Article Text

Abstract
Objective A comprehensive understanding of anticancer immune responses is paramount for the optimal application and development of cancer immunotherapies. We unravelled local and systemic immune profiles in patients with colorectal cancer (CRC) by high-dimensional analysis to provide an unbiased characterisation of the immune contexture of CRC.
Design Thirty-six immune cell markers were simultaneously assessed at the single-cell level by mass cytometry in 35 CRC tissues, 26 tumour-associated lymph nodes, 17 colorectal healthy mucosa and 19 peripheral blood samples from 31 patients with CRC. Additionally, functional, transcriptional and spatial analyses of tumour-infiltrating lymphocytes were performed by flow cytometry, single-cell RNA-sequencing and multispectral immunofluorescence.
Results We discovered that a previously unappreciated innate lymphocyte population (Lin–CD7+CD127–CD56+CD45RO+) was enriched in CRC tissues and displayed cytotoxic activity. This subset demonstrated a tissue-resident (CD103+CD69+) phenotype and was most abundant in immunogenic mismatch repair (MMR)-deficient CRCs. Their presence in tumours was correlated with the infiltration of tumour-resident cytotoxic, helper and γδ T cells with highly similar activated (HLA-DR+CD38+PD-1+) phenotypes. Remarkably, activated γδ T cells were almost exclusively found in MMR-deficient cancers. Non-activated counterparts of tumour-resident cytotoxic and γδ T cells were present in CRC and healthy mucosa tissues, but not in lymph nodes, with the exception of tumour-positive lymph nodes.
Conclusion This work provides a blueprint for the understanding of the heterogeneous and intricate immune landscape of CRC, including the identification of previously unappreciated immune cell subsets. The concomitant presence of tumour-resident innate and adaptive immune cell populations suggests a multitargeted exploitation of their antitumour properties in a therapeutic setting.
- colorectal cancer
- single-cell immunophenotyping
- mass cytometry
- innate lymphoid cells
- tissue-resident memory T cells
- immune landscape
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- colorectal cancer
- single-cell immunophenotyping
- mass cytometry
- innate lymphoid cells
- tissue-resident memory T cells
- immune landscape
Significance of this study
What is already known on this subject?
The colorectal cancer (CRC) immune microenvironment is a strong determinant of the clinical prognosis of patients, both in the context of natural disease progression and response to immunotherapy.
To date, most studies have focused on the investigation of the role of cytotoxic T cells in CRC, while an unbiased, comprehensive analysis that describes both innate and adaptive components of CRC immunity is largely lacking.
Mass cytometry allows a detailed single-cell characterisation of the immune landscape across multiple lineages to characterise antitumour immune cell responses and develop novel immunotherapeutic strategies.
What are the new findings?
A previously unappreciated innate lymphocyte population (Lin-–CD7+CD127–CD56+CD45RO+) is enriched in CRC tissues, displays cytotoxic activity and is particularly frequent in mismatch repair (MMR)-deficient cancers.
Tumour-resident immune cell populations across the adaptive (cytotoxic and helper) and innate (γδ) T cell compartments with highly similar activated (HLA-DR+, CD38+, PD-1+) tissue-resident (CD103+, CD69+) memory (CD45RO+) phenotypes are infrequent in colorectal healthy mucosa, tumour-draining lymph nodes and peripheral blood.
PD-1+ γδ T cells are enriched in MMR-deficient cancers and may be targeted by PD-1 checkpoint blockade in patients with CRC.
Non-activated counterparts of tumour-resident cytotoxic and γδ T cells are present in cancer and healthy colorectal tissues, but not detected in tumour-associated lymph nodes that are traditionally viewed as key players in the origin of antitumour immune responses.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Our work provides a comprehensive blueprint of antitumour immune responses across the adaptive and innate immune compartments in CRC. This will facilitate the development of novel immunotherapies exploiting the full potential of tumour-resident immune cell populations.
