Article Text

Abstract
Objective Increased de novo fatty acid (FA) synthesis and cholesterol biosynthesis have been independently described in many tumour types, including hepatocellular carcinoma (HCC).
Design We investigated the functional contribution of fatty acid synthase (Fasn)-mediated de novo FA synthesis in a murine HCC model induced by loss of Pten and overexpression of c-Met (sgPten/c-Met) using liver-specific Fasn knockout mice. Expression arrays and lipidomic analysis were performed to characterise the global gene expression and lipid profiles, respectively, of sgPten/c-Met HCC from wild-type and Fasn knockout mice. Human HCC cell lines were used for in vitro studies.
Results Ablation of Fasn significantly delayed sgPten/c-Met-driven hepatocarcinogenesis in mice. However, eventually, HCC emerged in Fasn knockout mice. Comparative genomic and lipidomic analyses revealed the upregulation of genes involved in cholesterol biosynthesis, as well as decreased triglyceride levels and increased cholesterol esters, in HCC from these mice. Mechanistically, loss of Fasn promoted nuclear localisation and activation of sterol regulatory element binding protein 2 (Srebp2), which triggered cholesterogenesis. Blocking cholesterol synthesis via the dominant negative form of Srebp2 (dnSrebp2) completely prevented sgPten/c-Met-driven hepatocarcinogenesis in Fasn knockout mice. Similarly, silencing of FASN resulted in increased SREBP2 activation and hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (HMGCR) expression in human HCC cell lines. Concomitant inhibition of FASN-mediated FA synthesis and HMGCR-driven cholesterol production was highly detrimental for HCC cell growth in culture.
Conclusion Our study uncovers a novel functional crosstalk between aberrant lipogenesis and cholesterol biosynthesis pathways in hepatocarcinogenesis, whose concomitant inhibition might represent a therapeutic option for HCC.
- Hepatocellular carcinoma
- Fatty acid synthase
- Cholesterol biosynthesis
- HMG-CoA reductase
- Systems biology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Aberrant activation of fatty acid synthase (FASN) and related de novo lipogenesis is major metabolic event along hepatocellular carcinoma (HCC) development.
Targeting FASN-mediated de novo lipogenesis is able to inhibit HCC growth in vitro and in vivo.
Emerging evidence suggests the existence of additional mechanisms supporting HCC cell proliferation and survival in the absence of de novo lipogenesis.
What are the new findings?
Loss of Fasn and its mediated de novo lipogenesis delays but does not completely prevent oncogene-driven hepatocarcinogenesis in some mouse models.
Tumour development eventually occurs and is orchestrated by the upregulation of Srebp2-driven cholesterol biosynthesis in Fasn null mouse liver tumour samples.
Similar phenotypes are found in human HCC cell lines when they are genetically depleted of FASN. These results unveil a novel biochemical crosstalk between de novo lipogenesis and cholesterol biosynthesis pathways along hepatocarcinogenesis
Blocking cholesterol biosynthesis completely prevents tumour formation in liver-specific Fasn KO mice.
Concomitant targeting de novo lipogenesis and cholesterol biosynthesis are highly detrimental for the growth of human HCC cells.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Our findings strongly suggest that simultaneous targeting of de novo lipogenesis and cholesterol biosynthesis may be a highly effective strategy for the treatment and prevention of HCC.
Furthermore, the same molecular mechanisms might occur (and, thus, could be effectively targeted) in other tumour types as well.
Introduction
The global incidence of primary liver cancer has progressively increased over the last decades. Among primary liver malignancies, hepatocellular carcinoma (HCC) is the predominant type.1 HCC has limited treatment options. The standard care of drugs for the treatment of advanced HCC are the multikinase inhibitors sorafenib and regorafenib, but they display limited efficacy.2 Thus, there is an obvious unmet medical need for new drug targets for HCC treatment.
Metabolic reprogramming is now recognised as one of the cancer hallmarks, and a key event during tumour initiation and progression.3 4 Among these alterations, de novo lipogenesis consists of the synthesis of endogenous fatty acids (FAs) from acetyl-CoA. Increased de novo lipogenesis has been observed in multiple cancer types.5 In human HCC, data from our and other laboratories have documented the increased expression of major enzymes associated with de novo lipogenesis, including fatty acid synthase (FASN), in tumour lesions when compared with liver non-neoplastic counterparts.6 7 Ablation of Fasn completely inhibited hepatocarcinogenesis in mice overexpressing an activated form of AKT, either alone or in combination with c-Met.8 9 However, emerging evidence suggests the existence of additional mechanisms supporting HCC cell proliferation and survival in the absence of de novo lipogenesis. Indeed, silencing of FASN delays, but does not completely inhibit, in vitro growth of HCC cell lines.7 10 Importantly, in a liver-specific Pten and Tsc1 double KO mouse HCC model, silencing of Fasn via AAV-shFasn significantly decreased, but did not completely prevent, HCC formation.11
Dysregulated cholesterol biosynthesis is another metabolic event frequently observed in HCCs. HMG-CoA reductase (HMGCR) is the rate limiting enzyme in cholesterol biosynthesis and the target of cholesterol-lowering drugs statins. Previous findings indicate the upregulation of HMGCR expression in human HCC samples.7 12 Noticeably, the use of statins has been associated with reduced risk of HCC development in large epidemiological studies, suggesting a potential tumour supporting role of the cholesterol biosynthesis pathway in this disease.13 14 Statins have also been shown to inhibit human HCC cell proliferation in culture as well as c-Myc-driven HCC development in mice.15–17
In this manuscript, we investigated the requirement of Fasn in sgPten/c-Met mouse HCC.18 Our results unveil novel biochemical crosstalk between de novo lipogenesis and cholesterol biosynthesis pathways along hepatocarcinogenesis.
Materials and methods
Hydrodynamic injection and mouse monitoring
Fasnfl/fl mice in C57BL/6 background were described previously.19 AlbCre mice in C57BL/6 background20 were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). Fasnfl/fl mice were crossed with AlbCre mice to eventually generate liver-specific Fasn knockout mice (FasnLKO mice). The physiological phenotype of FasnLKO mice has been extensively characterised.21 Both male and female mice were used in the study, and no sex-dependent differences were detected. Hydrodynamic transfection was performed as described.22 Mice were monitored weekly, euthanised and liver tissues collected when they developed large abdominal masses or by 40–50 weeks postinjection. The detailed mouse experiment data are available in online supplementary table S1. All mice were housed, fed and monitored in accordance with protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Supplemental material
Human tissue samples
Sixty-five frozen HCC and corresponding non-tumourous surrounding livers (SL) were used. Clinicopathological features of patients with liver cancer are summarised in online supplementary table S2. HCC specimens were collected at the Medical University of Greifswald (Greifswald, Germany). Informed consent was obtained from all individuals.
