Article Text

Abstract
Objective Fusobacteria are not common nor relatively abundant in non-colorectal cancer (CRC) populations, however, we identified multiple Fusobacterium taxa nearly absent in western and rural populations to be comparatively more prevalent and relatively abundant in southern Chinese populations. We investigated whether these represented known or novel lineages in the Fusobacterium genus, and assessed their genomes for features implicated in development of cancer.
Methods Prevalence and relative abundances of fusobacterial species were calculated from 3157 CRC and non-CRC gut metagenomes representing 16 populations from various biogeographies. Microbial genomes were assembled and compared with existing reference genomes to assess novel fusobacterial diversity. Phylogenetic distribution of virulence genes implicated in CRC was investigated.
Results Irrespective of CRC disease status, southern Chinese populations harboured increased prevalence (maximum 39% vs 7%) and relative abundances (average 0.4% vs 0.04% of gut community) of multiple recognised and novel fusobacterial taxa phylogenetically distinct from Fusobacterium nucleatum. Genomes assembled from southern Chinese gut metagenomes increased existing fusobacterial diversity by 14.3%. Homologues of the FadA adhesin linked to CRC were consistently detected in several monophyletic lineages sister to and inclusive of F. varium and F. ulcerans, but not F. mortiferum. We also detected increased prevalence and relative abundances of F. varium in CRC compared with non-CRC cohorts, which together with distribution of FadA homologues supports a possible association with gut disease.
Conclusion The proportion of fusobacteria in guts of southern Chinese populations are higher compared with several western and rural populations in line with the notion of environment/biogeography driving human gut microbiome composition. Several non-nucleatum taxa possess FadA homologues and were enriched in CRC cohorts; whether this imposes a risk in developing CRC and other gut diseases deserves further investigation.
- colonic bacteria
- colorectal cancer
- intestinal microbiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Fusobacterium nucleatum are specifically enriched in gut microbiomes of individuals with colorectal cancer (CRC).
The FadA adhesin and Fap2 lectin are implicated in the association between F. nucleatum and CRC.
What are the new findings?
Non-CRC southern Chinese populations carry multiple known and novel fusobacterial taxa phylogenetically distinct from F. nucleatum in their guts; these taxa are nearly absent in other surveyed populations.
Several fusobacterial taxa other than F. nucleatum are enriched in CRC cohorts relative to non-CRC controls.
Homologues of the FadA adhesin were detected in several species of Fusobacterium including F. varium and F. ulcerans, suggesting potential associations with CRC and/or disease.
How might it impact on clinical practice in the foreseeable future?
These findings indicate that CRC in southern Chinese populations may be linked to F. varium and other fusobacterial species in addition to F. nucleatum.
Use of microorganisms as disease biomarkers or targets for therapeutic intervention needs to be tailored according to discrepancies in gut microbiome composition among human populations.
Introduction
Fusobacterium nucleatum is a bacterial pathogen most well-known for its association with colorectal cancer (CRC) in humans. Irrespective of biogeography, multiple studies have consistently reported enrichment of F. nucleatum in the guts1–7 and tumour tissue8–10 of CRC subjects compared with non-CRC cohorts. Furthermore, the association between F. nucleatum and CRC has been demonstrated through cell model studies, implicating two proteins FadA11 12 and Fap213 14 in facilitating adherence, invasion and induction of oncogenic and inflammatory responses in CRC cells by F. nucleatum.
In contrast, relatively less is known about the biology of fusobacterial species other than F. nucleatum and their roles in human health, if any. According to the List of Prokaryotic names with Standing in Nomenclature (LPSN), there are 21 recognised species in the Fusobacterium genus at the time of writing. Apart from F. nucleatum, a few other species such as F. necrophorum,15 F. gonidiaformans,16 F. periodonticum,17 F. mortiferum, F. ulcerans and F. varium 18 have been reported in human-associated samples. For example, F. necrophorum are often associated with thrombophlebitis of the internal jugular vein (termed Lemierre’s syndrome), F. gonidiaformans found in urogenital and intestinal tracts,16 F. periodonticum in oral cavities associated with squamous cell carcinoma,19 F. ulcerans in skin ulcers20 and F. varium in human guts associated with ulcerative colitis (UC).21 22 Apart from cases of disease, their prevalence and relative abundances in guts of healthy individuals are relatively low, often below detection thresholds23–28 consistent with the notion that the presence of Fusobacterium in human guts is specifically associated with CRC.26
We initially produced shotgun metagenomes using stools collected from 556 self-reported healthy individuals recruited for establishing a gut microbiota databank in Hong Kong (HKGutMicMap project). These data were analysed together with publicly-available stool metagenomes of other healthy subjects from Hong Kong,2 29 Austria,4 China,30 31 Denmark,32 France, Germany,5 Israel,33 Spain,32 Sweden34 35 and the USA,3 27 as well as several rural populations from El Salvador, Peru,36 Fiji,37 Mongolia38 and Tanzania39 40 to assess variation in gut microbial community composition across different biogeographies. We serendipitously observed a consistent increase in prevalence and relative abundance of multiple fusobacterial species in the Hong Kong, Chinese and Spanish but not American, European and other rural cohorts, concordant with the idea of variation in human gut microbiomes primarily driven by environment/geography.41 Here, we reconstructed fusobacterial genomes from the Hong Kong gut metagenomes and showed that F. varium, F. ulcerans, F. mortiferum and other as yet uncharacterised fusobacterial taxa are prevalent in this population. We then investigated whether these genomes contained characteristics that could indicate potential associations with cancer and/or disease. Findings reported here suggest that the fusobacterial lineages prevalent in the Chinese gut possess genomic potential to facilitate development of CRC and possibly other diseases.