Introduction
T cell checkpoint blockade immunotherapies have revolutionised cancer treatment following the clinical success achieved with therapeutic antibodies targeting CTLA-4 and the PD-1/PD-L1 axis in patients with cancer. These strategies reinvigorate antitumour T cell responses and are particularly effective in cancers with high mutation burden like melanomas, non-small cell lung cancers and DNA mismatch repair (MMR)-deficient cancers.1–5 MMR deficiency occurs in approximately 15%–20% of colorectal cancers (CRCs) and leads to the widespread accumulation of somatic mutations in tumours, including insertions and deletions at DNA microsatellite sequences.6 7 Such a theoretically immunogenic profile is corroborated by the presence of numerous intraepithelial lymphocytes in these cancers, in contrast to MMR-proficient cancers.8 9 Nevertheless, not all MMR-deficient CRCs respond to immune checkpoint blockade, while MMR-proficient CRCs are insensitive to this therapy.
To understand the mechanisms that determine responses to current immunotherapies and for the design of alternative approaches, it is crucial to characterise the cancer microenvironment with multidimensional approaches that allow the simultaneous identification and characterisation of immune cell populations across multiple lineages.10 11 Mass cytometry allows a detailed single-cell characterisation of adaptive and innate immune landscapes, thereby providing a unique platform to discriminate immune cell subsets that can be exploited in an immunotherapeutic setting.
We performed an in-depth characterisation of immune landscapes across CRC tissues, tumour-associated lymph nodes, colorectal healthy mucosa and peripheral blood samples from 31 patients with CRC by high-dimensional single-cell mass cytometry. We revealed tumour tissue-specific immune signatures across the adaptive and innate compartments and discovered a previously unappreciated innate immune cell population implicated in antitumour immunity that strongly differentiated immunogenic (MMR-deficient) from non-immunogenic (MMR-proficient) CRCs.
Materials and methods
Human samples
Primary CRC tissues (n=35, of which 22 MMR-proficient and 13 MMR-deficient) with matched tumour-associated lymph nodes (n=26), colorectal healthy mucosa (n=17) and presurgical peripheral blood samples (n=19) from 31 patients with CRC were processed for this study (online supplementary table S1). All patients were treatment-naïve except five patients with rectal cancer who received neo-adjuvant therapy (online supplementary table S1). One patient was diagnosed with multiple primary colorectal tumours (n=5) at different locations, all of which were included in the study (online supplementary table S1). No patient with a previous history of inflammatory bowel disease was studied. To account for tumour heterogeneity, macroscopic sectioning from the lumen to the most invasive area of the tumour was performed for further processing. This study was approved by the Medical Ethical Committee of the Leiden University Medical Center (protocol P15.282), and patients provided written informed consent. All specimens were anonymised and handled according to the ethical guidelines described in the Code for Proper Secondary Use of Human Tissue in the Netherlands of the Dutch Federation of Medical Scientific Societies.
Supplemental material
Tissue processing and mass cytometry antibody staining
Details on tissue processing and mass cytometry antibody staining are available in online supplementary methods and online supplementary table S2.
Supplemental material
Supplemental material
Mass cytometry data analysis
Mass cytometry experiments were performed with a discovery and validation cohort of patients with CRC. The discovery cohort consisted of 19 CRC tissues, 17 tumour-associated lymph nodes, 4 colorectal healthy mucosa and 9 peripheral blood samples. Single, live CD45+ cells were gated in Cytobank12 (online supplementary figure S1). CD45+ cells were sample-tagged, hyperbolic ArcSinh transformed with a cofactor of 5 and subjected to dimensionality reduction analysis in Cytosplore.13 Of the 39 antibodies included in the panel, 36 showed clear discrimination between positive and negative cells (online supplementary figure S1). Major immune lineages (figure 1A,B) were identified at the overview level of a 5-level hierarchical stochastic neighbour embedding (HSNE) analysis14 15 on CD45+ data from all samples (8.9×106 cells) with default perplexity and iterations (30 and 1000, respectively). Naive and memory CD4+ and CD8+/γδ T cell, B cell, Lin–CD7+ innate lymphoid cell (ILC) and myeloid cell lineages were analysed in a data-driven manner up to a maximum number of 0.5×106 landmarks.15 Clustering of the data was performed by Gaussian mean shift (GMS) clustering in Cytosplore, and an algorithm was run that merged clusters showing high similarity in ArcSinh5-transformed median expression of all markers (<1). Hierarchical clustering on cell frequencies was performed in Matlab using Spearman’s rank correlation.

