Article Text

Abstract
The myeloid differentiation factor Schlafen4 (Slfn4) marks a subset of myeloid-derived suppressor cells (MDSCs) in the stomach during Helicobacter-induced spasmolytic polypeptide-expressing metaplasia (SPEM).
Objective To identify the gene products expressed by Slfn4+-MDSCs and to determine how they promote SPEM.
Design We performed transcriptome analyses for both coding genes (mRNA by RNA-Seq) and non-coding genes (microRNAs using NanoString nCounter) using flow-sorted SLFN4+ and SLFN4– cells from Helicobacter-infected mice exhibiting metaplasia at 6 months postinfection. Thioglycollate-elicited myeloid cells from the peritoneum were cultured and treated with IFNα to induce the T cell suppressor phenotype, expression of MIR130b and SLFN4. MIR130b expression in human gastric tissue including gastric cancer and patient sera was determined by qPCR and in situ hybridisation. Knockdown of MiR130b in vivo in Helicobacter-infected mice was performed using Invivofectamine. Organoids from primary gastric cancers were used to generate xenografts. ChIP assay and Western blots were performed to demonstrate NFκb p65 activation by MIR130b.
Results MicroRNA analysis identified an increase in MiR130b in gastric SLFN4+ cells. Moreover, MIR130b colocalised with SLFN12L, a human homologue of SLFN4, in gastric cancers. MiR130b was required for the T-cell suppressor phenotype exhibited by the SLFN4+ cells and promoted Helicobacter-induced metaplasia. Treating gastric organoids with the MIR130b mimic induced epithelial cell proliferation and promoted xenograft tumour growth.
Conclusion Taken together, MiR130b plays an essential role in MDSC function and supports metaplastic transformation.
- gastric cancer
- helicobacter felis
- gastric metaplasia
- gastric inflammation
- interferon-alpha
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The myeloid differentiation factor Schlafen4 (Slfn4) marks a subset of myeloid-derived suppressor cells (MDSCs) in the stomach during Helicobacter-induced spasmolytic polypeptide-expressing metaplasia, which is an early committed step appearing prior to gastric cancer.
What are the new findings?
MIR130b produced by SLFN4+ MDSCs plays an essential role in MDSC function and the levels in the blood correlate with metaplastic changes in the stomach.
How might it impact on clinical practice in the foreseeable future?
MIR130b has potential as an early non-invasive diagnostic and therapeutic biomarker for metaplastic changes in the stomach that could progress to gastric cancer.
Introduction
Metaplastic changes in the stomach typically follow chronic inflammation initiated by Helicobacter and precede neoplastic transformation. We previously reported that a subset of Hedgehog(Hh)-Gli1-dependent immune cells are recruited to the gastric epithelium during Helicobacter infection in mice and polarise into myeloid-derived suppressor cells (MDSCs),1 an event that coincides with parietal cell atrophy and spasmolytic polypeptide-expressing metaplasia (SPEM).2 MDSCs are a heterogeneous population of immature myeloid cells,3 and a subset increase their expression of Schlafen4 (SLFN4). SLFNs are a family of molecules strongly induced by type 1 interferons (IFNα), which has been implicated in lymphoid and myeloid cell development and differentiation.4 In particular, SLFN4 is a myeloid cell differentiation factor that regulates myelopoiesis.5 We previously demonstrated that SLFN4+ cells originate in the bone marrow, migrate to the stomach during Helicobacter infection and require Hh signalling. By sorting fluorescently tagged SLFN4+ cells from the stomachs of Helicobacter-infected mice, we showed that recruited SLFN4+ cells acquire their T cell suppressor phenotype 4–6 months following infection.6 Similar to murine SLFN4, protein expression of the human homologue SLFN12L increases in patients with Helicobacter pylori infection with intestinal metaplasia and also marks a population of MDSCs.6
Ostensibly, the immune suppressive function of MDSCs emerges to dampen the active inflammatory process on presumed resolution of the Helicobacter infection. However, inherent in resolving the mucosal damage, secretion of pro-proliferative signals from the myeloid cells creates a permissive environment for hyperplasia, metaplasia and eventually tumour formation.7 To define the underlying mechanism that drives the emergence of this myeloid cell subpopulation and investigate its role in tumour progression, we performed transcriptome analyses for both coding genes (mRNA by RNA-Seq) and non-coding genes (microRNAs using NanoString nCounter) using flow-sorted SLFN4+ and SLFN4– myeloid cells from the stomachs, bone marrow and spleens of Helicobacter-infected mice. MicroRNAs (miRNAs) are endogenous small, non-coding RNAs that negatively regulate target gene expression. Secreted miRNAs are recognised as mediators of intercellular communication.8 Given their stability in the blood, they can serve as stable circulating biomarkers of disease.9 Here, we show that SLFN4+-MDSCs isolated from the metaplastic mouse stomach express MiR130b. Moreover, we identified elevated levels of MIR130b in the serum of both Helicobacter-infected mice and human patients that correlated with the respective metaplastic changes in the stomach.
Results
RNA profiling of SLFN4+ and SLFN4– cells
We flow-sorted Slfn4-tdTomato+ (SLFN4+) and Slfn4-tdTomato- (SLFN4–) myeloid cells (CD11b+) from the stomachs, bone marrow and spleens of Slfn4-CreERT2:Rosa-LSL-tdTomato mice infected with Helicobacter felis (H. felis) for 6 months. The cells were profiled using two high throughput genome-wide analyses (RNAseq and miRNA Nanostring nCounter).