Materials and methods
HKGutMicMap cohort sample collection and DNA sequencing
Subjects were recruited from the Hong Kong public as part of the HKGutMicMap study to generate gut microbiome profiles representative of the local, non-disease population. A research associate measured parameters such as body weight, waist circumference, body height and blood pressure and subjects were provided stool collection kits for self-collection. They were asked to deliver fresh stools to the laboratory within 2 hours of defecation. Faecal specimens were stored at −80°C until further processing. DNA was extracted from 0.1 g homogenised fractions of stool using the DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany) following manufacturer’s instructions. The concentration of extracted DNA was determined using the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, Massachusetts) and normalised to 20 ng/µL with 10 mM Tris-HCl. Normalised DNA samples were sent to a sequencing service provider (Novogene HK Company Limited, Wan Chai, Hong Kong) for library preparation and paired-end shotgun metagenomic sequencing (Illumina NovaSeq 6000). A mock community sequencing control (ZymoBIOMICS Microbial Community DNA Standard, catalogue number D6305, Zymo Research, Irvine, California) was included.
Prevalence and relative abundances of fusobacterial species based on gut metagenomes
To examine prevalence and relative abundances of fusobacterial species in guts of human populations from different geographical backgrounds, we included non-CRC gut metagenome sequence data generated by previous studies in Hong Kong,2 29 China,30 31 USA,3 27 Austria,4 Denmark,32 France, Germany,5 Spain,32 Israel,33 Sweden,34 35 El Salvador, Peru,36 Fiji,37 Mongolia38 and Tanzania37 40 (online supplementary table S1). The Hong Kong, Austria, France, Germany and one USA cohort3 were comprised of CRC and non-CRC subjects. These gut metagenome data sets were chosen because they have been binned for microbial genomes.42–44 Together with data generated from the HKGutMicMap cohort (present study), raw sequences were quality-filtered using Trimmomatic V.0.38 to remove adapter and low quality regions. Next, microbial community compositional profiles were inferred from quality-filtered sequences (forward reads) using MetaPhlAn245 V.2.6 with the v20 database. For each fusobacterial species identified by MetaPhlAn2, their prevalence rates were calculated based on the number of samples each species was detected in (ie, relative abundance >0%) divided by the total number of samples in the respective cohorts.
Supplemental material
Binning fusobacterial population genomes from metagenomes
To explore genomic diversity of fusobacterial species in the Hong Kong population, we assembled metagenomes from the Hong Kong cohorts and binned population genomes (termed metagenome-assembled genomes (MAGs)) from each de novo assembly. Overlapping sequence pairs in the quality-filtered data were first merged to produce longer sequences, and then assembled together with unmerged pairs using MEGAHIT46 V.1.1.1. Sequence coverage profiles were then obtained by mapping quality-filtered reads to their respective assemblies using BWA-MEM47 V.0.7.17. With these coverage information, MAGs were binned from each of the metagenomes using MetaBAT48 V.2.10.2, MetaBAT V.2.12.1 and MaxBin49 V.2.2.5. A non-redundant set of MAGs were calculated by merging output from the three sets of bins using DASTool50 V.1.1.0. The resulting non-redundant MAGs were quality-checked using the lineage workflow in CheckM51 V.1.0.13. MAGs with >90% completeness and <5% contamination were retained, and their taxonomy was inferred using Genome Taxonomy Database52 (GTDB) toolkit (GTDB-Tk) V.0.2.2 database release 86_2.
Construction of fusobacterial phylogenetic trees
We downloaded fusobacterial reference genomes from the National Centre for Biotechnology Information (NCBI) RefSeq database (release 89), and MAGs from recent publications that have assembled microbial genomes from the human metagenome data sets used above.42–44 These genomes were checked for completeness and contamination using CheckM, and only those with >90% completeness and <5% contamination were retained. We constructed two phylogenetic trees of the Fusobacterium genus—one using a dereplicated set of fusobacterial reference genomes and MAGs to highlight existing genomic diversity, and the other using all genomes to explore distribution of putative virulence protein homologues in this genus (described in section below). We included Cetobacterium, a member of the Fusobacteriaceae family as outgroup taxa in both trees. For the first tree, genomes were dereplicated using dRep V.1.4.3 based on genome distances and average nucleotide identities (ANI),53 and a concatenated amino acid alignment of 120 phylogenetically informative single-copy bacterial marker genes was generated using GTDB-Tk. A maximum-likelihood tree was constructed based on this alignment using RAxML54 V.8.2.11, and node support estimated from 100 bootstraps. For the second tree without genome dereplication, a concatenated amino acid alignment consisting of all genomes was generated and used to infer a bootstrapped phylogeny as per the first tree.
Annotating genes in fusobacterial genomes
First, protein-coding sequences in all MAGs and reference genomes were translated into amino acid sequences using Prodigal55 V.2.6.3. Amino acid sequences were aligned against the UniRef100 database (March 2018) using DIAMOND56 V.0.9.24 (≥30% sequence identity and ≥70% alignment length between query and reference) to identify gene families, and aligned counts were collated according to Kyoto Encyclopaedia of Genes and Genomes orthology to infer presence/absence of gene families. ANI comparisons were performed using FastANI57 V.1.1.
To explore presence/absence of two known CRC-associated fusobacterial genes FadA and Fap2, we annotated fusobacterial genomes using eggNOG-mapper58 V.2.0.1 with reference to eggNOG database V.5.0. The presence/absence of FadA and Fap2 homologues was visualised on a phylogenetic tree consisting of all 663 fusobacterial genomes. Amino acid gene trees were constructed for both FadA and Fap2 by aligning the putative homologues using MAFFT59 V.7.407 and inferring a maximum likelihood tree using RAxML. Both trees were midpoint-rooted using GenomeTreeTk V.0.0.53 (https://github.com/dparks1134/ GenomeTreeTk).