Supplemental material
Statistical analysis
Data were analysed using the Prism 6 software (GraphPad, San Diego, California, USA). Comparisons between two groups were performed with two-tailed unpaired t-test. Statistical differences among the various groups were assessed with the Tukey-Kramer’s test. Kaplan-Meier method and log-rank test were used for survival analysis. All graphs are the mean±SEM P values <0.05 were considered to be statistically significant.
Additional information can be found in online supplementary materials and methods.
Supplemental material
Results
Ablation of Fasn delays sgPten/c-Met-induced hepatocarcinogenesis in mice
We discovered that lipogenic proteins are strongly induced in hepatocellular lesions in sgPten/c-Met mice.18 To investigate the requirement of Fasn and mediated de novo lipogenesis in sgPten/c-Met-driven hepatocarcinogenesis, conditional Fasn KO mice (Fasnfl/fl mice) were employed.19 Specifically, two distinct approaches were applied. In the first approach, we bred AlbCre mice with Fasnfl/fl mice to eventually generate FasnLKO mice. sgPten and c-Met plasmids were injected into FasnLKO mice (n=10) as well as control Fasnfl/fl mice (n=4) (figure 1A). All sgPten/c-Met-injected Fasnfl/fl mice developed high burden of liver tumours by 8–10 weeks postinjection and were required to be euthanised, in agreement with previous data (online supplementary table S1).18 In contrast, at this time point, all sgPten/c-Met-injected FasnLKO mice appeared to be healthy with no palpable abdominal masses. However, hepatocarcinogenesis was delayed but not abolished by Fasn deletion in sgPten/c-Met-injected FasnLKO mice, which exhibited a high burden of malignant lesions within 14~40 weeks postinjection (figure 1B and online supplementary table S1). Histologically, sgPten/c-Met co-expression led to the formation of pure HCCs in the liver of Fasnfl/fl and FasnLKO mice (figure 1C). Immunohistochemical analysis (IHC) showed that all HCCs from wild-type mice expressed high levels of Fasn protein, which will be referred to as ‘Fasn(+) HCC’, whereas all HCC cells in FasnLKO mice showed no Fasn expression, which will be referred to as ‘Fasn(−) HCC’ (figure 1A and C). The levels of ectopically expressed human c-Met as well as the loss expression of Pten protein were equivalent in Fasn(+) HCCs and Fasn(−) HCCs (figure 1C,D).
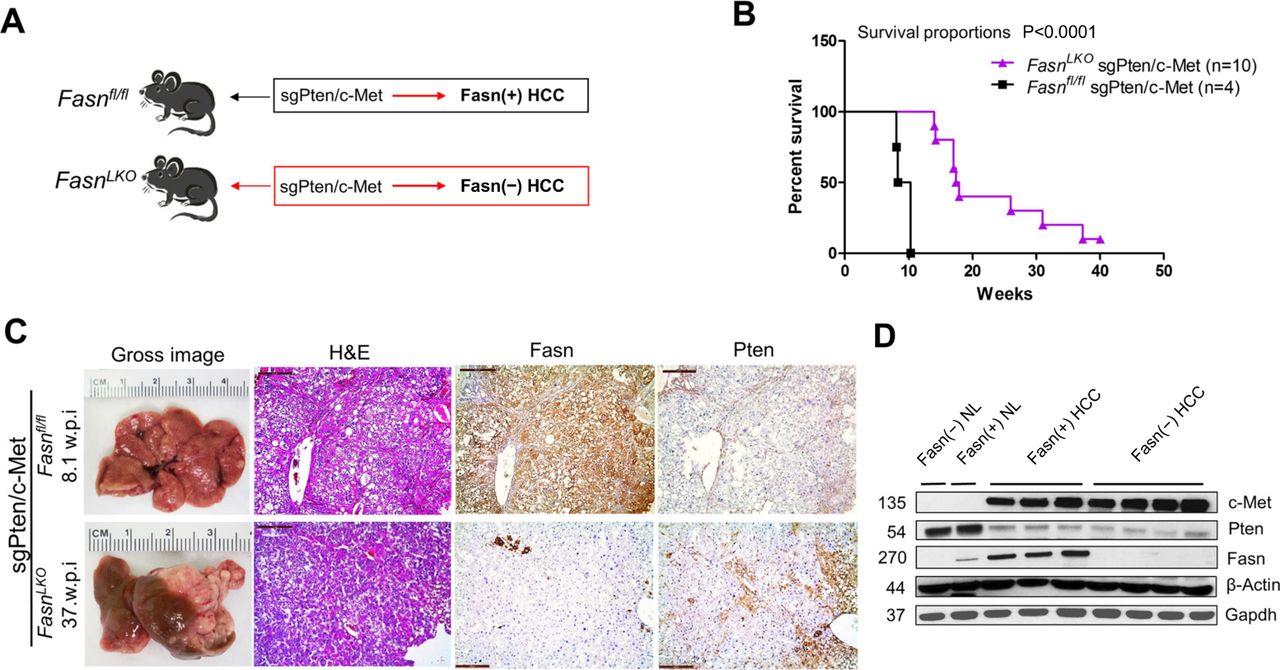
Genetic ablation of Fasn in the mouse liver delays sgPten/c-Met-driven hepatocellular carcinoma (HCC) formation. (A) Scheme of the experiment. (B) Survival analysis of sgPten/c-Met-induced HCC in Fasnfl/fl mice (n=4) and in FasnLKO mice (n=6). (C) Gross images, H&E and immunohistochemistry of FASN and Pten expression in sgPten/c-Met HCC from Fasnfl/fl mice (upper panels) and FasnLKO mice (lower panels); w.p.i., weeks postinjection; scale bar: H&E, Fasn and Pten; 200 µm; Ki67, 100 μm. (D) Western blot analysis of c-Met, Pten and Fasn expression in liver tissues. β-Actin and Gapdh were used as loading controls. NL, normal liver tissues.
In the second approach, we hydrodynamically injected sgPten, c-Met and Cre plasmids into Fasnfl/fl mice (sgPten/c-Met/Cre; n=6), thus allowing the deletion of Pten together with overexpression of c-Met oncogenes and deletion of Fasn in the same subset of mouse hepatocytes. As a control, sgPten, c-Met and pT3EF1α (empty vector) were co-injected into Fasnfl/fl mice (sgPten/c-Met/pT3, n=5) (online supplementary figure S1A). All sgPten/c-Met/pT3 mice developed liver tumours by 9–12 weeks postinjection (online supplementary figure S1B), whereas macroscopic and/or histopathological alterations were detected in the livers of sgPten/c-Met/Cre mice between 20 and 51 weeks postinjection (online supplementary figure S1B and online supplementary table S1). Histological and biochemical analyses revealed that HCC lesions from both cohorts expressed the transfected c-Met and displayed loss of Pten. Fasn expression was confirmed to be lost in sgPten/c-Met/Cre HCCs (online supplementary figure S1C).