Tumour-resident immune cell populations derive from multiple lineages. (A,B) HSNE embedding showing 7.5×104 landmarks representing immune cells (8.9×106 cells) isolated from CRC tissues (n=19), tumour-associated lymph nodes (n=17), colorectal healthy mucosa (n=4) and peripheral blood (n=9) samples from the discovery cohort. Colours represent the different tissue types (A) and the relative expression of indicated immune lineage markers (B). Arrows indicate the HSNE location of phenotypically distinct tumour-resident immune cell populations. (C) Example of an HSNE analysis of 7.4×102 landmarks representing 1.1×106 cells from the memory CD8+/γδ T cell compartment as identified in (A). All landmarks are selected and embedded at the next, more detailed levels showing a finer granularity of structures with 5.0×103 landmarks at level 2, to 3.0×104 landmarks at level 3 and 1.6×105 landmarks at level 4. Phenotypically distinct immune cell clusters were identified by unsupervised GMS clustering based on the density features. Black dots indicate the centroids of the identified clusters. (D,E) t-SNE embedding showing 1079 cells from CRC tissues (n=7) analysed by single-cell RNA-sequencing. Colours represent the different clusters (D) and the log-transformed expression levels of indicated immune lineage markers (E). Each dot represents a single cell. CRC, colorectal cancer; GMS, Gaussian mean shift; HSNE, hierarchical stochastic neighbour embedding; LN, lymph node; PBMC, peripheral blood mononuclear cell; t-SNE, t-distributed stochastic neighbour embedding.
The validation cohort consisted of 16 CRC tissues, 9 tumour-associated lymph nodes, 13 colorectal healthy mucosa and 10 peripheral blood samples. Single, live CD45+ cells were hyperbolic ArcSinh transformed with a cofactor of 5 and classified into the preidentified immune cell clusters of the discovery cohort based on similarity in marker expression. To obtain consistent cell clusters across both cohorts, a linear discriminant analysis classifier was trained using the cell clusters of the discovery cohort and was used to automatically predict the cluster label for each cell in the validation cohort.16 To account for technical variation, a peripheral blood mononuclear cell (PBMC) reference sample was included in every mass cytometry experiment. ComBat was applied to align the PBMC reference samples and corresponding patient samples to correct for batch effects.17
Single-cell RNA-sequencing
CD45+ cells from seven tumours (four MMR-deficient and three MMR-proficient) were MACS-sorted with anti-CD45-PE antibodies (clone 2D1, Thermo Fisher Scientific) and anti-PE microbeads (Miltenyi Biotec). Single-cell RNA-sequencing libraries were prepared using the Chromium Single Cell 3' Reagent Kit, V.2 Chemistry (10× Genomics) according to the manufacturer’s protocol. Libraries were sequenced on a NovaSeq6000 using paired-end 2×150 bp sequencing (Illumina). Downstream analysis was performed using the Seurat R package according to the author’s instructions.18 Briefly, cells with fewer than 200 expressed genes, and genes that were expressed in less than 3 cells were excluded. Furthermore, cells with outlying percentages of differentially expressed mitochondrial genes (>0.20) and cells with outlying numbers of expressed genes (>5000) were excluded. This resulted in a final dataset of 1079 cells expressing a total of 1972 variable genes. Cells were preprocessed using principal component analysis, clustered using graph-based community detection19 and visualised by t-distributed stochastic neighbour embedding (t-SNE).20 Differentially expressed genes were identified for each cell cluster and visualised in violin plots. In addition, CD45+ cells from one MMR-deficient tumour with high numbers of Lin–CD7+CD127–CD56+CD45RO+ ILCs were sorted on a FACS Aria II sorter (BD Biosciences) (online supplementary table S3). A similar single-cell RNA-sequencing analysis pipeline was performed while sequencing was performed on a HiSeq4000 (Illumina). Cut-offs for outlying percentages of differentially expressed mitochondrial genes (>0.05) and cells with outlying numbers of expressed genes (>5500) were used. Here, a final dataset of 795 cells expressing a total of 1814 variable genes was obtained.