Isolation of Fusobacterium from stools
Frozen stools were thawed and diluted in brain heart infusion culture medium. Dilutions were inoculated onto blood agar plates and anaerobically cultured at 37°C for 2 days. Colonies were identified using a MALDI Biotyper (Bruker, Billerica, Massachusetts). Colonies identified as Fusobacterium were subcultured onto fresh blood agar plates. Genomic DNA was extracted from pure cultures using a Gentra Puregene Yeast/Bact. DNA isolation Kit (QIAGEN, Hilden, Germany), and sent to Novogene HK for library preparation and paired-end shotgun metagenomic sequencing (Illumina NovaSeq 6000). Trimmomatic quality-filtered reads were assembled using MEGAHIT V.1.1.1 and annotated using eggNOG-mapper V.2.0.1 with reference to eggNOG database V.5.0.
Data availability
Raw sequence data generated for this study are available in the Sequence Read Archive under BioProject accession PRJNA557323.
Results
The HKGutMicMap cohort
At the time of analysis, the HKGutMicMap cohort representing the general population in Hong Kong consisted of 556 subjects with shotgun metagenome data. These subjects were self-reported as healthy with no chronic disease. There were 294 females to 262 males, and their median age at time of sample collection was 51 years (SD 16.3 years). The median body mass index was 22.7 kg m-2 (SD 3.4). These and other parameters such as body weight, blood pressure and waist circumference are listed in online supplementary table S2.
F. mortiferum, F. ulcerans and F. varium are prevalent in Chinese populations irrespective of CRC disease status
In total, 3157 stool metagenomes comprising non-CRC and CRC subjects were included in this study. These metagenomes represent populations from China (Hong Kong, Shenzhen and Zhejiang), USA, Austria, Denmark, France, Germany, Spain, Sweden, Israel, El Salvador, Peru, Fiji, Mongolia and Tanzania. To assess the distribution of fusobacterial species across biogeography of these populations, quality-filtered sequences from each metagenome were mapped to lineage-specific marker genes using MetaPhlAn2 to produce prevalence and relative abundance estimates.
In non-CRC subjects (n=2515), overall gut microbial community composition significantly differed among cohorts (p<0.05, permutational multivariate analysis of variance; figure 1A, online supplementary figure S1). At the phylum level, the three Chinese and USA cohorts had higher relative abundances of Bacteroidetes compared with Firmicutes (63% vs 30%), whereas Firmicutes were more relatively abundant in the other cohorts compared with Bacteroidetes (55% vs 29%). Peru was the exception with Actinobacteria being the most dominant phylum (60%) (online supplementary table S3). In addition, Spirochaetes were detected at >1% relative abundance only in El Salvador, Fiji and Tanzania cohorts, compared with an average 0.004% in the Western and Chinese cohorts. Fusobacterium were also more relatively abundant in the Chinese and Spanish (average 0.47%) compared with other cohorts (0.01%). We were interested in the comparatively higher relative abundances of Fusobacterium since F. nucleatum has been widely implicated in CRC. Within the fusobacterial genus, F. mortiferum, F. nucleatum, F. ulcerans and F. varium were more prevalent and relatively abundant in the Chinese cohorts relative to others including Spain (p<0.001, Kruskal-Wallis test adjusted for false discovery rate) (table 1, figure 1B).
Supplemental material

Microbial community composition of the typical human gut. (A) Average relative abundances of microbial phyla detected in human stool metagenomes from the HKGutMicMap cohort (this study) and previously described non-colorectal cancer (CRC) individuals from various geographical backgrounds. (B) Average relative abundances of fusobacterial species. The stacked bars represent cohorts from: Hong Kong (HKGutMicMap and two others),2 29 Austria,4 China,30 31 Denmark,32 France, Germany,5 Israel,33 Spain,32 Sweden34 35 and the USA,3 27 as well as several rural populations from El Salvador, Peru,36 Fiji,37 Mongolia38 and Tanzania.39 40 Relative abundances were calculated using MetaPhlAn2 on quality-filtered metagenome sequences. Values shown in (B) for fusobacterial species are percentages of the total community. For case-control studies with CRC cohorts,2–5 29 only non-CRC individuals were included in the calculation of relative abundances.
Prevalence and average relative abundances of fusobacterial species in non-CRC subjects
In CRC subjects (n=642), average fusobacterial relative abundances in subjects from Hong Kong were higher than the USA, German and Austrian, but not the French cohort (online supplementary figure S2). F. nucleatum was detected in all six cohorts as expected for CRC. F. varium, F. ulcerans and F. mortiferum were more prevalent in Hong Kong (online supplementary table S4). In addition, the French were enriched in F. gonidiaformans and F. necrophorum relative to the others. F. ulcerans was also present in the Austrian cohort, however, its prevalence was still six fold higher in Hong Kong. These findings indicate that F. mortiferum, F. ulcerans and F. varium are typically more common and detected at higher relative abundances in the guts of Hong Kong populations compared with some North Americans and Europeans, irrespective of CRC disease status.
Several fusobacterial species other than F. nucleatum are enriched in CRC
F. nucleatum and more broadly the Fusobacterium genus have been shown to be enriched in the guts of CRC patients compared with non-CRC controls,6 7 although associations between CRC and other fusobacterial species have not been specifically mentioned. Since F. mortiferum, F. ulcerans and F. varium were more prevalent and relatively abundant in the guts of Chinese populations, we wanted to know whether their distributions and abundances were changed in association with CRC akin to F. nucleatum. We compared the prevalence and relative abundances of the fusobacterial species between CRC and non-CRC subjects in studies with case-control cohorts, and found that values for F. gonidiaformans and F. nucleatum were increased in all six CRC cohorts, and F. periodonticum and F. varium in five of six CRC cohorts compared with non-CRC cohorts (online supplementary tables S4,S5). A generalised linear model taking into account cohorts indicated that relative abundances of F. nucleatum and F. varium were significantly associated with CRC disease (p<0.05), although the prevalence of F. varium was not as striking as F. nucleatum.