Supplemental material
Mutations of PIK3CA occur in ~4% of human HCCs.23 We previously showed that PIK3CAH1047R cooperates with c-Met or NRasV12 to promote HCC development in mice (PIK3CA/c-Met or PIK3CA/Ras).24 To determine whether the delayed hepatocarcinogenesis induced by loss of Fasn could be observed in other murine HCC models, we deleted Fasn in PIK3CA/c-Met and PIK3CA/Ras mice. We found that in all cases, ablation of Fasn resulted in significant delay but not suppression of tumour development (online supplementary figure S2–S5).
Altogether, our data indicate that Fasn and its mediated de novo lipogenesis are key metabolic events during hepatocarcinogenesis. However, oncogene-expressing hepatocytes could eventually overcome the loss of Fasn and progress into HCC.
Expression analysis of Fasn regulated genes and pathways in sgPten/c-Met-driven hepatocarcinogenesis
As loss of PTEN is more commonly seen than PIK3CA mutations in human HCC,23 we used the sgPten/c-Met HCC model for the subsequent functional studies. Normal liver tissues from Fasnfl/fl mice (Fasn(+) NL) and FasnLKO mice (Fasn(−) NL) were used as controls when necessary. Using Ki67 IHC, we found that tumour cells proliferated at a higher rate in Fasn(+) HCC than Fasn(−) HCC (online supplementary figure S6). Next, we analysed the major signalling pathways downstream of Pten and c-Met. Using Western blot analysis, we found that p-ERK levels did not differ in Fasn(+) and Fasn(−) HCC tissues, indicating that Fasn does not affect Ras/MAPK cascade during hepatocarcinogenesis (online supplementary figure S7). Of note, p-AKT at S473 was mildly decreased in Fasn(−) HCCs, whereas p-AKT at T308 was significantly downregulated. The decreased p-AKT activity may be due to the decreased expression of rapamycin-insensitive companion of mammalian target of rapamycin (Rictor) (online supplementary figure S7), a key component of mTORC2, consistent with our previous study showing that FASN regulates Rictor stability.9 Decreased AKT activity led to reduced expression of some AKT downstream effectors, including phosphorylated glycogen synthase kinase-3beta (p-GSK3β) and phosphorylated proto-oncogene serine/threonine-protein kinase (p-c-Raf), but not phosphorylated forkhead box protein O1 (p-Foxo1) and phosphorylated proline-rich AKT1 substrate 1 (p-PRAS40), in Fasn(−) HCCs (online supplementary figure S7). In addition, mTOR pathway activity, as illustrated by the levels of p-RPS6 and p-4EBP1, was similar in Fasn(+) and Fasn(−) HCCs (online supplementary figure S7).
To further understand how Fasn regulates hepatocarcinogenesis, we performed global gene expression analysis. Four samples from each group (Fasn(+) NL; Fasn(−) NL; Fasn(+) HCC and Fasn(−) HCC) were subjected to microarray experiments. Hierarchical cluster analysis showed distinct gene expression patterns in NL and HCC with or without Fasn, indicating that cancer-related gene signature was dominating in expression analysis (figure 2A and online supplementary dataset S1A). To identify genes that might contribute to Fasn-mediated tumourigenesis, we specifically searched for genes differentially expressed in Fasn(+) versus Fasn(−) HCC samples. We identified 284 differentially expressed genes (log2FoldChange>1 or log2FoldChange<−1, padj<0.05), including 98 upregulated genes and 186 downregulated genes in Fasn(−) HCCs (online supplementary dataset S1B). Interestingly, 32 of the differentially expressed genes were related to lipid catabolism (figure 2B and online supplementary dataset S1C). Pathway analysis of the differentially expressed genes revealed that Srebp signalling pathway was in the top category of the upregulated pathways between Fasn(−) HCC versus Fasn(+) HCC. Genes downstream of Srebp transcription factors, including those involved in triglyceride biosynthesis, FA biosynthesis and mevalonate pathway, were all found to be upregulated in Fasn(−) HCCs (figure 2C). Indeed, expression levels of Acly and Acaca, which are related to FA biosynthesis, Elovl6, an enzyme for very long FA elongation, Hmgcr, squalene monooxygenase (Sqle) (padj=0.057), diphosphomevalonate decarboxylase (Mvd), which are involved in the mevalonate and cholesterol biosynthesis pathways, were highest in Fasn(−) HCCs and cytochrome P450 family 7 subfamily a member 1 (Cyp7a1) (padj=0.205) and cytochrome P450 family 7 subfamily b member 1 (Cyp7b1), two key enzymes responsible for cholesterol ester (CE) degradation, were lowest in the same tumours (figure 2B and D). In contrast, several enzymes involved in FA oxidation, including acetyl-CoA acyltransferase 1a (Acaa1a), acetyl-CoA acyltransferase 1b (Acaa1b) and hydroxysteroid 17-beta dehydrogenase 4 (Hsd17b4) (padj=0.060) were downregulated (online supplementary dataset S1C; figure 2B,C). Carnitine palmitoyltransferase 1a (Cpt1a) is the master regulator of FA oxidation, and its levels were similar in Fasn(−) HCC and Fasn(+) HCC. Thus, we analysed Cpt1a protein expression (online supplementary figure S7), and found that Cpt1a was expressed at lower levels in Fasn(−) HCC, supporting the decreased FA oxidation in Fasn(−) tumour cells.