RNAseq identified over 5000 differentially expressed genes (fold change >2) from stomach SLFN4+ (ST4+) compared with bone marrow SLFN4+ (BM4+) and splenic SLFN4+ cells (SP4+) as well as stomach SLFN4– cells (ST4–) (online supplementary figure S1A). The hierarchical clustering heat map displayed distinct transcriptome profiles for ST4+ cells (online supplementary figure S1B), demonstrating that a distinct gene expression signature was acquired in the stomach. Many of the highly induced transcripts identified in the ST4+ cells were consistent with MDSC polarisation (online supplementary figure S1C). A subset of these genes was validated by qPCR (online supplementary figure S1D-L). Of note, MDSCs express high levels of inducible nitric oxide synthase (NOS2) and arginase I (ARG1), which consume L-arginine in the microenvironment to produce nitric oxide and reactive oxygen species.10 Reduced L-arginine blocks TCR- ζ chain synthesis and T cell proliferation. Transcripts encoding these two major effector enzymes, especially NOS2, were highly expressed in the gastric ST4+ myeloid cells, but were significantly less in the bone marrow and spleen, consistent with our prior findings that acquisition of MDSC function occurs in the stomach.6
Supplemental material
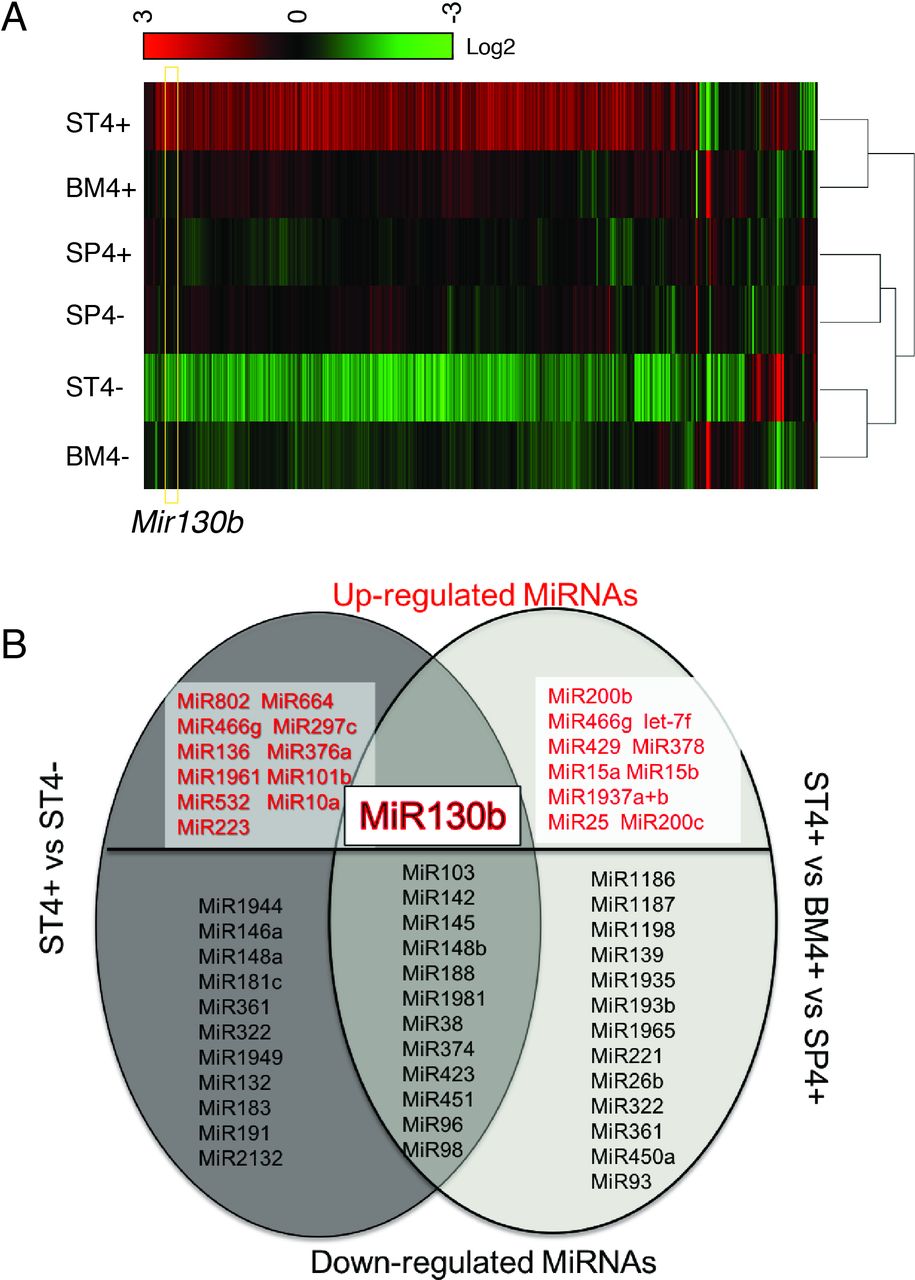
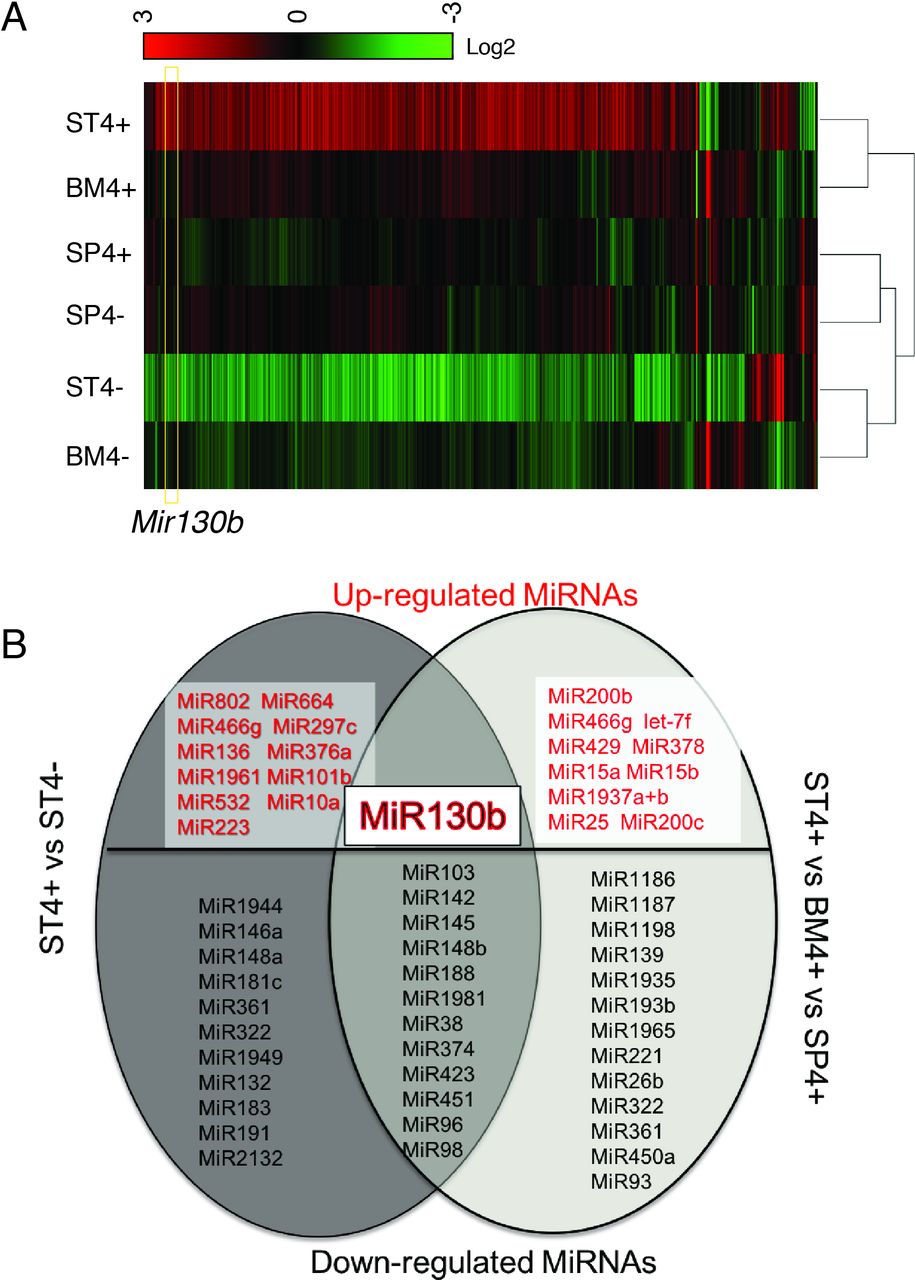
MiRNA analysis of SLFN4+ cells with miRNA profiling. WT chimeric mice reconstituted with Slfn4-tdT bone marrow were treated with Tx 2 weeks prior to euthanisation. SLFN4+ versus SLFN4– cells sorted from stomach (ST4+/–), spleen (SP4+/–) and bone marrow (BM4+/–) were profiled by the miRNA NanoString nCounter Array. (A) Cluster analysis of differentially expressed miRNAs. (B) A Venn diagram of the upregulated and downregulated miRNAs in ST4+ versus ST4– and ST4+ versus SP4+ versus BM4+ after miRNA profiling. Each sample was pooled from five mice. N=2 separate flow sorts. One-way ANOVA test. Cut-off value: p<0.05. ANOVA, analysis of variance.
