study of 285 patients with PSC, raised serum IgG4 was found in 33 (12%), and these patients underwent steroid treatment.59 Although improvement in bilirubin occurred in 90% (despite cirrhosis in 50%), relapse occurred in 50% after an initial favourable response, and complications from steroids, or progression of liver disease, were common.
Recommendation 3: We recommend that other organ involvement (in particular, pancreatic manifestations of IgG4-RD) may provide important information to distinguish IgG4-SC from PSC (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 4: We recommend that IgG4-SC should be diagnosed according to the recommendations of the international consensus guidelines (strength of recommendation: STRONG; quality of evidence: MODERATE).
Treatment
There are few randomised placebo-controlled data to guide treatment of IgG4-SC (or IgG4-RD in general), but large case series have reported rapid and favourable disease control after an initial course of steroids. A common steroid regimen used in the UK is oral prednisolone 40 mg daily for 2–4 weeks, subsequently reduced by 5 mg every week over approximately 8–12 weeks. The effect is measured by clinical response (eg, resolution of jaundice, liver biochemistry, etc) and radiological findings such as resolution of mass lesions and improvement in cholangiopathy. Lack of objective improvement in radiological abnormalities on repeat imaging at weeks 4–8 suggests either an incorrect diagnosis or a fibrotic, non-inflammatory phase of disease. Although serum IgG4 often falls in response to steroids, its level is not used to monitor or plan further treatment. Relapse after cessation of steroid treatment may occur in at least 60% of patients with IgG4-SC, and is more common in those with multiorgan involvement. To date, practice in Europe and North America has often been to introduce an immunomodulator (eg, azathioprine, mercaptopurine or mycophenolate) with further steroids if there is evidence of relapse,266 272 or a high risk of relapse. There is no clear consensus on dosing regimens, including the need to maintain low dose steroids in those receiving azathioprine (usually at a dose of 2 mg/kg/day). Japanese experts favour maintenance steroid treatment, and a recent randomised study (in IgG4-RD, rather than specifically IgG4-SC) showed lower rates of relapse at 3 years in those treated with maintenance prednisolone 5–7.5 mg (23%) than in those given an initial steroid withdrawal regimen (58%).273 Emerging evidence from case series suggest that >95% of these patients with IgG4-RD will respond to biological therapy using anti-CD20 monoclonal antibodies such as rituximab.272 Rituximab is likely to be the preferred treatment for patients who fail to respond to first- or second-line treatment or whose disease flares on withdrawal of steroids, particularly in those with multisystem or complex disease. Small studies have suggested the efficacy of rituximab in patients with IgG4-SC.274 275 Given the complexity of management it is advisable for patients with possible IgG4-SC to be referred to specialists or centres with experience of the disease to establish the diagnosis, plan management and recruit into trials.
Recommendation 5: We recommend that patients with active IgG4–SC should be given corticosteroids as first-line treatment (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 6: We recommend that all patients with IgG4-SC, including those with multiorgan involvement in IgG4-RD, should be considered for continued immunosuppressive therapy (strength of recommendation: STRONG; quality of evidence: MODERATE).
Recommendation 7: We recommend that patients with complex IgG4-SC and those with suspected malignancy should be referred to a specialist MDM for review (strength of recommendation: STRONG; quality of evidence: LOW).
Audits and future studies
Research in the field of IgG4-SC/IgG4-RD has increased significantly over the past 10 years, initially in small case series and uncontrolled trials in specialist units. Most centres will have an insufficient number of patients to affect their clinical service, for audit purposes or to undertake independent clinical trials. Gastroenterology units should be encouraged to link patient databases in order to improve knowledge of the natural history and response to treatments, and to improve future clinical trials.
Research and clinical audit topics that may require more clarity include:
The aetiopathogenesis of IgG4-RD.
Development of a robust database to map the epidemiology, progression and management of IgG4-RD in the UK and to assess causes of death.
The optimal medical management for patients with IgG4-SC.
Reporting of patient perceptions of problems and risks of living with IgG4-RD.
Conclusions
IgG4-SC falls within the spectrum of a multiorgan fibroinflammatory disease (IgG4-RD). It requires distinction from other causes of biliary stricturing, particularly PSC and biliary malignancy, as improvement may be seen with steroids and other immunosuppressant agents. The risk of cancer appears to be low, unlike for patients with PSC. There are insufficient data to recommend surveillance for cancer. Patients with IgG4-SC should be considered for referral to specialist centres.
Acknowledgments
Thank you to Mr Zohur Miah for help with reviewing and formatting the document and figures.
References
Footnotes
Contributors The guidelines were conceived by SPP. MHC was the primary author of the draft documents and coordinated the amendments made after review of the writing committee. Additional regular review of the content was provided by SPP, DT, GGJW, GMH and MW. All authors contributed to authorship of the guidelines as members of the writing committee and agreed the content for publication. Final versions were submitted by MHC and SPP who take responsibility as guarantors for the overall content.
Funding SPP is supported by the National Institute for Health Research, University College London Hospitals Biomedical Research Centre. GMH is supported by the National Institute for Health Research. No other funding was received for the production of these guidelines.
Competing interests DT – involvement in clinical trials in PSC (NGM, Takeda, Shire), funding from British Liver Trust, PSC Partners, PSC Support, EASL, SMR – Involvement in clinical trials in PSC and sits on the advisory board for FALK and Intercept. GH - medical adviser for PSC Support, speaker fees and educational support from Falk Pharma.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.