Supplemental material
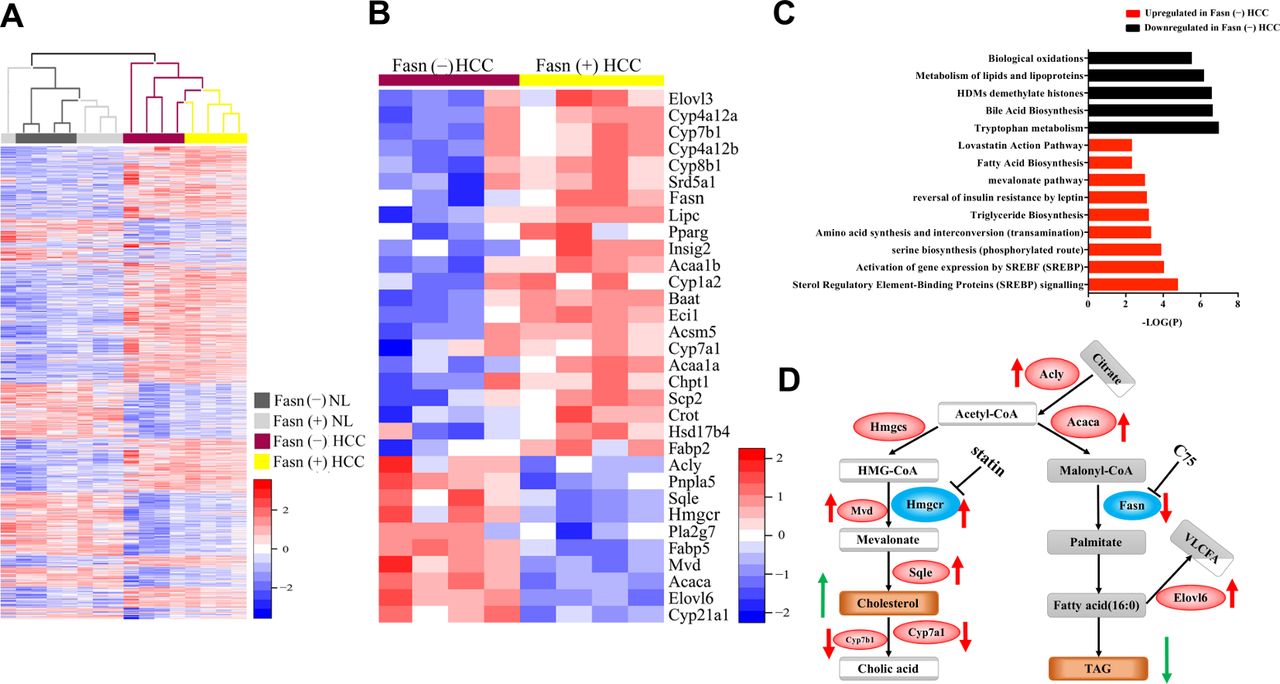
Microarray data analysis of fatty acid synthase (Fasn)(+) and Fasn(−) normal liver and hepatocellular carcinoma (HCC). (A) Heat map of four groups of samples (n=4/each) from Fasn(+) NL and HCC as well as Fasn(−) NL and HCC (|log2 FC|>1, padj<0.05). (B) Heat map of lipid catabolism-related genes in Fasn(−) HCC and Fasn(+) HCC. (C) Pathway analysis of the genes that show differences (P<0.05) between Fasn(−) and Fasn(+) HCC. (D) Summary of the signalling pathways regulating cholesterol and fatty acid synthesis in Fasn(−) HCC vs Fasn(+) HCC; red arrows indicate deregulated genes, while green arrows indicate deregulated metabolites. NL, normal liver tissues.
To further validate our observation, we performed qRT-PCR analysis in Fasn(+) versus Fasn(−) HCCs. Expression levels of acetyl-CoA carboxylase alpha (Acaca), ATP citrate lyase (Acly), Hmgcr, Sqle, Mvd and Elovl6 were homogeneously upregulated, whereas Fasn expression was strongly downregulated, in Fasn(−) HCCs (online supplementary figure S8).
Since FA synthesis and cholesterol production are controlled by SREBP1 and SREBP2 transcription factors, respectively,25 their expression was evaluated. Based on microarray data, Srebf1/Srebf2 mRNA levels did not change significantly in the various cohorts of liver samples. Consistently, total Srebp1 and Srebp2 protein levels did not change (figure 3A). However, using nuclear extracts, we found that nuclear levels of Srebp1 had a trend to increase and Srebp2 levels were significantly higher in Fasn(−) HCC (figure 3B,C). Using immunohistochemistry, we confirmed the increased nuclear Srebp1 and Srebp2 indexes in Fasn(−) HCCs (figure 3D,E).

Expression analysis of fatty acid synthase (Fasn) regulated genes and pathways in Fasn(+) and Fasn(−) mouse hepatocellular carcinoma (HCC) samples. (A) Western blot analysis of total sterol regulatory element binding protein (Srebp)1 and Srebp2 protein levels in the liver tissues; β-actin and Gapdh were used as loading controls. (B) Western blot analysis of nuclear (n-) Srebp1 and Srebp2 in liver tissues; histone H3 and β-tubulin were used as loading controls for nuclear and cytoplasmic fractions, respectively. (C) Quantification of western blot analysis data of nuclear (n-) Srebp1 and Srebp2 in Fasn(+) and Fasn(−) HCC. (D) Immunohistochemical staining of Srebp1 and Srebp2 expression of NL, Fasn(+) HCC and Fasn(−) HCC. Red arrows indicate nuclear staining of Srebp1 and Srebp2. (E) Quantification of the percentage of nuclear (n-) Srebp1 and Srebp2 levels using immunostaining in Fasn(+) and Fasn(−) HCC tissues. Scale bar: 100 µm. NL, normal liver tissues.
Altogether, expression analysis demonstrates that loss of Fasn leads to feedback activation of Srebp2, resulting in the increased expression of genes involved in cholesterol biosynthesis.
Upregulation of cholesterol esters in Fasn(−) HCC tissues
We investigated how the loss of Fasn modulates lipid metabolisms along hepatocarcinogenesis. We performed lipidomic analysis of Fasn(+) NL, Fasn(−) NL, Fasn(+) HCC and Fasn(−) HCC (n=6/each cohort). Lipid classes were analysed, including neutral lipids, phospholipids, and sphingolipids (online supplementary dataset S2). As shown on the heat map, the alterations of lipids were more profound between Fasn(−) versus Fasn(+) HCC, and less remarkable between Fasn(−) and Fasn(+) NL (figure 4A,B). Among the major lipids, CE levels were higher in Fasn(+) HCC than in normal liver tissues and highest in Fasn(−) HCC. In contrast, triacylglycerols (TAG), diacylglycerols (DAG) and free fatty acids (FFA) were all highest in Fasn(+) HCC, whereas loss of Fasn led to their decrease (figure 4A,B). When analysing lipid composition based on the mole percentage of total lipids, TAG, which represents ~70% of total lipids in Fasn(+) HCCs, dropped to ~40% in Fasn(−) HCC. In striking contract, CE were only ~5% of total lipids in Fasn(+) HCCs, but raised to ~20% in Fasn(−) HCC (figure 4C). Overall, when specifically comparing lipid profiles between Fasn(+) HCC and Fasn(−) HCC, TAG, DAG and FFA levels were low, whereas CE levels highest in Fasn(−) HCC (figure 4D). Consistently, Oil Red O staining, which mainly stains TAG droplets, revealed extensive positivity in Fasn(+) HCC, and loss of staining in Fasn(−) HCC (online supplementary figure S9A).