Population genomes from Chinese gut metagenomes reveal expanded diversity in the Fusobacterium genus
At the time of writing, there were 157 fusobacterial genomes in the NCBI RefSeq database (release 89), of which 65 (41.4%), 36 (22.9%) and 17 (10.8%) are classified as F. nucleatum, F. necrophorum and F. periodonticum, respectively (online supplementary table S6). The other 18 recognised fusobacterial species according to the LPSN and any novel taxa yet to be classified are represented by the remaining 39 genomes. Since MetaPhlAn2 indicated that fusobacterial species such as F. mortiferum, F. ulcerans and F. varium were more prevalent in the guts of Chinese populations, we wanted to explore and expand known genomic diversity of these less characterised fusobacterial lineages. Using metagenomes from the Hong Kong cohort (inclusive of non-CRC and CRC subjects), we binned 171 high quality fusobacterial MAGs (>90% complete,<5% contamination based on CheckM’s lineage workflow) (online supplementary table S7). Another four high quality fusobacterial MAGs we previously binned from gut metagenomes of patients from a clinic were also included in this study (annotated in online supplementary table S7). In addition, recent efforts in characterising genomic diversity of human microbiomes42–44 have yielded an additional 336 high quality fusobacterial MAGs (online supplementary table S7). Together with 152 high quality fusobacterial genomes from RefSeq R89, we first dereplicated these 663 genomes and inferred a genome tree to establish their phylogenetic relationships to one another. Dereplication was performed to highlight existing genome diversity in the Fusobacterium genus, resulting in a phylogenetic tree comprising 218 unique fusobacterial genomes. Taxonomic information for all MAGs and reference genomes inferred according to the GTDB release 86_v2 was then appended onto the phylogenetic tree (figure 2).
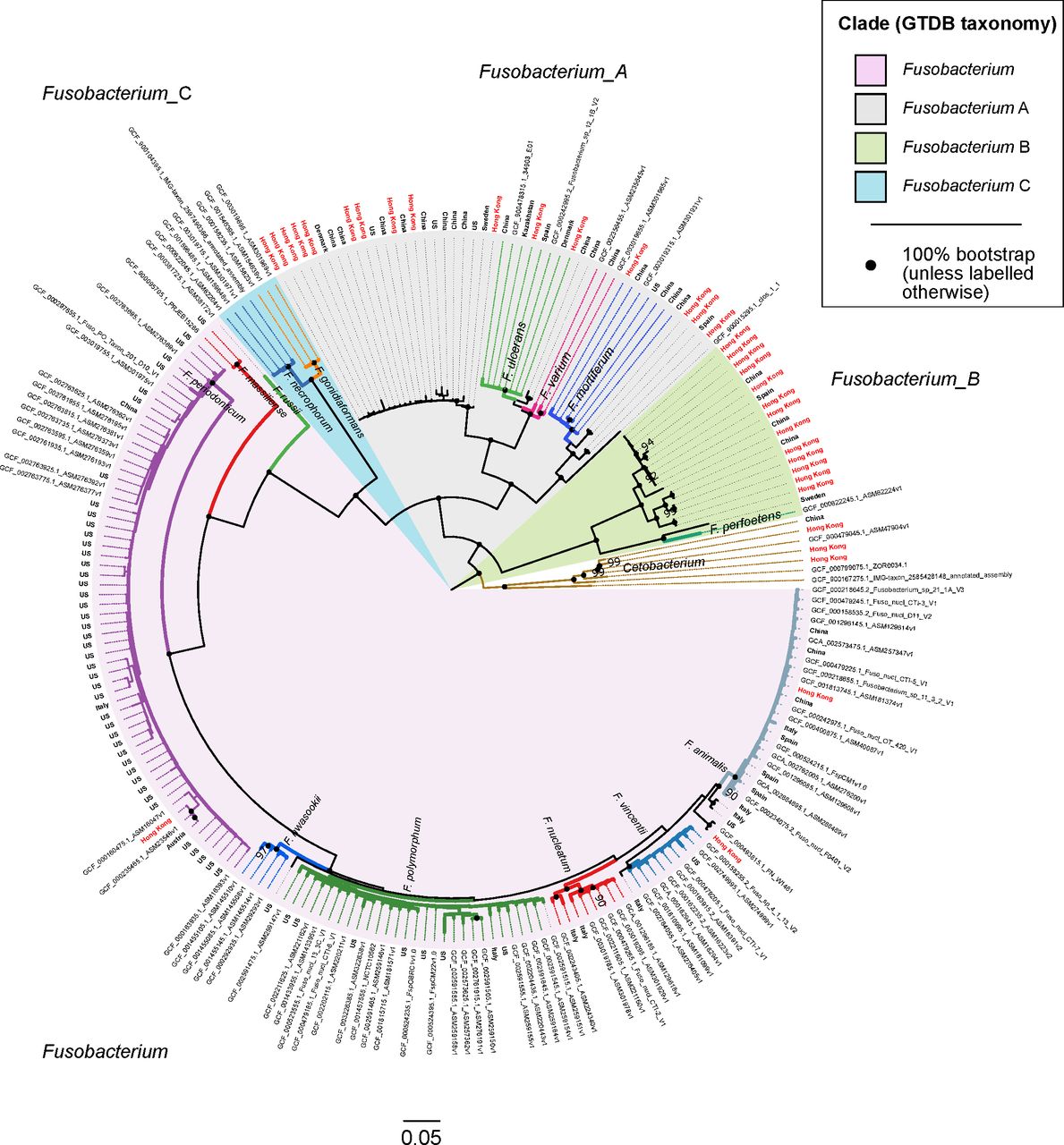
Phylogenetic tree showing evolutionary relationships among 218 fusobacterial genomes. Seven Cetobacterium genomes serve as outgroup to root the tree. Genomes in this figure are from a dereplicated set of 676 fusobacterial and Cetobacterium genomes assembled from gut metagenomes from Hong Kong (HKGutMicMap, Yu et al 2 and Coker et al 29) and other regions,42–44 and reference genomes downloaded from RefSeq (release 89). Reference genomes obtained from RefSeq are labelled with their corresponding accession numbers, while metagenome-assembled genomes have branch labels showing their country of origin (those from Hong Kong are in red text). All 676 genomes were >90% complete and had <5% contamination based on the lineage workflow in CheckM,51 and were dereplicated using dRep53 to highlight existing genome diversity of the Fusobacterium genus in this figure. A concatenated amino acid alignment was produced to infer taxonomy of the genomes according to the genome taxonomy database (GTDB),52 and subsequently used to construct maximum likelihood trees using RAxML.54 Four major monophyletic clades in the Fusobacterium genus are shaded and denoted with suffixes according to the GTDB. Branch colours are intended to delineate species boundaries (indicated by labels) and do not represent any taxa in particular; genomes without species designations have black branches. Black circles at nodes represent 100% bootstrap support unless otherwise indicated (no less than 90% bootstrap). Scale bar indicates number of amino acid substitutions per site.