Flow cytometry
Single-cell suspensions of CRC tissues (n=8, of which 5 MMR-deficient and 3 MMR-proficient) were stimulated in IMDM/L-glutamine medium (Lonza) complemented with 10% human serum with 20 ng/mL PMA (Sigma-Aldrich) and 1 µg/mL ionomycin (Sigma-Aldrich) for 6 hours at 37°C. Ten μg/mL brefeldin A (Sigma-Aldrich) was added for the last 4 hours. A flow cytometry antibody panel was designed to detect granzyme B/perforin, IFN-γ and TNF-α production by ILC, T cell and γδ T cell populations (online supplementary table S3). In addition, FOXP3 expression by ICOS+ regulatory T cells was assessed in single-cell suspensions of CRC tissues (n=4, of which 1 MMR-deficient and 3 MMR-proficient). Details on flow cytometry antibody staining are available in online supplementary methods.
Immunohistochemical staining
Details on immunohistochemical detection of MMR proteins and human leucocyte antigen (HLA) class I expression of CRC tissues are available in online supplementary methods.
Multispectral immunofluorescence
A six-marker immunofluorescence panel was applied to 5 µm frozen tissue sections of 4 MMR-deficient and 4 MMR-proficient colorectal tumours, as described previously.21 Details on immunofluorescence antibody staining are available in online supplementary methods and online supplementary table S4. For each tumour, five different tissue sections were imaged at 20× magnification with the Vectra 3.0 Automated Quantitative Pathology Imaging System (Perkin Elmer). InForm Cell Analysis software (Perkin Elmer) was used for image analysis and spectral separation of dyes, by using spectral libraries defined with single-marker immunofluorescence detection. Tissue segmentation was trained manually with DAPI to segment images into tissue and ‘no tissue’ areas. All images were visually inspected for the number of CD3–TCRαβ–-CD127-–CD7+CD45RO+ ILCs and cell counts were normalised by tissue area (number of cells per mm2).
Statistical analysis
Data were presented as median±IQR range. Group comparisons were performed with Mann-Whitney U test, Kruskal-Wallis test with Dunn’s test for multiple comparisons or Friedman test with Dunn’s test for multiple comparisons (GraphPad Prism V.7), as indicated. In the correlation analysis, p values were adjusted for multiple testing using the Benjamini-Hochberg procedure. P<0.05 were considered statistically significant.
Results
Tumour-resident immune cell populations derive from multiple lineages
Mass cytometric analysis of 36 immune cell markers was performed on single-cell suspensions isolated from cancer and healthy tissues of patients with CRC. To decipher their immune composition, we performed HSNE analysis in Cytosplore on all acquired CD45+ cells of the discovery cohort (8.9×106 cells in total) (figure 1A). Based on the density features of the HSNE-embedded landmarks, seven major immune cell clusters were identified by unsupervised GMS clustering, which corresponded to naive and memory (based on CD45RO and CCR7 expression) CD4+ and CD8+/γδ T cells, B cells, Lin–CD7+ ILCs and myeloid cells (figure 1A,B). Memory CD4+ and CD8+ T cells, as well as myeloid cells, were dominant immune lineages in the tumour microenvironment, while B cells, Lin–CD7+ ILCs and naive CD4+ and CD8+ T cells were present at a lower extent (online supplementary figure S2). The HSNE analysis also unveiled the presence of several tumour tissue-specific, phenotypically distinct landmarks within the memory CD4+ T cell, CD8+/γδ T cell, Lin–CD7+ ILC, and myeloid cell compartments (figure 1A).