Using the Nanostring nCounter profiling, we found that differences in microRNA expression in ST4+ cells differed from the BM4+ and SP4+ gene signatures (figure 1A). Thirty-five differentially expressed miRNAs (>2-fold) were identified by comparing ST4+ cells to ST4– cells. Moreover, 36 transcripts were differentially expressed by comparing ST4+ to the BM4+ and SP4+ cells (figure 1B). Of the 13 miRNAs that overlapped between the two groups, 12 miRNAs were significantly suppressed while only Mir130b was induced (figure 1B).
MiR130b correlates with mouse SLFN4 and human SLFN12L expression
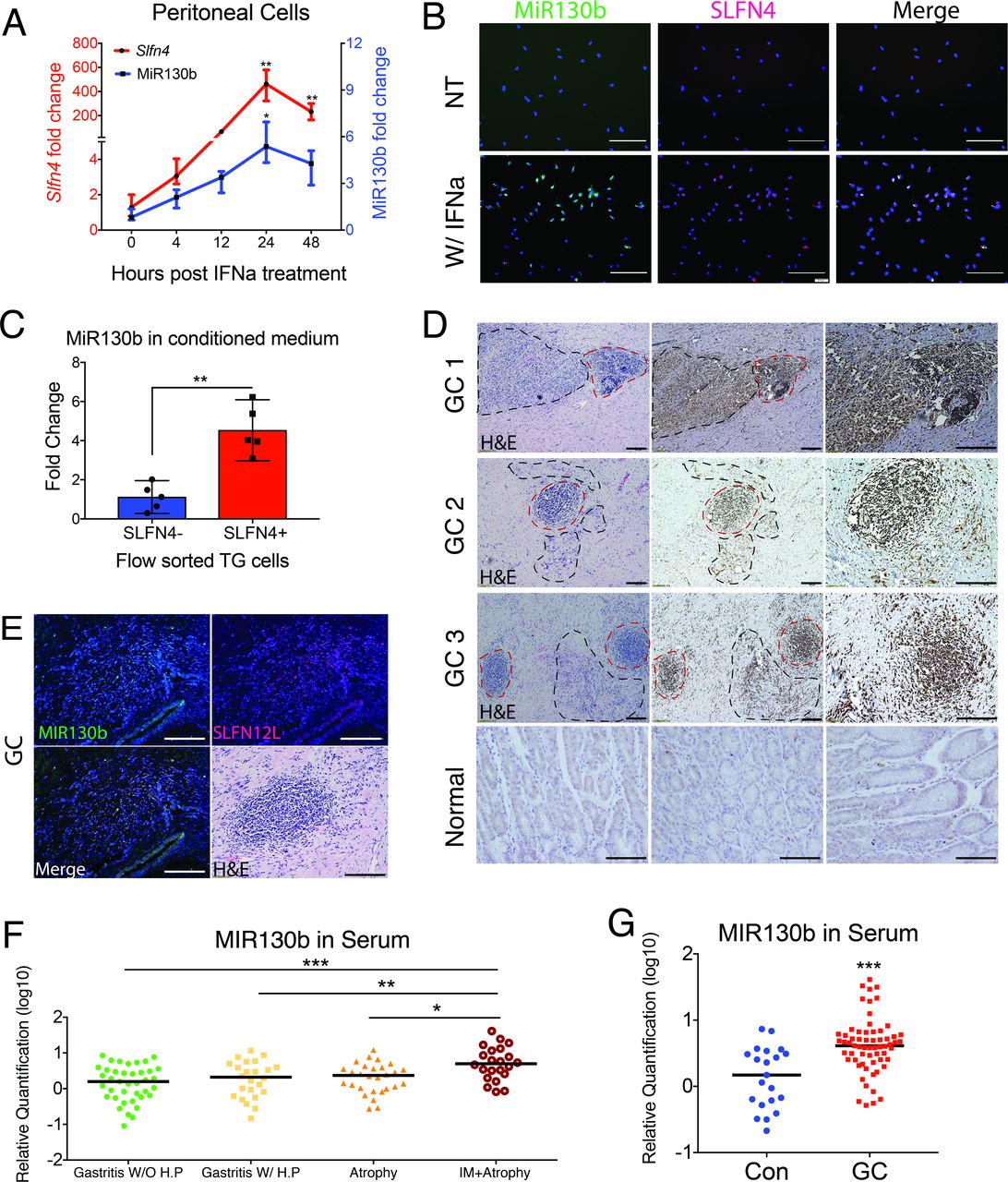
To determine whether MiR130b expressed in the gastric SLFN4+-MDSCs correlated with its expression in serum, MiR130b expression was measured in gastric extracts and serum over 6 months following H. felis infection. MiR130b increased in both the sera and gastric tissues from infected mice (online supplementary figure S2), which coincided with Slfn4 mRNA induction and the appearance of Helicobacter-induced SPEM as previously reported.6 Thioglycollate (TG)-elicited peritoneal cells were treated ex vivo with IFNα to induce myeloid cell polarisation6 and peak expression of both Slfn4 and MiR130b within 24 hours (figure 2A). MiR130b in situ hybridisation (ISH) was performed showing MiR130b localisation to the SLFN4+ myeloid cells (figure 2B). To determine if these SLFN4+ MDSCs released MiR130b, SLFN4+ and SLFN4– cells were flow-sorted after treating naïve TG peritoneal myeloid cells with IFNα for 24 hours. The amount of MiR130b found in the media of the SLFN4+ cells was 4.5-fold higher than the amount present in the SLFN4– media (figure 2C). Since Slfn4 is a Gli1-dependent gene inducibly regulated by IFNα,1 6 we showed that MiR130b expression in SLFN4+ cells was also Gli1-dependent by treating the peritoneal myeloid cells isolated from Gli1–/–:Slfn4-tdTomato+ vs Slfn4-tdTomato+ mice with IFNα (online supplementary figure S3A).