Four major monophyletic lineages (termed clades) were resolved within the Fusobacterium genus congruent with taxonomic inferences produced by GTDB (denoted with suffixes Fusobacterium, Fusobacterium_A, Fusobacterium_B and Fusobacterium_C) (figure 2). The clade denoted as Fusobacterium was comprised of F. nucleatum including its traditional subspecies animalis, vincentii, nucleatum and polymorphum, F. hwasookii, F. periodonticum, F. massiliense and F. russii. Fusobacterium_A was comprised of F. ulcerans, F. varium, F. mortiferum and various unclassified fusobacterial genomes; Fusobacterium_B comprised of F. perfoetens and other unclassified genomes; Fusobacterium_C of F. gonidiaformans and F. necrophorum. Lineages within the Fusobacterium_A and Fusobacterium_B clades were highly represented by genomes derived from Hong Kong and Chinese metagenomes (48 of 67 genomes; many genomes from RefSeq do not have accompanying geographic information and were assumed to be of non-Chinese origin) (figure 2). In contrast, the Fusobacterium and Fusobacterium_C clades were more represented by genomes from other regions (only 7 of 151 genomes were from Chinese sources). MAGs from Chinese populations collectively increased phylogenetic diversity of the overall tree by 14.3% based on branch lengths, indicating that the Chinese gut harbours novel fusobacterial diversity not yet represented by reference genomes. To demonstrate that these novel fusobacteria were indeed more prevalent in Chinese populations, we mapped sequences from all non-CRC samples to the dereplicated set of 218 fusobacterial genomes and counted the proportion of aligned sequences in each cohort. The Chinese had 10–100-fold higher proportions of reads mapped to Fusobacterium_A genomes compared with other cohorts (online supplementary table S8, figure S3), consistent with MetaPhlAn2 estimates of higher relative abundances of Fusobacterium_A lineages in Chinese samples. Similarly, in CRC samples the Hong Kong cohort generally showed 10–100-fold higher proportions of reads mapped to Fusobacterium_A genomes compared with Austrian, French, German and USA samples (online supplementary table S9, figure S4).
Circumscribing new species in the Fusobacterium genus
Using the 218 dereplicated fusobacterial genomes, we performed pairwise ANI comparisons to establish species boundaries with reference to intraspecies and interspecies cutoffs derived from published studies (intraspecies >95% ANI; interspecies 78%–95%).59 60 With these cutoffs, we identified (i) six putative species in the Fusobacterium_B clade not including F. perfoetens, (ii) three species basal to F. mortiferum, (iii) one species sister to F. ulcerans, (iv) one species sister to the F. ulcerans and F. varium lineage, (v) one species basal to the lineage containing F. polymorphum, nucleatum, vincentii and animalis and (vi) two species basal/sister to F. animalis (online supplementary figure S5, table S10). These genomes share <95% ANI to any circumscribed fusobacterial taxa, and could represent novel or one of the 18 recognised species in the LPSN yet to have genome representation. In addition to drawing species boundaries, we could infer the degree of intraspecies genome similarity by comparing the initial number of 663 genomes to the resulting number of dereplicated genome clusters. For example, we observed that F. mortiferum was highly clonal despite its high prevalence in Chinese populations, whereas F. periodonticum genomes were more variable and formed more clusters of unique genomes compared with F. mortiferum (figures 2 and 3, online supplementary table S11).