All seven major immune lineages were analysed in detail by hierarchical exploration of the data by HSNE. As an example, the embedding of the memory CD8+/γδ T cell compartment is shown in figure 1C. Altogether, analysis of these seven major immune lineages yielded 220 distinct immune cell clusters, of which 2 consisted of less than 100 cells and were excluded from further analysis. All acquired CD45+ cells of the validation cohort (6.6×106 cells in total) were subsequently classified into these preidentified immune cell clusters based on their phenotype (see Methods section).
The mass cytometric analysis was accompanied by single-cell RNA-sequencing of CD45+ cells from 7 CRC tissues. Seven immune cell clusters could be detected based on transcriptomic profiles (figure 1D), corresponding to B cells, CD8+ and CD4+ T cells, ILCs, myeloid cells, proliferating cells and plasma B cells (figure 1D,E).
Activated CD8+ and γδ T cells are tumour tissue-specific and enriched in mismatch repair-deficient colorectal cancers
Hierarchical clustering analysis revealed that memory CD8+/γδ T cell phenotypes clustered in a tissue-specific manner (figure 2A). Two CD8+CD103+PD-1+ populations (#60 and 96), distinguished by CD161 expression, were present in tumour tissues (constituting up to 28.2% of CD45+ cells) and infrequent in all other samples (figure 2B,C), with the exception of one lymph node sample that was found to be infiltrated by tumour cells on histological examination (data not shown). These CD8+CD103+PD-1+ cells were further characterised by the coexpression of CD69, FAS, HLA-DR and CD38 (figure 2B). Interestingly, the CD161– counterpart of CD8+CD103+PD-1+ T cells (#60) was particularly abundant in MMR-deficient tumours as compared with MMR-proficient tumours (online supplementary figure S3). Within the CD8+CD103+PD-1+CD38+ subset, we observed coexpression of CD39 (online supplementary figure S3), a marker that has recently been found to identify tumour-reactive CD8+ T cells.22 23 Next to these tumour-resident cells, a cluster (#61) with a similar phenotype but lacking HLA-DR, PD-1, FAS and possessing a lower expression of CD38 was present in both tumour and healthy colorectal samples (figure 2B,C) and may represent a non-activated counterpart. Single-cell RNA-sequencing revealed that CD8+ T cells in colorectal tumours expressed cytolytic molecules (eg, GZMA, GZMB, GZMH, PRF1) (figure 2D). Furthermore, they displayed expression of the immune checkpoint molecule LAG3 (figure 2D).