Supplemental material

Lipidomic and fatty acid composition of cholesterol ester (CE) data from normal liver and hepatocellular carcinoma (HCC) tissues with or without fatty acid synthase (Fasn). (A) Heat map of average levels of lipid species concentrations (μM) in liver tissues. (B) Amount of lipid species in liver tissues illustrated as concentration (μM) of each lipid species. Lipid species with very low concentration are shown in the right corner of the panel. (C) Amount of lipid species in liver tissues illustrated as percentage of each lipid species in the total lipid pool; lipid species with very low percentage of concentration are shown in the right corner of the panel; Fasn(−) HCC vs Fasn(+) HCC, *P<0.05 and **P<0.01. (D) Summary of various lipid species differences between Fasn(−) and Fasn(+) HCC. (E) Heat map of species of CE containing different lengths of fatty acids based on the concentrations (µM) in liver tissues. (F) Concentration (μM) of CE containing different lengths of fatty acids in the liver tissues; CE species with very low concentration are shown in the right corner of the panel. (G) The percentage of CE with different lengths of fatty acids relative to total amount of CE in the liver tissues; Fasn(−) HCC vs Fasn(+) HCC, *P<0.05 and **P<0.01. (H) Classification of sources of fatty acids from diet or de novo lipogenesis based on the length of side chains and lipidomic data. NL, normal liver tissues.
Next, we analysed the FA composition of the various lipid species. Consistent with the loss of Fasn in Fasn(−) HCC, the concentration of C14:0 (myristic acid) and C16:0 (palmitic acid), which are end-products of Fasn, were lower in Fasn(−) HCC than Fasn(+) HCC (figure 4E). In the case of CE, except for C14:0 and C16:0, the levels of most long-term FAs, such as C18:1 and C18:2, were highest in Fasn(−) HCC (figure 4E–G). The detailed analyses of FA composition in other lipid species, including TAG, DAG, FFA, phosphatidylcholine, phosphatidylethanolamine and sphingomyelin, are shown in online supplementary figure S10–S15.
When analysing the source of FAs in HCCs, we identified an overall significantly higher contribution of dietary FA in Fasn(−) HCC (figure 4H). The results are consistent with the hypothesis that when de novo lipogenesis is inhibited, FA uptake is a major compensatory mechanism for obtaining FA required for tumour growth.
Deregulation of cholesterol biosynthesis pathway is a major event in Fasn(−) HCC
We combined the data from genomics and lipidomics studies. At the gene expression level, loss of Fasn in sgPten/c-Met HCC cells resulted in a compensatory increased expression of genes associated with FA and cholesterol biosynthesis downstream of Srebp1/Srebp2 (figure 2D). However, due to the limitation of FAs, TAG levels decreased in Fasn(−) HCC (figure 2D). On the other hand, increased Srebp2 led to the augmented expression of cholesterol biosynthesis genes, including Hmgcr, Sqle and Mvd (figure 2D), resulting in highest CE levels in Fasn(−) HCC (figures 2D and 4D).
In summary, using a systems biology approach by combining genomics and lipidomics analyses, we discovered that loss of de novo lipogenesis may trigger the activation of Srebp2 and increased cholesterol biosynthesis along hepatocarcinogenesis.
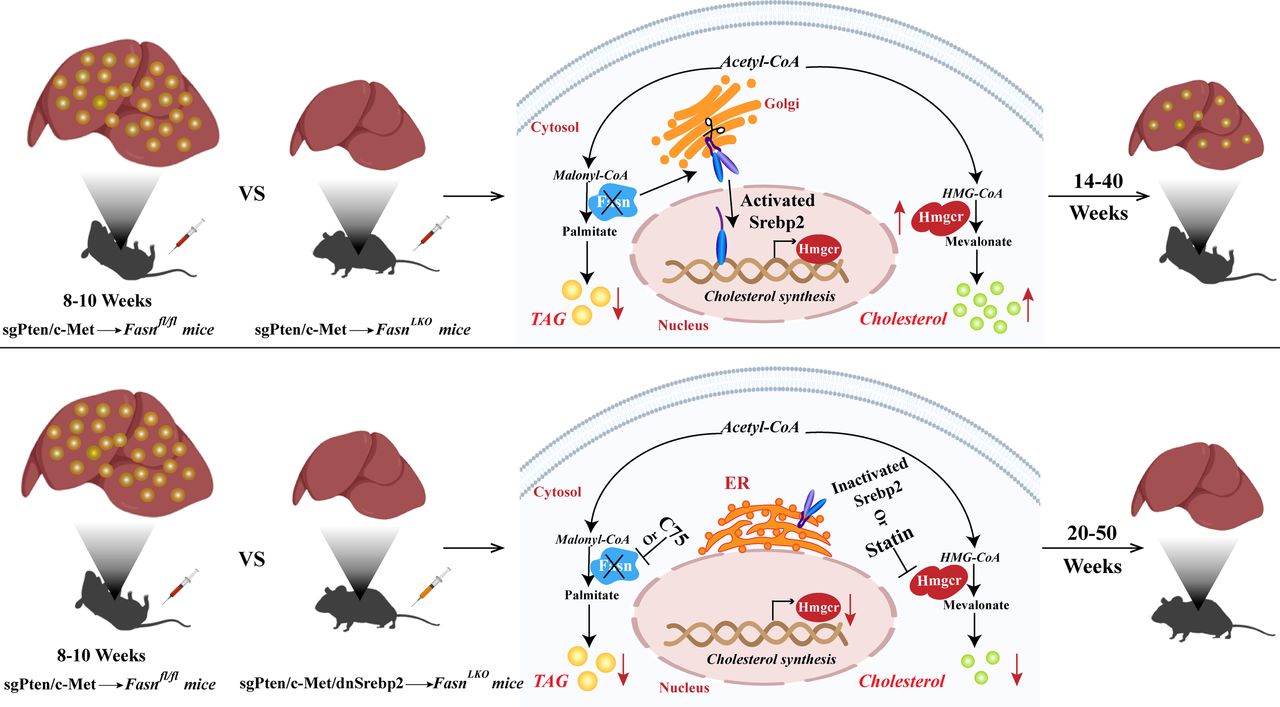
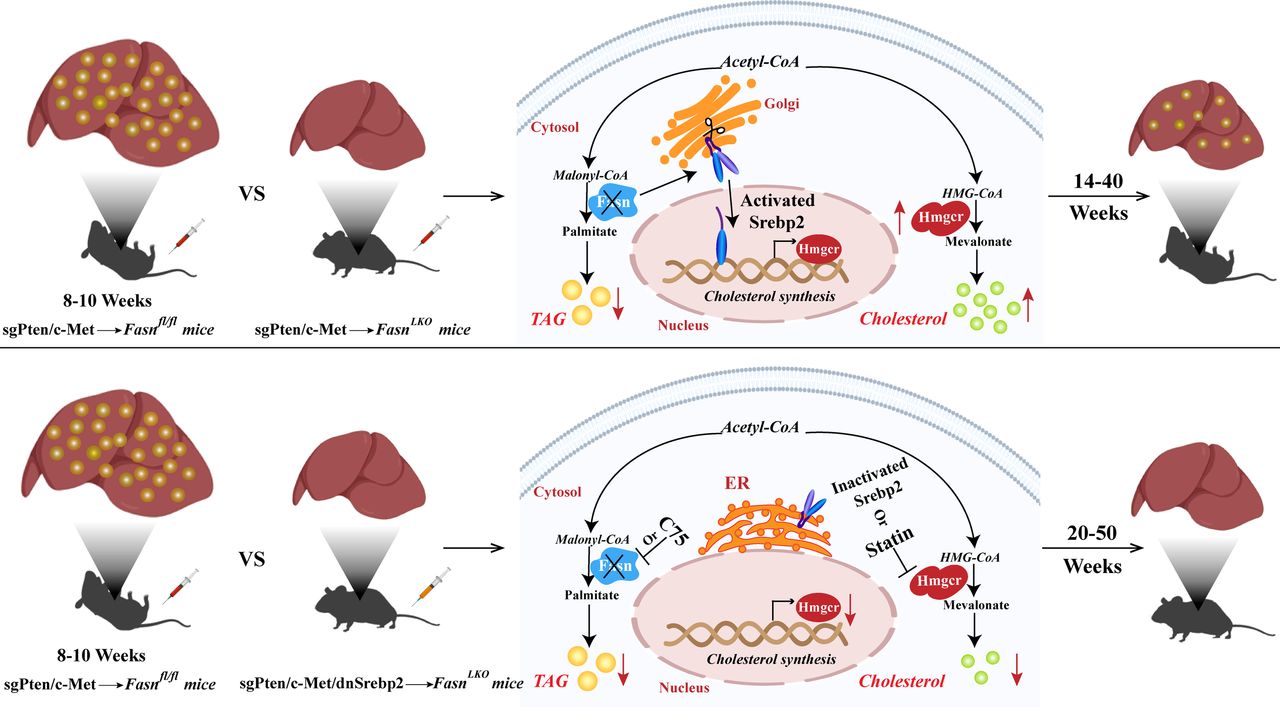
Co-expression of a dominant negative Srebp2 (dnSrebp2) completely blocks sgPten/c-Met-induced HCC formation in FasnLKO mice