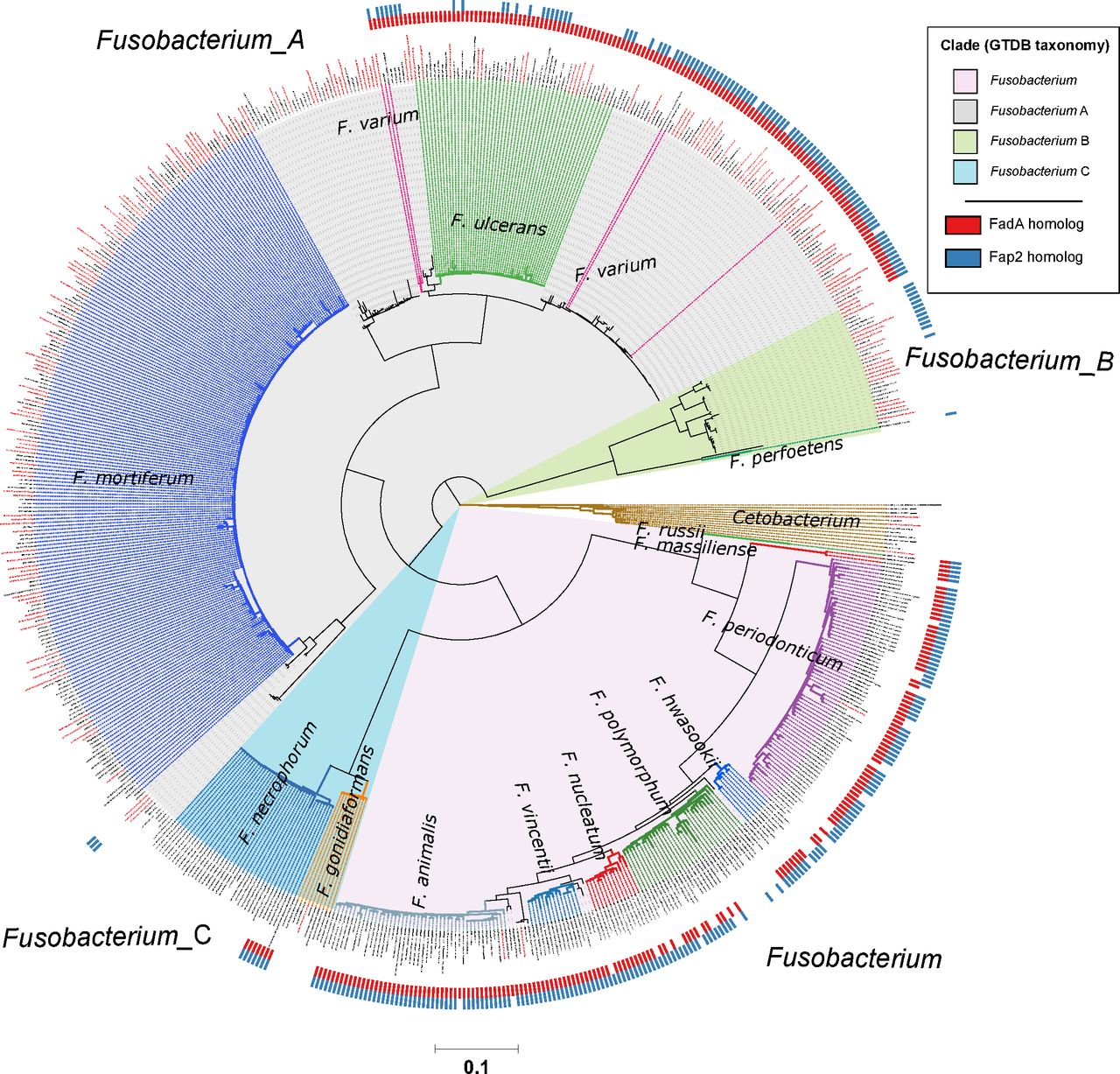
Distribution of FadA and Fap2 homologues in the Fusobacterium genus. Red and blue ticks next to branch tips indicate detection of FadA and Fap2 homologues, respectively, in the corresponding genomes. Homologues were identified using the eggNOG-mapper58 with reference to the eggNOG database V.5.0. This phylogenetic tree consists of 663 fusobacterial and 13 Cetobacterium genomes assembled from gut metagenomes from Hong Kong (HKGutMicMap cohort from this study, Yu et al 2 and Coker et al 29 cohorts) and other regions,42–44 and reference genomes downloaded from RefSeq (release 89). Reference genomes obtained from RefSeq are labelled with their corresponding accession numbers, while metagenome-assembled genomes are labelled with bin IDs. Genomes from Hong Kong have labels in red. All genomes were >90% complete and had <5% contamination based on the lineage workflow in CheckM.51 A concatenated amino acid alignment was produced to infer taxonomy of the genomes according to the genome taxonomy database (GTDB),52 and subsequently used to construct maximum likelihood trees using RAxML.54 Four major monophyletic clades in the Fusobacterium genus are shaded and denoted with suffixes according to the GTDB. Branch colours are intended to delineate species boundaries (indicated by labels) and do not represent any taxa in particular; genomes without species designations have black branches. Scale bar indicates number of amino acid substitutions per site.
Fusobacterial genome features possibly associated with disease
Previous functional analyses of CRC gut metagenomes have revealed features such as shifts towards amino acid degradation and trimethylamine (TMA) production via choline metabolism.6 7 61 We annotated the fusobacterial MAGs and observed that while they did not contain key genes involved in TMA production (TMA-lyase (cutC, K20038), and L-carnitine/gamma-butyrobetaine antiporter (caiT, K05245)), several orthologues such as proline iminopeptidase (K01259), glutamate formiminotransferase (K00603) and tryptophanase (K01667) were prevalent in genomes from the Fusobacterium clade (online supplementary table S12). Moreover, Fusobacterium clade genomes possess genes that may be involved in the catabolism of amino acids and production of glucose (phosphoenolpyruvate carboxykinase K01610, fructose-bisphosphate aldolase K01623, oxaloacetate decarboxylase K01571), as well as several other features that could be linked to cancer such as iron scavenging (K07230, K07243, K11707, K11708, K11709, K11710),62 ceramide glucosyltransferase (K00720) involved in production of glycosylated sphingolipids63 and para-aminobenzoate synthetase (K01664, K01665) in the production of folate.64 Likewise, urease (K01428–K01430)65 was detected in Fusobacterium_A and Fusobacterium_B clades but not Fusobacterium. Some of these features are consistent with those identified in CRC gut microbiota metagenomes, though it is important to point out that these findings do not imply that fusobacteria contribute wholly to the altered microbial functional signature in CRC guts6 7 as they are typically <1% relative abundance. In addition, the distribution of features by clades suggest that disease associations, if any, likely vary among the fusobacterial lineages.