MIR130b correlates with mouse SLFN4 and human SLFN12L expression. Thioglycollate-elicited peritoneal cells collected from Slfn4-tdT mice were treated with IFNα (800 U/mL) or were NT. (A) Time course of mRNA expression for Slfn4 and MiR130b were analysed by qPCR and B) by FISH for MiR130b (green) colocalising with immunofluorescent staining for Slfn4-tdT (red). N=5 expts. Scale bar=50 µm. One-way ANOVA followed by Tukey’s multiple comparisons test on log-transformed values. P values are relative to UI or time 0. (C) Flow-sorted SLFN4+ versus SLFN4– cells after IFNα treatment were cultured for 24 hours; then, conditioned media was collected. MiR130b secreted into the media was determined by qPCR. Mann-Whitney U test was performed. (D) Combined MIR130b ISH and IHC detection in normal tissue and gastric cancer tissue collected from patients with gastric cancer. Dotted black lines mark cancer cells and dotted red lines mark immune cells positive for MIR130b expression. Representative staining from three out of five GC samples. (E) FISH with MIR130b primers (green) followed by immunofluorescent staining for SLFN12L (red), merged view and H&E stained tissue. Scale bar=100 µm. Serum MIR130b levels were determined by qPCR in (F) patients with gastritis, n=27–31 per group for four groups of patients: Gastritis without active Helicobacter pylori infection (IgG+,CLO-); Gastritis with active H. pylori infection (CLO+); with atrophy and with intestinal metaplasia. P values are relative to gastritis without active H. pylori infection group and in (G) patients with gastric cancer, n=21–63 per group. Significance was determined using Kruskal-Wallis ANOVA with Dunn’s test of multiple comparisons. *P<0.05, **P<0.01, ***P<0.001. Horizontal lines represent the median and IQR. ANOVA, analysis of variance; NT, not-treated.

MiR130b is essential for SLFN4+-MDSC activity. (A) TG-elicited PCs collected from WT mice were transfected with MiR130b mimic, MiR130b antisense (a-130b), Slfn4 siRNA or scrambled control (Scr) and then treated with IFNα (800 U/mL) or PBS for 24 hours. Differential gene expression was evaluated by qPCR. N=5 expts. (B) Protocol to collect SLFN4+ cells for coculture with T cells. TG-elicited PCs from Slfn4-tdT mice were treated with IFNα for 24 hours and then flow-sorted for SLFN4+ and SLFN4– cells. Sorted cells were then transfected with MiR130b mimic, antisense or Scr. These sorted cells and NT peritoneal cells were cocultured with activated T cells for 72 hours. (C) CFSE-based T cell suppression assay was quantified by flow cytometry. The top four representative histograms show proliferation of control groups: NO CFSE, without anti-CD3/28 microbeads activation, with CD3/28 only and cocultured with Tregs (CD4+CD25+). The median percentage of proliferating T cells is shown in the representative histograms and plotted for n=4 expts in the (D) bar graph. One-way ANOVA followed by Tukey’s multiple comparisons test on log-transformed values. The mice infected with Helicobacter felis for 4 months were treated with MIR130b antisense or scrambled control using Invivofectamine. Three weeks postinjection, (E) CD45+CD8+ cytotoxic T cells in the stomach were detected by flow cytometry and (F) metaplastic change was shown by H&E, CD44 variant 9 (red), GSII (green) and clusterin (green) staining. N=3 expts. *P values are relative to IFNα-treated group. #P values are relative to CON or scrambled. * or #p<0.05, ** or ##p<0.01, *** or ###p<0.001, ####p<0.0001. NS, not significant. The median and IQR is shown. ANOVA, analysis of variance; NT, non-treated; PCs, peritoneal myeloid cells; TG, thioglycollate; UI, uninfected.
MIR130b was also highly expressed in human gastric cancer cells and surrounding immune cells compared with normal tissue (figure 2D). Since SLFN12L is the closest human homologue of mouse Slfn4 and also marks human MDSCs,6 we colocalised SLFN12L protein with MIR130b by ISH in the gastric cancer tissue (figure 2E). MIR130b and SLFN12L also coexpressed in the HL-60 human myeloid line after coculturing with H. pylori (online supplementary figure S3B). Knockdown of GLI1 prevented SLFN12L and MIR130b mRNA expression in the presence of H. pylori (online supplementary figure S3B, C). Moreover, similar to Slfn4, IFNα strongly induced SLFN12L (online supplementary figure S3D, E). HL-60 cells treated with H. pylori with or without the GLI1 inhibitor GANT61 or GLI1 siRNA blocked both MIR130b and SLFN12L induction, indicating that MIR130b expression correlates with Slfn4 and SLFN12L myeloid expression in a GLI1-dependent manner.
We used qPCR to determine the levels of MIR130b in archived sera collected from 115 Vietnamese patients with gastritis, atrophy or atrophy plus intestinal metaplasia (online supplementary table S1 and figure 2F) and found significantly elevated serum levels of MIR130b only in patients with atrophy and metaplasia. Furthermore, analysis of a cohort of Chinese gastric cancer patients similarly demonstrated elevated serum levels of MIR130b (online supplementary table S2 and figure 2G). Therefore, as observed in Helicobacter-induced SPEM in mice, elevated MIR130b levels were observed in the serum of patients with intestinal metaplasia and gastric cancer.
MIR130b is essential for SLFN+-MDSC activity
Slfn4 is highly induced in peritoneal myeloid cells treated with IFNα and acquire the ability to suppress T cell proliferation.6 Therefore, to examine the functional effect of the microRNA on SLFN4+ cells, we compared the effect of knocking down endogenous MiR130b during IFNα induction of Slfn4 to MiR130b overexpression with the mimic. Knocking down Slfn4 with siRNA abolished the induction of MiR130b; however, knocking down MiR130b did not inhibit Slfn4 induction by IFNα, indicating that Slfn4 was required for MiR130b induction (figure 3A). IFNα, but not treatment with the MiR130b mimic induced Arg1 and Nos2 expression. However, either Slfn4 or MiR130b antisense abolished the IFNα induction of these two mRNAs, demonstrating a requirement for MiR130b and Slfn4 in the expression of enzymes characteristic of MDSCs function. To determine directly whether SLFN4+-MDSCs require MiR130b for T cell suppressor function, we flow-sorted SLFN4+ and SLFN4– cells from TG peritoneal myeloid cells treated with IFNα and cocultured them with T cells after transfecting with MiR130b mimic, antisense or scrambled oligos (figure 3B). As expected, Tregs (splenic CD4+CD25+) inhibited T cell proliferation (figure 3C,D). Flow-sorted SLFN4+ cells inhibited T cell proliferation by 60% (from 57.4% to 22%). However, no significant T cell suppression was observed when SLFN4– cells or untreated TG peritoneal myeloid cells were cocultured with activated T cells (figure 3, NT). Transfecting SLFN4– cells with the MiR130b mimic was not sufficient to impart T cell suppressor function to these cells. However, SLFN4+ cells transfected with the MiR130b antisense oligo exhibited only minimal T cell suppression. Therefore, MiR130b was required but not sufficient for the SLFN4+ cells to exhibit the MDSC T cell suppressor phenotype. To further test the effect of MiR130b, we knocked down MiR130b in vivo by IP injecting mice infected with Helicobacter for 4 months with antisense MiR130b using Invivofectamine reagent. Knockdown of MiR130b in the stomach was validated by qPCR 3 weeks after transfection (online supplementary figure S4). Suppression of MiR130b was sufficient to restore CD8+ cytotoxic T cell infiltration of the stomach (figure 3E), demonstrating impaired MDSC function. Less SPEM developed after knocking down MiR130b in vivo as shown by reduced Clusterin and coexpression of CD44 with GSII at the base of gastric glands (figure 3F) as well as less Tff2, Clusterin (Clu) and CD44v9 mRNA without significant recovery of parietal and chief cell markers H/K-ATPase α subunit (Atp4a), Gif, respectively (online supplementary figure S5).