Based on these observations, we hypothesised that increased Srebp2 might compensate for the loss of Fasn and related mediated de novo lipogenesis in promoting HCC development in mice. Thus, it would be expected that blocking Srebp2 prevents sgPten/c-Met-induced hepatocarcinogenesis in FasnLKO mice. To test this hypothesis, we co-expressed sgPten/c-Met oncogenes with dnSrebp225 into FasnLKO mice (sgPten/c-Met/dnSrebp2; figure 5A). Additional FasnLKO mice were hydrodynamically injected with sgPten/c-Met and pT3 empty vector as control (sgPten/c-Met/pT3, figure 5A). All sgPten/c-Met/pT3-injected FasnLKO mice were harvested between 15 and 31 weeks postinjection and all but one mouse exhibited high liver tumour burden (figure 5B and online supplementary table S1). In contrast, sgPten/c-Met/dnSrebp2 mice were aged until 50 weeks postinjection and appeared healthy, with no liver tumour nodules visible (figure 5B and online supplementary table S1). Histological evaluation showed HCC lesions in sgPten/c-Met/pT3 liver tissues, and normal liver in sgPten/c-Met/dnSrebp2 mice (figure 5C). Furthermore, we analysed apoptosis, inflammation and fibrosis in liver tissues from the different cohorts of injected FasnLKO mice. Immunostaining of α-smooth muscle actin and Sirius Red staining revealed more stromal cells in HCC lesions than those in liver tissues from uninjected FasnLKO mice or sgPten/c-Met/dnSrebp2 FasnLKO mice (online supplementary figure S16). Other cellular processes, such as inflammation and apoptosis, were not significantly altered in HCC lesions or sgPten/c-Met/dnSrebp2 liver tissues (online supplementary figures S17 and S18).

dnSrebp2 inhibits sgPten/c-Met-induced hepatocellular carcinoma (HCC) formation in fatty acid synthase (Fasn) LKO mice. (A) Study design; (B) tumour development incidence curves in sgPten/c-Met/pT3 (n=6) and sgPten/c-Met/dnSrebp2-injected FasnLKO mice (n=10); (C) gross images, H&E, Fasn and Ki67 immunohistochemical analysis of sgPten/c-Met/pT3-injected (upper panel) and sgPten/c-Met/dnSrebp2-injected (lower panel) FasnLKO mice. w.p.i., weeks postinjection; scale bars: H&E and Fasn; 200 µm; Ki67, 100 μm.
Altogether, the results indicate that co-expression of dnSrebp2 effectively blocks sgPten/c-Met-induced HCC formation in FasnLKO mice. Concomitant inhibition of Srebp2-driven cholesterol biosynthesis and Fasn-mediated de novo lipogenesis might completely prevent HCC formation in vivo.
Functional interplay between de novo lipogenesis and cholesterol biosynthesis pathways in human HCC cell lines
We investigated whether the same molecular crosstalk occurs in human HCC using a panel of HCC cell lines (online supplementary figure S19). FASN gene was silenced in human HCC cells using specific small interfering RNA (siRNA). Knockdown of FASN resulted in increased nuclear SREBP2 protein expression (figure 6A) and activity (online supplementary figure S20). Furthermore, FASN depletion led to the increased expression of HMGCR in a SREBP2-dependent manner, as silencing of SREBP2 blocked siFASN-induced HMGCR upregulation (figures 6B and online supplementary figure S21). To further validate this observation, FASN was deleted via CRISPR/Cas9-based gene-editing technology in the SNU449 HCC cell line (online supplementary figure S22). Two single cell clones, sgFASN.18 and sgFASN.32, were selected; western blot analysis and genomic sequencing confirmed FASN deletion (figures 6C and online supplementary figure S22). In both clones, deletion of FASN resulted in nuclear localisation of SREBP2 (figure 6C,D), as well as increased HMGCR mRNA expression (figure 6E). Furthermore, consistent with previous studies, sgFASN.18 and sgFASN.32 showed decreased cell proliferation (online supplementary figure S23).