Homologues of colorectal cancer-associated fadA and Fap2 are present in several fusobacterial species
Previous cell model studies have identified two proteins in F. nucleatum that allow the bacterium to potentiate CRC, namely the FadA adhesin9 10 and Fap2 lectin.11 12 To identify whether fusobacterial species other than F. nucleatum also possess similar genes that may allow them to interact with CRC cells, we annotated all 663 fusobacterial genomes with reference to the eggNOG database and searched for putative homologues. A phylogenetic tree incorporating all 663 genomes was constructed to visualise distribution of these homologues within the Fusobacterium genus. For FadA, a total of 999 homologues (online supplementary table S13) were identified in 311 genomes including all lineages belonging to the Fusobacterium clade, a monophyletic subset of F. necrophorum, and in F. varium, F. ulcerans and several uncharacterised monophyletic taxa in the Fusobacterium_A clade (figure 3). These FadA homologues possibly comprise three or more protein families as shown by a protein tree constructed from amino acid alignments. Sequences from the Fusobacterium_A clade were distinct compared with those from the Fusobacterium clade, while homologues from F. necrophorum were placed together with the Fusobacterium homologues (figure 4). These observations indicate that FadA homologues from F. varium, F. ulcerans and uncharacterised Fusobacterium_A lineages could have distinct roles compared with homologues found in the Fusobacterium clade. For Fap2, we identified 754 putative homologues in 288 genomes (online supplementary table S14). Lineages in which Fap2 homologues were identified largely overlapped with FadA, encompassing members of the Fusobacterium clade, a monophyletic subset of F. necrophorum genomes, several F. varium and F. ulcerans, and in a subset of Fusobacterium_B clade genomes (figure 3, online supplementary figure S6). Overall distribution of the FadA and Fap2 homologues in the Fusobacterium genus suggests that the potential association with CRC may be present in several distinct fusobacterial lineages. Since some of these fusobacterial species were increased in relative abundance and prevalence in CRC compared with non-CRC subjects, the detection of FadA and Fap2 homologues suggest that species such as F. varium may have the ability to potentiate disease akin to F. nucleatum.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
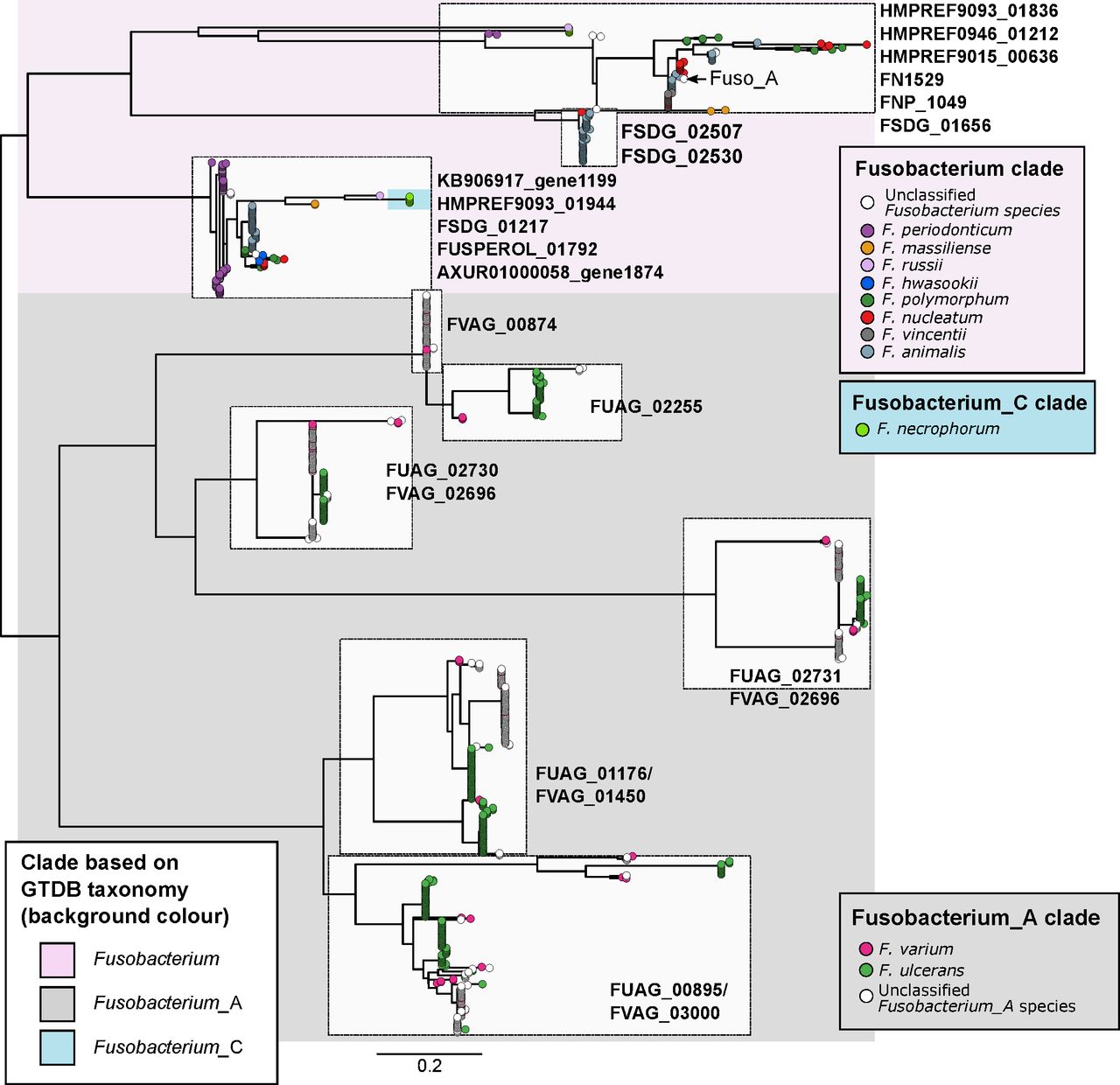
Phylogenetic relationships of FadA protein homologues identified in fusobacterial genomes. Figure shows a maximum-likelihood tree of aligned amino acid sequences of FadA homologues rooted at the midpoint. Each tip represents a homologue and is coloured according to species of the genome homologues were found in. Text labels next to tree tips indicate the corresponding seed orthologues in the eggNOG database. Background shading is according to the four major monophyletic clades identified in the genome-based phylogenetic tree in figure 2. Scale bars indicate amino acid substitutions per site.