Activated CD8+ and γδ T cells are tumour tissue-specific and enriched in mismatch repair-deficient colorectal cancers. (A) HSNE embedding of 1.6×105 landmarks representing the memory CD8+/γδ T cell compartment (1.1×106 cells) from the discovery cohort of patients with CRC coloured by tissue type (first plot) and relative expression of indicated markers. (B) A heatmap showing median marker expression values (left) and frequencies of selected memory CD8+/γδ T cell clusters (right). Hierarchical clustering was performed on cluster frequencies using Spearman’s rank correlation. Colour bars indicate tissue type. (C) Frequencies of selected memory CD8+/γδ T cell clusters among CRC tissues (n=35, MMR-deficient (n=13) and MMR-proficient (n=22)), colorectal healthy mucosa (n=17), tumour-associated lymph nodes (n=26) and peripheral blood (n=19) as percentage of total CD45+ cells (upper panel) and memory CD8+ or γδ T cells (lower panel). Cluster IDs correspond to the ones in (B). Bars indicate median±IQR. Each dot represents an individual sample. Data from 22 independent experiments with mass cytometry. *P<0.05, ***p<0.001, ****p<0.0001 by Kruskal-Wallis test with Dunn’s test for multiple comparisons. (D) Violin plot showing log-transformed expression levels of the top 20 differentially expressed genes within CD8+ T cells (n=217) analysed by single-cell RNA-sequencing on CD45+ cells from CRC tissues (n=7) (figure 1D). Each dot represents a single cell. CRC, colorectal cancer; HSNE, hierarchical stochastic neighbour embedding; LN, lymph node; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient; PBMC, peripheral blood mononuclear cell.
Strikingly, a TCRγδ+CD103+PD-1+ population (#99) was almost exclusively found in MMR-deficient tumours, constituting up to 8.4% of CD45+ cells (figure 2B,C). These γδ T cells had a phenotype similar to the CD8+CD103+PD-1+ cells, as defined by coexpression of CD69, FAS, CD38 and HLA-DR (figure 2B). An HLA-DR–PD-1– counterpart of these cells (#97 and 101) was also observed in colorectal healthy mucosa and MMR-proficient tumours and may represent a non-activated form of the CD103+PD-1+ γδ T cells in the tumour microenvironment (figure 2B,C). We analysed the cytotoxic potential of the tumour-resident γδ T cells by flow cytometry and determined that these were capable of producing IFN-γ and granzyme B/perforin on stimulation with PMA/ionomycin (online supplementary figure S4).
ICOS+ and activated CD4+ T cells are dominant, tumour tissue-specific T cell populations in both mismatch repair-deficient and repair-proficient colorectal cancers
Next, we determined the cell surface phenotype of memory CD4+ T cells in patients with CRC. Memory CD4+ T cells also distributed in a tissue-specific manner (figure 3A). Here, a large population of CD4+ICOS+CD27– cells (#20 and 58) constituted up to 21.1% of CD45+ cells in CRCs, while being absent in all other tissues with the exception of tumour-positive lymph node samples (figure 3B,C). Part of this population coexpressed CD161 and PD-1 (#58), whereas the other part was negative for these markers but expressed high levels of CD25 (#20), indicative of a regulatory-like phenotype (figure 3B). Flow cytometry analysis confirmed the expression of FOXP3 in 91%–98% of ICOS+ CD4+CD45RO+CD25+CD127low T cells in colorectal tumours (online supplementary figure S5). Interestingly, the ICOS+ CD4+ T cells were present in MMR-deficient as well as MMR-proficient tumours to a similar extent (figure 3B,C).

ICOS+ and activated CD4+ T cells are dominant, tumour tissue-specific T cell populations in both mismatch repair-deficient and repair-proficient colorectal cancers. (A) HSNE embedding of 3.1×105 landmarks representing the memory CD4+ T cell compartment (2.0×106 cells) from the discovery cohort of patients with CRC coloured by tissue type (first plot) and relative expression of indicated markers. (B) A heatmap showing median marker expression values (left) and frequencies of selected memory CD4+ T cell clusters (right). Hierarchical clustering was performed on cluster frequencies using Spearman’s rank correlation. Colour bars indicate tissue type. (C) Frequencies of selected memory CD4+ T cell clusters among CRC tissues (n=35, MMR-deficient (n=13) and MMR-proficient (n=22)), colorectal healthy mucosa (n=17), tumour-associated lymph nodes (n=26) and peripheral blood (n=19) as percentage of total CD45+ cells (upper panel) and memory CD4+ T cells (lower panel). Cluster IDs correspond to the ones in (B). Bars indicate median±IQR. Each dot represents an individual sample. Data from 22 independent experiments with mass cytometry. *P<0.05, ***p<0.001, ****p<0.0001 by Kruskal-Wallis test with Dunn’s test for multiple comparisons. (D) Violin plot showing log-transformed expression levels of the top 20 differentially expressed genes within CD4+ T cells (n=245) analysed by single-cell RNA-sequencing on CD45+ cells from CRC tissues (n=7) (figure 1D). Each dot represents a single cell. CRC, colorectal cancer; HSNE, hierarchical stochastic neighbour embedding; LN, lymph node; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient; PBMC, peripheral blood mononuclear cell .