Silencing of fatty acid synthase (FASN) induces activation of sterol regulatory element binding protein 2 (SREBP2) and HMG-CoA reductase (HMGCR) expression in human hepatocellular carcinoma (HCC) cells. (A) Silencing of FASN with siFASN led to increased nuclear SREBP2 expression in human HCC cell lines; (B) qRT-PCR analysis of mRNA expression of FASN, SREBP2 and HMGCR in SNU475 cells treated with various small interfering RNA (siRNA). Similar results were obtained using Hep40, Hep3B, SNU475 and MHCC97H HCC cell lines (online supplementary figure S20). P<0.05 scrambled control (siSc); scramble siRNA; (a) vs control; (b) vs siSc; (c) vs siFASN; (d) vs siHMGCR; (e) vs siSREBP2; (f) vs siFASN+siHMGCR; (g) vs siFASN+siSREBP2. (C) Western blot analysis of FASN, full length (f-) and nuclear (n-) SREBP2 expression in control, sgFASN.18 and sgFASN.32 subclones of SNU449 cells. (D) Immunofluorescence of SREBP2 in control, sgFASN.18 and sgFASN.32 subclones of SNU449 cells; scale bar: 10 µm. (E) qPCR analysis of FASN and HMGCR miRNA expression in control, sgFASN.18 and sgFASN.32 subclones of SNU449 cells.
Based on these data, we hypothesised that concomitant inhibition of FASN-mediated de novo lipogenesis and SREBP2/HMGCR-mediated cholesterol biosynthesis might show stronger antitumour growth activity in human HCC cells. We assessed the cell growth rates in a panel of human HCC cells treated with siFASN and siHMGCR or siSREBP2, either alone or in combination (figures 7A and online supplementary figure S24). While silencing of either FASN, HMGCR or SREBP2 alone led to decreased growth in all cell lines, co-transfecting HCC cells with siFASN and siHMGCR or siSREBP2 resulted in significantly stronger growth restraint (figures 7A and online supplementary figure S24). At the molecular level, suppression of FASN led to the upregulation of HMGCR and SREBP2 mRNAs as well as of SREBP2 transcriptional activity and its targets genes (mevalonate kinase [MVK], SQLE and squalene synthase). Knockdown of either HMGCR or SREBP2 instead resulted in downregulation of SREBP2 gene targets while triggering FASN upregulation. To further substantiate this observation, we applied a pharmacological approach, by treating HCC cells with the FASN inhibitor C75 and the HMGCR inhibitor atorvastatin, alone or in combination (figures 7B and online supplementary figure S25). Consistently, we found that concomitant treatment of cells with C75 and atorvastatin led to increased growth decline (figures 7B and online supplementary figure S25). As these HCC cell lines have different levels of AKT/mTORC2 activity (online supplementary figure S19), the results suggest that combined inhibition of de novo lipogenesis and cholesterogenesis augments growth inhibition in HCC cells regardless of mTORC2 status.

Concomitant targeting of fatty acid synthase (FASN)-mediated de novo lipogenesis and HMGCR-mediated cholesterol biosynthesis results in increased growth inhibition in human hepatocellular carcinoma (HCC) cell lines. (A) Cell proliferation (upper panel) and apoptosis (lower panel) in Hep40 and SNU475 HCC cells treated with various small interfering RNA (siRNA). Similar results were obtained using Hep3B and MHCC97H HCC cells (online supplementary figure S24). P<0.05 (a) vs control; (b) vs siSc; (c) vs siFASN; (d) vs siHMGCR; (e) vs siSREBP2; (f) vs siFASN+siHMGCR; (g) vs siFASN+siSREBP2. siSc, scrambled control. (B) Cell proliferation (upper panel) and apoptosis (lower panel) in Hep40 and SNU475 HCC cells treated with C75, atorvastatin or C75+atorvastatin. Similar results were obtained using Hep3B, PLC and MHCC97H HCC cells (online supplementary figure S25). P<0.05 a) vs dimethyl sulfoxide (DMSO); (b) vs C75 and (c) vs atorvastatin.
Collectively, our findings demonstrate that loss FASN triggers the compensatory activation of SREBP2/HMGCR metabolic cascade in human and mouse HCC cells (figure 8). Concomitant targeting of FASN and HMGCR may be required for the effective prevention or treatment HCC (figure 8).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme summarising the compensatory activation of cholesterol biosynthesis following fatty acid synthase (FASN) depletion along hepatocarcinogenesis. In the sgPten/c-Met mouse model, all sgPten/c-Met-injected Fasnfl/fl mice develop high burden of liver tumours by 8–10 weeks postinjection, with hepatocellular carcinoma (HCC) tissues showing high triglyceride and cholesterol levels. Hepatocarcinogenesis is instead delayed but not abolished by Fasn deletion in sgPten/c-Met-injected FasnLKO mice. These mice exhibit a high burden of malignant lesions within 14~40 weeks postinjection, with decreased triglyceride levels and increased cholesterol esters. Mechanistically, loss of Fasn promotes nuclear localisation and activation of sterol regulatory element binding protein 2 (Srebp2), which upregulates the key enzyme Hmgcr of cholesterol biosynthesis, resulting in cholesterol accumulation and, eventually, HCC formation. Noticeably, blocking of cholesterol synthesis via the dominant negative form of Srebp2 (dnSrebp2) completely prevents sgPten/c-Met-driven hepatocarcinogenesis in Fasn knockout mice. Even when these mice are aged until 50 weeks postinjection, they appear healthy, with no liver tumour nodules visible. Thus, concomitant inhibition of FASN and HMGCR is likely to prevent triglyceride and cholesterol accumulation, leading to strong suppression of hepatocarcinogenesis. Up red arrows and down red arrows indicate upregulation and downregulation, respectively.