To check that the fusobacterial MAGs recovered from metagenome data are representative of actual genomes, we isolated and sequenced genomes of eight fusobacteria obtained from five stool samples. These genomes were classified as Fusobacterium_A (seven genomes) and F. ulcerans (one), and scored >99% ANI to MAGs recovered from Hong Kong gut metagenomes (online supplementary table S15). Moreover, they contained FadA and Fap2 homologues consistent with their phylogeny, providing confidence that MAGs recovered here indeed represent real microbial genomes. Nevertheless, we recognise that MAG validation with only eight isolates is inadequate, and more work is needed to verify MAGs representing other fusobacterial lineages.
Discussion
While it has been established in human populations from various geographical backgrounds that F. nucleatum is associated with CRC,1–5 less is known about the distribution of other fusobacterial species in human guts. Here, we showed that fusobacterial lineages such as F. ulcerans, F. varium, F. mortiferum and multiple uncharacterised taxa are more prevalent in the guts of non-CRC Chinese and Spanish cohorts compared with counterparts from several geographical regions. While these non-nucleatum taxa may simply reflect biogeographical differences in human gut microbiome composition, we saw two lines of evidence suggesting that they could possess oncogenic and/or disease-causing potential: (i) increased prevalence and relative abundances in CRC compared with non-CRC cohorts (online supplementary table S5) and (ii) detection of virulence gene homologues in multiple monophyletic lineages (figure 3). Taken together, these evidence suggest that F. periodonticum, a subset of F. necrophorum, F. varium and F. ulcerans together with their uncharacterised sister lineages, F. hwasookii, F. massiliense and F. russii might have a role in the development of CRC. These implicated lineages are consistent with a set of ‘active versus passive invader’ species proposed by Manson McGuire and colleagues based on genomic features such as genome size, presence of FadA-related proteins, expanded number of membrane protein-encoding genes and MORN2 protein domains.18 In addition, their link to disease is supported by independent microbial community data and cell model studies. For example, a recent microbial community composition survey indicated that F. periodonticum in the oral cavity is associated with oral squamous cell carcinoma.19 Another example is of F. necrophorum, in which blood culture-based surveys of fusobacterial infections have indicated that this species was the second most common isolate after F. nucleatum.66 67 As for F. varium and F. ulcerans, less is known about their distribution and association with cancers or disease. Gut microbiota surveys in Japanese cohorts have indicated that F. varium is associated with UC,21 22 and a genome sequencing study of F. varium strain Fv113-g1 isolated from a UC patient reported expression of FadA homologues in monocultures simulating in vivo conditions within the human gut.68 Our data on FadA homologues show that sequences from the Fusobacterium_A clade (F. varium, F. ulcerans and other uncharacterised sister taxa) are not identical to sequences from the Fusobacterium clade (in which the CRC-associated F. nucleatum is located) (figure 4), thereby suggesting distinct functions or targets among these homologues. While the presence/absence of these gene homologues do not directly translate to invasiveness,69 we postulate that Fusobacterium_A taxa and their copies of the FadA homologue can be risk factors for diseases other than CRC.
In light of the inference that Fusobacterium_A taxa are prevalent in Chinese populations and could be potential risk factors of disease in humans, a limitation of this study is the lack of published data or cultured isolates to validate our observations. Findings reported here imply that disease-association is possible in this clade, from which we have isolated eight Fusobacterium_A members with genomes that match MAGs recovered from metagenomes. The immediate next step is to test these isolates in cell and animal model experiments to determine whether they have the potential to facilitate CRC or other diseases akin to F. nucleatum. Specifically, the role of virulence gene homologues such as FadA and Fap2 can be studied via knockout/knockdown experiments to assess their impact on disease outcomes. Following this, further isolation and testing of other uncharacterised fusobacterial lineages will provide a more comprehensive understanding of the biology and disease associations outside the F. nucleatum complex.
In conclusion, while fusobacterial species other than F. nucleatum have not been identified as risk factors likely owing to their almost non-existence in western populations and ubiquity in non-CRC southern Chinese populations, our findings suggest that there is potential in some of these prevalent but overlooked fusobacterial lineages to facilitate CRC. If any positive associations are confirmed, individuals carrying the corresponding taxa in their guts should be assessed for predispositions to disease. Findings reported here underscore the variability in gut microbiota composition across populations, and support ongoing efforts to characterise microbial diversity of the human microbiome.
Acknowledgments
We thank staff and students involved in the HKGutMicMap project for coordinating collection, processing and maintaining inventory of samples, and Jin Yan Lim and Geicho Nakatsu for downloading and organising metagenome data and metadata.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors YKY designed the study, analysed data and wrote the manuscript; ZC performed laboratory work; MCSW recruited subjects and edited the manuscript; MH revised the manuscript; JY, SCN and JJYS recruited subjects and acquired data; FKLC initiated the subject recruitment drive and provided funding; PKSC obtained funding, designed recruitment plan, recruited subjects, supervised the study and edited the manuscript.
Funding This study was supported by a seed fund for gut microbiota research provided by the Faculty of Medicine, The Chinese University of Hong Kong.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval This study has been approved by the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (reference number 2016.707). Written informed consent was obtained from all participants prior to collecting stool samples.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA557323. Gut metagenome and fusobacterial isolate genome sequence data are available in the Sequence Read Archive (SRA) under BioProject accession PRJNA557323.