In addition, CD4+CD103+PD-1+ cells (#85 and 86), which constituted up to 23.8% of CD45+ cells, were also enriched in tumour tissues (figure 3B,C). Strikingly, several features of these cells mirrored our observations in the CD8+/γδ compartment, including an activated tissue-resident phenotype defined by coexpression of CD69, FAS, CD38 and HLA-DR (figure 3B). Moreover, expression of CD161 also subdivided CD4+CD103+PD-1+ T cells into a positive (#85) and negative (#86) population, where CD161– cells were more abundant in MMR-deficient as compared with MMR-proficient tumours (online supplementary figure S3). In contrast to the tumour-resident CD8+ and γδ T cells, a non-activated counterpart could not be detected for these cells.
While ICOS+ regulatory T cells (Tregs) were tumour tissue-specific, ICOS–CD25+CD127– Tregs (#13–73) were found in both tumour-associated lymph nodes and CRC tissues (figure 3B,C). Last, immune cell populations such as CD4+CD27+CD127+ central memory (CCR7+CD45RO+) cells (#1–37) were more abundant in peripheral blood and lymph nodes (figure 3B,C). The expression of ICOS on CD4+ T cells was confirmed by single-cell RNA-sequencing, which also revealed the expression of TNFRSF4 (OX40R) and TNFRSF18 (GITR) (figure 3D). t-SNE analysis revealed the coexpression of all three immunotherapeutic targets by CD4+ T cells (online supplementary figure S6).
CD127–CD56+CD45RO+ ILCs are the prevalent ILC population in mismatch repair-deficient colorectal tumours