MiR130b promotes gastric epithelial cell proliferation and xenograft tumour formation. Gastric organoids from wildtype mice were (A) transfected with MiR130b mimic, MiR130b antisense (a-130b) or scrambled control (Scr) or (B) cocultured with flow-sorted SLFN4– or SLFN4+ cells transfected with a-130b or Scr in the transwell system for 48 hours. Organoids were then stained with EdU antibody. EdU+ cells were visualised by (A) confocal or (B) quantified by flow cytometry. Quantified data were plotted in bar graphs. N=4 expts. Xenograft assays were performed by injecting subcutaneously into the right flank of the NSG mice 3 human gastric cancer patient derived organoid lines (TGO1=Diffuse type; TGO2=intestinal type; TGO4=poorly differentiated with signet ring cells) transfected with MiR130b mimic, antisense or Scr. (C) Right flank of mice at the end of the xenograft assay. Tumour height (h), length (l) and width (w) from three different xenograft (D–F) were measured every 4 days using a calliper. Tumour volume was plotted over a 36-day time course post-transplant. *Significance compared with Scr group. (G) MiR130b expression determined by RT-qPCR in NSG mouse serum and tumour tissue. One-way ANOVA followed by Tukey’s multiple comparisons test on log-transformed values. N=10 mice across three expts. *P<0.05, **P<0.01. Horizontal lines represent the median and IQR. ANOVA, analysis of variance; TGO, tumour gastric organoid.

MIR130b targets cylindromatosis (CYLD) and induces NFkb activity. (A) Microarray heat map of MIR130b candidate targets predicted by TargetScan or reported previously. Samples were pooled from five mice per expt. N=2 expts. (B) Hypothetical model illustrating the NFκB/MIR130b/CYLD axis and schematic of a typical MIR130b promoter. The predicted NFκB binding sites were located at −229 to −239, –474 to −484 and −1467 to −1477 upstream of the transcription start site. NFκB upregulates the transcription of MIR130b, which targets and decreases CYLD expression and disrupts the negative feedback of CYLD on NFκB activation. This axis constitutively activates NFκB. (C) Chromatin immunoprecipitation (ChIP) assay using HL-60 cells showed that the NFκB physically bound to the promoters13 as indicated (online supplementary methods). Lane 1, input chromatin prior to immunoprecipitation. Lane 2, immunoprecipitation with a non-specific antibody (IgG). Lanes 3 and 4, immunoprecipitation with NFκB P65 antibody after the cells were transfected with lane three scrambled control (Scr) or lane 4 MIR130b mimic. Lane 5, immunoprecipitation with RNA polymerase II (RNAP II) antibody as a positive control. (D) HL-60 cells were treated with TNFα, MIR130b mimic, antisense (a-130b) or TNFα plus MIR130b antisense (TNFα/a-130b). NFkb p65 expression was determined by Western blot. GAPDH as a loading control. N=5 expts. (E) Western blot of CYLD and NFkB-p65 expression in cytoplasmic and nuclear fractions of xenograft tumours from scrambled control (Scr), mimic and a-130b groups. (F) Bar graphs show quantitation using ImageJ. α-tubulin and histone-3 served as cytoplasmic and nucleic loading control, respectively. N=3 expts. One-way ANOVA followed by Tukey’s multiple comparisons test on log-transformed values. *P<0.05, the median±IQR. ANOVA, analysis of variance.
MIR130b promotes gastric epithelial cell proliferation and xenograft tumour formation
Since MIR130b was detected in the media of cultured SLFN+-MDSCs, we tested whether MIR130b exerts a proliferative effect on gastric epithelial cells in addition to regulating MDSC activity. Gastric cell proliferation was measured after transfecting AGS cells, a human gastric cancer cell line, and non-transformed mouse or human gastric organoids with MIR130b mimic or antisense. The MIR130b mimic induced AGS cell proliferation, while addition of MIR130b antisense decreased proliferation (online supplementary figure S6). Proliferating cells within gastric organoids from normal mice or human subjects were labelled with EdU and quantified by immunofluorescent staining and flow cytometry. MIR130b mimic doubled the number of proliferating cells, while the antisense did not significantly affect proliferation (figure 4A, online supplementary figure S7). Mouse gastric organoids were then cocultured with flow-sorted SLFN4+ or SLFN4– cells prepared after treating the TG peritoneal myeloid cells from the Slfn4-tdTomato mice with IFNα cultured in transwells. The 0.4 µm pore size of the insert membrane permitted diffusion of soluble factors from myeloid cells in the upper chamber to the lower chamber containing mouse organoids while preventing the transfer of any cells. After 48 hours, mouse gastric organoids cocultured with IFNα-induced SLFN4+ cells exhibited a greater number of proliferating cells than organoids cultured with SLFN4– cells (figure 4B). Knocking down MIR130b significantly reduced this induction. Thus, SLFN4+-MDSCs secreted MiR130b, which was capable of promoting gastric epithelial proliferation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