Negative prognostic role of cholesterol pathway activation in human hepatocellular carcinoma
Finally, we evaluated the mRNA levels and activity of SREBP2 in a human HCC cohort whose clinicopathological data were available (n=65; online supplementary table S3). We found that SREBP2 mRNA expression did not significantly differ between HCC and corresponding SL (online supplementary figure S26A). In contrast, SREBP2 transcriptional activity was significantly higher in HCC than in SL (online supplementary figure S26A). Furthermore, SREBP2 activity was significantly more elevated in HCC samples with poorer prognosis (HCCP) when compared with those with better prognosis (HCCB). When assessing the relationship between SREBP2 and patients’ clinicopathological data, we found that higher activity of the SREBP2 gene correlates with lower HCC survival rate (P<0.0001; online supplementary figure S26B). This association remained strongly significant after multivariate Cox regression analysis (p<0.0001; online supplementary table S3), thus implying SREBP2 activity as an independent prognostic factor for HCC. No relationship between the transcriptional activity of SREBP2 and other clinicopathological parameters of the patients, including age, gender, aetiology, the presence of cirrhosis, tumour size and tumour grade were detected (online supplementary table S3). A similar correlation with tumour aggressiveness and patients’ survival was detected when assessing the levels of SREBP2 target HMGCR in the same sample collection (online supplementary figure S26C). In addition, a significant, positive correlation between the transcription activity of SREBP2 and the mRNA levels of HMGCR was identified (p<0.0001; online supplementary figure S26D). To further validate these results, we analysed the mRNA expression of additional SREBP2 target genes, including MVK, lanosterol synthase (LSS), and 24-dehydrocholesterol reductase (DHCR24). Noticeably, mRNA expression of these genes correlated with SREBP2 activity in human HCC samples, but not SREBP2 mRNA levels (online supplementary figure S27A–C). Importantly, high levels of MVK, LSS and DHCR24 were all associated with poor patients’ prognosis (online supplementary figure S27D).
Supplemental material
Altogether, these data demonstrate that upregulation of SREBP2 and its transcriptional programme is a molecular feature of aggressive HCC in humans.
Discussion
A major finding of the present investigation is that loss of Fasn only delays tumour initiation, but sgPten/c-Met overexpression could still induce HCC formation in Fasn knockout mice. Mechanistically, Fasn(−) HCC showed the feedback activation of Srebp1/Srebp2, in accordance with previous studies showing that FAs are key regulators of Srebp1/Srebp2.26 The precise mechanism(s) whereby ablation of Fasn increases Srebp1/Srebp2 activation requires further investigation. Our preliminary observations indicate that the regulation presumably occurs at the protein level, by regulating Srebp1/Srebp2 nuclear localisation and consequent transcriptional activity. Increased Srebp1/Srebp2 activity resulted in the upregulation of genes involved in de novo lipogenesis and cholesterol biosynthesis. However, due to the loss of Fasn, de novo lipogenesis rate was clearly repressed, as levels of myristic acid and/or palmitic acid were low in all lipid species, and TAG levels were decreased in Fasn(−) HCC tissues. The net outcome of increased SREBP2 activity was the increased expression of genes promoting cholesterogenesis, leading to the enhanced CE levels in Fasn(−) HCC. To further substantiate that this phenotype is not limited to sgPten/c-Met HCC model, we investigated the expression of Hmgcr, Mvd and Sqle in Fasn(+) and Fasn(−) HCCs from PIK3CA/c-Met and PIK3CA/Ras mice. Consistently, elevated expression of these cholesterol biosynthesis genes occurred only in Fasn(−) tumour samples (online supplementary figure S28). To the best of our knowledge, this is the first observation of crosstalk between FA synthesis and cholesterol biosynthesis cascades along carcinogenesis. It is worth to note that this biochemical crosstalk is specific for HCC. Indeed, gene expression data from Fasn(+) and Fasn(−) normal liver tissues did not reveal the upregulation of SREBP1/2 cascade in Fasn(−) normal liver (online supplementary dataset S3, online supplementary figure S29–S31). Instead, ‘response to stress’, ‘regulation of p38MAPK cascade’ and ‘de novo protein folding’ were among the most significantly upregulated pathways in Fasn(−) normal liver (online supplementary figure S31), suggesting that Fasn may function to protect normal liver from stress. Clearly, additional studies are required to define the contribution of Fasn on normal liver physiology. Furthermore, it remains to be defined how the loss of FASN triggers SREBP1/SREBP2 activity in human and mouse HCC cells. Importantly, in a recent study, it was discovered that SREBP2 is negatively regulated by the p53 tumour suppressor; and suppression of cholesterogenesis restricted HCC development induced by p53 loss in mice.27 These data support the major role of cholesterol biosynthesis pathway in hepatocarcinogenesis. Additional studies are needed to delineate the precise mechanism(s), whereby increased SREBP2/HMGCR and the related cholesterol biosynthesis contribute to hepatocarcinogenesis.
Supplemental material
The present findings have important clinical implications both in terms of prognosis prediction and innovative therapies. Indeed, our data suggest that SREBP2 activity and cholesterol biosynthesis might be a novel, negative prognostic marker in this tumour type. In terms of therapy perspectives, since the loss of FASN triggers the compensatory upregulation of HMGCR and cholesterol biosynthesis in vitro and in vivo, concomitant inhibition of FASN and HMGCR is likely to prevent this metabolic adaptation, leading to strong decrease of tumour growth in patients with HCC. Our in vivo experiments using dnSrebp2 (figure 5) and in vitro studies using siRNAs against FASN and HMGCR or SREBP2 support this hypothesis (figure 7). Altogether, our data provide important preclinical evidence supporting the testing of co-administration of FASN and HMGCR inhibitors for HCC therapy and prevention.
Acknowledgments
The authors would like to thank Dr Clay F Semenkovich (Washington University, St. Louis, Missouri, USA) for providing Fasnfl/flmice, Dr Almut Schulze (Comprehensive Cancer Center Mainfranken, Würzburg, Germany) for the dnSrebp2 plasmid, Dr Binbin Liu (Liver Cancer Institute and Zhongshan Hospital of Fudan University, China) for the MHCC97H cell line and Dr Brian Carr (Dokuz Eylul University, Turkey) for the Hep40 cell line.
References
Footnotes
Contributors Study concept and design: LGC, XC, DC; performing the experiments: LC, WC, YQ, JZ, XS, LL, JJ, AC, CC; data analysis: LC, WC, YL, JW, ZM, WK, ZT, ZZ, GS, SR, FD, ME, TO, MP, CC, GP; drafting of the manuscript: LC, WC; revising of the manuscript: LGC, XC, DC; LGC, XC and DC obtained the funding for the study; all authors have access to the study data and have reviewed and approved the final manuscript.
Funding This work is supported by National Key R&D Program of China (grant no 2018YFA0506903), National Science Foundation of China (grant no 91857108 and 81470839), National Science and Technology Major Projects for Major New Drugs Innovation and Develop (grant no 2018ZX09711003-004-002), Tsinghua University Initiative Scientific Research Program (grant no 20161080086) to LGC; NIH grants R01CA136606 to XC; P30DK026743 for UCSF Liver Center; Grant from the Italian Association Aainst Cancer (AIRC; grant no IG 19175) to DC; as well as National Science Foundation of China (grant no 81872253) to LL.
Competing interests None declared.
Ethics approval This study was approved by the local ethical committee of the Medical University of Greifswald (approval # BB 67/10).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.