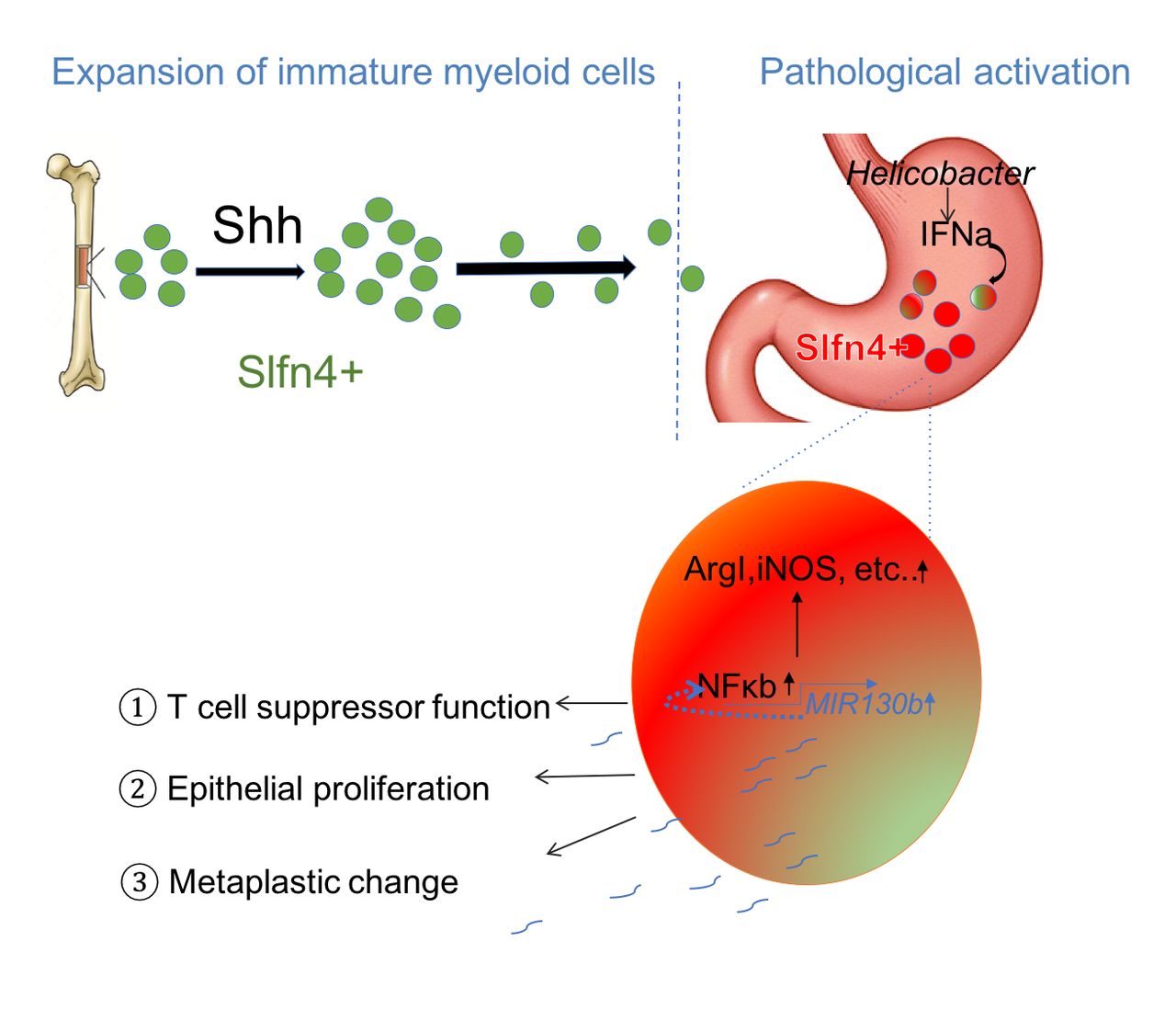
Schematic of SLFN4+-MDSCs polarised in the stomach during Helicobacter infection. Acute SHH release by parietal cells into the circulation is sensed by BM-derived SLFN4+ myeloid cells (green), which are home to the infected stomach. Eventually, the SLFN4+ myeloid cells become activated and polarised to MDSCs (red) and accumulate in response to tissue IFNα produced during chronic Helicobacter infection. There is a feedback loop between NFκb activation and MIR130b expression in SLFN+-MDSCs (enlarged red cell). MiR130b produced by SLFN+-MDSCs (1) regulate T cell suppressor function, (2) affect epithelial cell proliferation, (3) promote metaplastic changes. MDSCs, myeloid-derived suppressor cells; SHH, Sonic hedgehog.
Next, we examined the effect of MIR130b using a xenograft tumour model generated using patient-derived organoids from three human gastric cancers (diffuse, intestinal, signet ring). Organoids were pretreated with either scrambled, MiR130b mimic or antisense oligos before injecting into the right flank of immune-deficient NSG mice. Xenograft tumour volumes from the different organoid lines are shown in figure 4D–F. Tumour growth was observed in the scrambled group. However, the mimic-treated group doubled their tumour size while few antisense-treated organoids developed into tumours (figure 4C,D). To rule out the possibility that organoids in the antisense group were not viable, a cell viability assay was performed on organoids transfected with mimic, antisense or scrambled sequences prior to xenograft transplantation. All groups exhibited 60%–70% viability after transfection (online supplementary figure S8). Immunofluorescence using an antibody specific for human histone H3 confirmed engraftment of human-derived cells (online supplementary figure S9). Although differences in the MIR130b serum levels were not statistically significantly, the microRNA levels in xenograft tumours were significantly higher in the mimic group and knocked down in the antisense group (figure 4G). Collectively, MIR130b exhibited a pro-proliferative effect on gastric epithelial cells while reducing MIR130b tissue levels exerted a remarkable antitumour effect.
MIR130b targets cylindromatosis (Cyld) and induces NFκb activity
TargetScan V.7.0 was used to predict direct downstream gene targets that MIR130b potentially regulates. A heatmap was generated showing MIR130b gene targets from the public database that overlapped with transcripts differentially expressed in the RNA-seq data (figure 5A). The tumour suppressor genes Runx3, Trp53inp1, Cebpε, Pten and Cyld are previously reported MIR130b targets in various tissues11–17 and were expressed in the ST4+ cells at much lower levels than the BM4+ and SP4+ cells. Transfecting SLFN4+ peritoneal myeloid cells with the MIR130b mimic or antisense oligos showed that the mimic significantly suppressed Cyld and Trp53inp1, suggesting that they might be the relevant direct targets of MIR130b in these myeloid cells (online supplementary figure S10). Indeed, the cylindromatosis gene (CYLD) encodes a deubiquitinating enzyme that inhibits ubiquitination of Iκb and retains the NFκb heterodimer p65/p50 in the cytoplasm (figure 5B). Cyld was recently shown to be a bona fide target of MIR130b and that the NFκb subunit p65 was a potential regulator of the MIR130b locus.13 18 Therefore, we determined whether there is a feedback loop between NFκb activation and MIR130b expression, by using the human myeloid HL-60 cell line. The three predicted NFκb binding sites in the MIR130b promoter region are shown13 (figure 5B). To determine whether NFκb bound to these promoter sites, HL-60 cells were transfected with scrambled or MIR130b mimic prior to performing chromatin immunoprecipitation (ChIP). The Iκb promoter was used as a positive control for NFκb binding while GAPDH was used as a negative control. NFκb physically bound to the MIR130b promoter, and the binding activity was enhanced by overexpressing MIR130b (figure 5C). Interestingly, the NOS2 promoter is also a NFκb target.19 Accordingly, MIR130b overexpression also induced binding of NFκb to both the Nos2 and Iκb promoters, suggesting that MIR130b increases the DNA binding activity of NFκb (figure 5C). The MIR130b mimic also induced NFκb p65 expression, while knocking down endogenous MIR130b reduced basal levels of p65 and blocked induction by TNFα (figure 5D). These results demonstrated that NFκb induces MIR130b expression by directly binding to its promoter, and that MIR130b subsequently induces NFκb expression and activity.
Immunohistochemistry for p65 in the xenografts (online supplementary figure S11) and western blots of the cytoplasmic and nuclear extracts showed that NFκb translocates to the nucleus in the mimic-treated group (figure 5E,F). We observed higher expression of NFκb in both the cytoplasm and nucleus, which inversely correlated with CYLD expression, suggesting that increased NFκb activity contributed to accelerated tumour growth observed with the MIR130b mimic.
Discussion
SLFN4 is a known myeloid differentiation and T cell quiescence factor.1 4 6 We previously reported that Slfn4 marks a subset of CD11b+Gr-1+ MDSCs that coincide with Helicobacter-induced SPEM.6 Here, we demonstrate that SLFN4+-MDSCs modulate their suppressor function by producing MiR130b.
The MIR130 family has been linked to several types of cancers, including glioma,18 20 hepatocellular carcinoma,11 21 colorectal,17 pancreatic,22 renal cell,23 endometrial,24 bladder,13 breast15 as well as gastric cancer.25 26 MIR130b exerts its proneoplastic function by modulating multiple signalling pathways and suppressing expression of tumour suppressors such as TP53INP1,11 RUNX3,12 CYLD13 14 or PTEN,15 which promote self-renewal of tumour-initiating cells, proliferation, invasion and migration of various cancer cells. We show here that IFNα-polarised SLFN4+-MDSCs activate the NFκb pathway, which directly induces MIR130b expression (figure 6). Subsequently, MIR130b can sustain persistent activation of NFκb, as previously reported for bladder cancer.13 Combining our RNA-Seq with in vitro cell line analysis, we surmised that Cylindromatosis (CYLD) is one of the MIR130b gene targets that likely contributes to SLFN4+-MDSC function. CYLD is a deubiquitinase that can directly interact with an essential modulator of NFκb and the TNFR-associated factor TRAF2 to negatively regulate NFκb activity. It is noteworthy that there are NFκb response elements within the MIR130b promoter.13 Moreover, NFκb modulates multiple signalling pathways, perhaps explaining how MIR130b coordinately regulates several genes.
A by-product of the elevated tissue levels of MIR130b includes detectable levels in the circulation. While other sources of MIR130b likely exist, we observed in both mouse and human blood that elevated MIR130b levels correlate with preneoplastic events in the stomach. Moreover, MIR130b colocalised with human SLFN12L in immune cells adjacent gastric cancer cells, which coincided with its presence in the blood of these patients. This clinical evidence supports a close association between circulating MIR130b initially with gastric metaplasia that also remains elevated in gastric cancer. Since microRNAs remain stable in the circulation, they can function as biomarkers of disease.8 Indeed, lymphocytes secrete microvesicles laden with MIR130b that regulate recipient cells.27 Whether blood levels of circulating MIR130b prove to be a relevant biomarker awaits prospective validation.
Knockdown of MiR130b abolished the SLFN4+-MDSC T cell suppressive effect, suggesting that MiR130b is required for acquisition of MDSC function. Specifically, MiR130b suppression abolished Arg1 and Nos2 induction, required for MDSC activation. In addition to their immunosuppressive function, MDSCs also affect remodelling of the tumour microenvironment by producing a variety of cytokines and other molecules that can promote epithelial proliferation.28–30 Functional microRNAs can be exported and delivered to recipient cells typically by exosomes to regulate target cells. Treating primary myeloid cells, a gastric cell line and gastric organoids with MIR130b mimic showed that MIR130b promotes epithelial cell proliferation. A xenograft model of human-derived gastric cancer organoids further confirmed an essential role of MIR130b in promoting tumour formation. Since MIR130b directly stimulates epithelial cell proliferation, it might also contribute to metaplasia to cancer progression, in addition to creating a permissive immune microenvironment through T cell suppression.
In summary, we show here that MIR130b has potential as a diagnostic and therapeutic biomarker for metaplasia in the stomach that progresses to gastric cancer. Indeed, secreted microRNAs are recognised as mediators of intercellular communication and stable circulating biomarkers of disease.8 Moreover, some clinical trials have now emerged using miRNA-based therapeutics.31
Materials and methods
Transgenic mice
Gli1nLacZ/nLacZ (referred to Gli1–/–), SLFN4-CreERT2/Rosa26-tdTomato (SLFN4-tdT) and SLFN4-CreERT2/Rosa26-tdTomato/Gli1–/– (SLFN4-tdT/Gli1–/–) transgenic mouse lines have been previously described.1 6 All mice were cohoused under the same specific pathogen-free conditions and were maintained on a genetic background. The University of Michigan and the University of Arizona Institutional Animal Care and Use Committee approved all mouse protocols used in this study.
Patient samples
Expression of serum MIR130b was determined by qPCR from two cohorts of deidentified human patients: 115 subjects with gastritis from the Institute of Biotechnology in Vietnam (online supplementary table S1) (IRBMED; ID: HUM00108090). Detection of H. pylori infection was performed using H. pylori IgG ELISA and campylobacter-like organism (CLO) test (for active infection). The pathological diagnosis was determined by a GI pathologist blinded to the clinical diagnosis. Eighty-four samples were from Xiangya Hospital (China) (IRBMED; ID: HUM00113773), containing 21 healthy controls and 63 patients with gastric cancer (online supplementary table S2). Colocalisation of MIR130b determined by FISH and SLFN12L determined by immunofluorescence was performed in gastric cancer specimens and normal tissue collected from five deidentified patients who underwent surgery at Xiangya Hospital in 2016 (IRBMED; ID: HUM00113773).
Single cell preparation for transcriptome analysis
Cells isolated from stomach, spleen and bone marrow for RNAseq and microRNA array were prepared as described previously.6 The single cell suspension from different tissues was flow-sorted to collect Cd11b+SLFN4+ and Cd11b+SLFN4– cells based on tdTomato expression, using an iCyt Synergy Flow Sorter (Sony Biotechnology). To obtain sufficient mRNA for analysis, cells isolated and sorted from five mice infected with H. felis (as described previously6 for 6 months were pooled for one experiment. All six groups (ST4+, ST4–, BMS4+, BM4–, SP4+, SPS4–) of cells were from two independent experiments. Total RNA was extracted in TRIzol LS Reagent (Invitrogen) and purified using the miRNeasy Mini kit (Qiagen). The RNA integrity was assessed using the Bioanalyzer 2100 system (Agilent Technologies).
Nanostring nCounter miRNA assay for miRNA profiling
MiRNA profiling was performed at the UCLA Center for Systems Biomedicine, using the NanoString nCounter Mouse V.1.5 miRNA Expression Assay Kit (NanoString), in which more than 600 pairs of probes specific for a set of miRNAs was combined with a series of internal controls to form the Mouse miRNA Panel Code Set. Details are in the Supplementary Methods.
In situ hybridisation (ISH) detection of miRNA
ISH detection of MiR130b was performed using cells cultured on coverslips or paraffin tissue sections. The sections were deparaffinized in xylene and rehydrated in serial ethanol solutions and DEPC-treated water, then digested with 20 µg/mL proteinase K for 5 min at 37°C. For both cells and tissues, a prehybridisation incubation was performed using a 50% formamide Denhardt’s solution for 1 hour at 55°C. The single strand DNA probes labelled with 5’ 6-FAM and 3’ DIG were diluted to a working concentration of 50 ng/mL with hybridisation buffer and incubated for 24 hours at 60°C. Samples were washed with 50% formamide in 5× sodium citrate saline (pH 7.0) buffer for 20 min at 40°C. Samples were rinsed with DEPC-treated water prior to the diaminobenzidine based immunohistochemistry (Abcam, ab4238) or immunofluorescent staining of SLFN4 or SLFN12L. Probes were purchased from Integrative DNA Technologies, sense probe as a negative control.
MiR130b-3p probe: 5’6-FAM/ATGCCCTTTCATCATTGCACTG/3’DIG.
MiR130b-3p control probe: 5’6-FAM/ CAGTGCAATGATGAAAGGGCAT /3’DIG.
Cell culture and treatment
TG-elicited peritoneal myeloid cells were prepared from the Slfn4-tdT mice, as described previously.6 Cells were treated with 100 nM Tamoxifen (Tx, dissolved in DMSO) for 24 hours to induce Cre recombinase activity and tdTomato expression ex vivo. The Hsa-MIR130b-3p miRNA mimic (50 nM, Applied Biological Materials, MCH01270) and the mimic negative control (MCH00000) were transfected into cells using Lipofectamine LTX with PLUS reagent (Thermo Fisher). The Mmu-Mir130b-3p miRNA antisense (CAGUGCAAUGAUGAAAGGGCAU) (10 nM, MSTUD0173, Sigma Aldrich), Slfn4 siRNA or the scrambled controls were transfected using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher) for 48 hours. To induce Slfn4 expression, the cells were treated with 800 U/mL recombinant IFNα (R&D, #12 125–1) for 24 hours or at different time points.
T cell suppression assay
Carboxyfluorescein diacetate succinimidyl ester based T cell suppression assay was performed as described previously.6 Peritoneal cells isolated from Slfn4-tdT mice were treated with IFNα (800 U/mL) for 24 hours and then flow-sorted for SLFN4+ (tdTomato, red) and SLFN4– cells (figure 3B). SLFN4+ and SLFN4– cells were then transfected with MiR130b mimic, antisense or scrambled control for 48 hours. Suppression of T cell proliferation was assayed after the addition of SLFN4+ or SLFN4– cells for 3 days at a T cell/SLFN4 cell ratio of 10:1. Cell proliferation was analysed using an Attune Acoustic Focusing cytometer (Applied Biosystem).
In vivo RNAi transfection
MiR130b antisense or scrambled control were combined with a lipid-based in vivo RNAi transfection reagent Invivofectamine 3.0 (Invitrogen) according to the manufacturer’s instruction. Three mice infected with H. felis for 4 months were injected intraperitoneally once with 1.5 mg/kg of the antisense or scrambled oligo complexes. Three weeks after the infection, the mice were necropsied and their stomachs were collected for histology. A single cell suspension was generated for flow cytometry.
Organoid/cell coculture
Mouse gastric organoids suspended in Matrigel were seeded in each well and then overlaid with gastric organoid media as described above. Peritoneal myeloid cells treated with IFNα as described above were flow- sorted into SLFN4+ or SLFN4– groups. Next 105 of either SLFN4+ or SLFN4– cells were plated onto the 24 mm polycarbonate membrane insert (0.4 µm pore diameter; Corning) with 0.5% FBS in growth media. To initiate the coculture, inserts were combined with the wells for an additional 48 hours.
Xenograft assay
Tumour derived organoid lines were generated using the stomach tissue from three patients with gastric cancer following a previously described protocol.32 TGO1 was from a diffuse type, TGO2 from an intestinal type and TGO4 from a poorly differentiated adenocarcinoma with diffuse and signet ring cell types. The organoids transfected with MIR130b mimic, antisense or scrambled and cultured for 48 hours were injected subcutaneously into the right flank of NOD Scid Gamma mice. Morphometrics (tumour height, length and width) were measured with a calliper every 4 days. Tumour volume was calculated using a published equation33. The cell viability assay was performed on organoids transfected with Mimic/antisense/scrambled prior to xenograft transplantation (online supplementary figure S6) by flow cytometry using LIVE/DEAD Viability/Cytotoxicity Kit (Thermo Fisher, #L3224).
Real-time quantitative PCR
Total RNA from cells was extracted in TRIzol (Invitrogen) and purified with the RNeasy Minikit (Qiagen). The qPCR was performed as described previously,6 using the primer sequences described in online supplementary table S3.
For detection of miR130b, total RNA was extracted from 200 µL serum using the miRNeasy Serum/Plasma Kit (Qiagen). Before RNA extraction, Caenorhabditis elegans miRNA cel-miR39 miRNA mimic (Cat#219610, Qiagen) was used to spike the serum samples as an endogenous control for normalisation during extraction. The stem-loop reverse transcription and subsequent qPCR was performed using the Hsa-MIR130b real-time RT-PCR detection and cel-miR-39–3 p calibration kit (#MBS8244866, Mybiosource) according to the manufacturer’s instructions. Stem-loop RT primers specific for MIR130b and cel-mir39-3p bind to the 3’ portion of miRNA for transcription with reverse transcriptase. The RT product was quantified using real-time PCR that includes a specific primer set and SYBR Green dye under the following conditions: initial denaturation at 95°C for 3 m followed by 40 cycles of denaturation at 95°C for 12 s; annealing and extension at 62°C for 40 s. Fluorescence was detected using the CFX96 real-time PCR detection system (Bio-RAD).
Statistics
For qPCR and proliferation experiments, statistical analysis for significance was performed on the log-transformed values using 1-way analysis of variance with Tukey’s posthoc test for multiple comparisons (GraphPad Prism). All data were expressed as the median with the IQR. P<0.05 were considered statistically significant. The number of samples per group and replicate experiments are indicated in the figure legends.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LD and QL contributed equally.
Contributors LD and QL designed research studies, conducted experiments, analysed data and wrote the manuscript. AM, YZ, JC and ZM performed xenograft-related experiments. EFK, SM, NR and DI performed microRNA analysis. ROR conducted organoid experiments. MTHH and RS performed mouse breeding and genotyping. JKG read the histology. GPP ran the Hp IgG analysis. GZ, NTHH and LCS collected and provided clinical samples. JLM designed research studies and wrote and edited the manuscript.
Funding Support from R01DK118563-01 (to JLM) and P01DK062041-15 (to JLM), the University of Michigan Digestive Disease Centre P30DK34933-31 and National Natural Science Foundation of China (No. 81974064). The miRNA profiling using NanoString technology was performed by the UCLA Integrated Molecular Technologies Core/Centre for system Biomedicine supported by the UCLA DDRC_CURE grant P30DK41301-26.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information, and are available upon reasonable request.