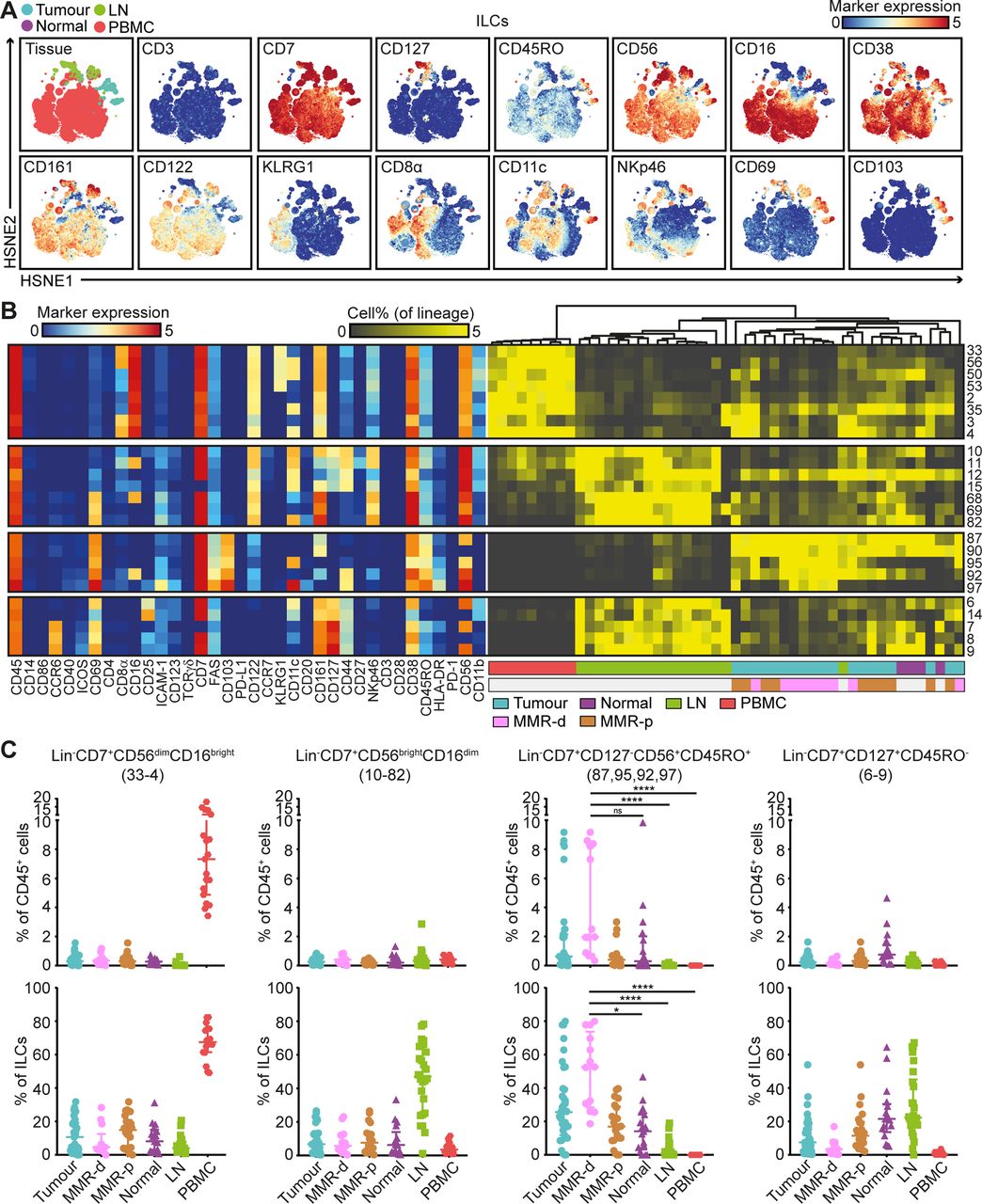
Mass cytometric profiles of the innate lymphoid compartment revealed the presence of three distinct Lin–CD7+ cell clusters: CD127–CD56+CD45RO–natural killer (NK) cells (90.4%), CD127+ ILCs (3.4%) and a cluster of CD127–CD56+CD45RO+ cells (6.2%) (figure 4A). Analysis of cluster frequencies demonstrated that CD56dimCD16bright NK cells (#33–4) were present in high frequencies in peripheral blood, whereas CD56brightCD16dim NK cells (#10–82) were the dominant NK-type in lymph node samples (figure 4B,C). CD127+ ILCs (#6–9) were more abundant in healthy mucosa, lymph nodes and MMR-proficient tumours and displayed a KLRG1– phenotype, characteristic of ILC3 cells (figure 4B,C). Strikingly, the CD127–CD56+CD45RO+ ILCs (#87, 95, 92, 97) were enriched in tumour tissues, accounting for up to 80% of the innate lymphoid compartment (figure 4B,C). Moreover, they were particularly abundant in MMR-deficient tumours, especially CD161– populations (#95, 92 and 97) (figure 4B,C). The CD127–CD56+CD45RO+ ILC population has recently been identified in human fetal intestine as intermediate-ILCs.24 Consistent with that work, hierarchical clustering positioned the CD127–CD56+CD45RO+ ILCs in between NK cells and CD127+ ILCs (figure 4B). We observed coexpression of CD69 and CD103 on all CD127-CD56+CD45RO+ ILCs, but differential expressions of CD16, ICAM-1, FAS, CD11c, CD161, CD44 and HLA-DR, indicative of further heterogeneity within this cell cluster (figure 4B).
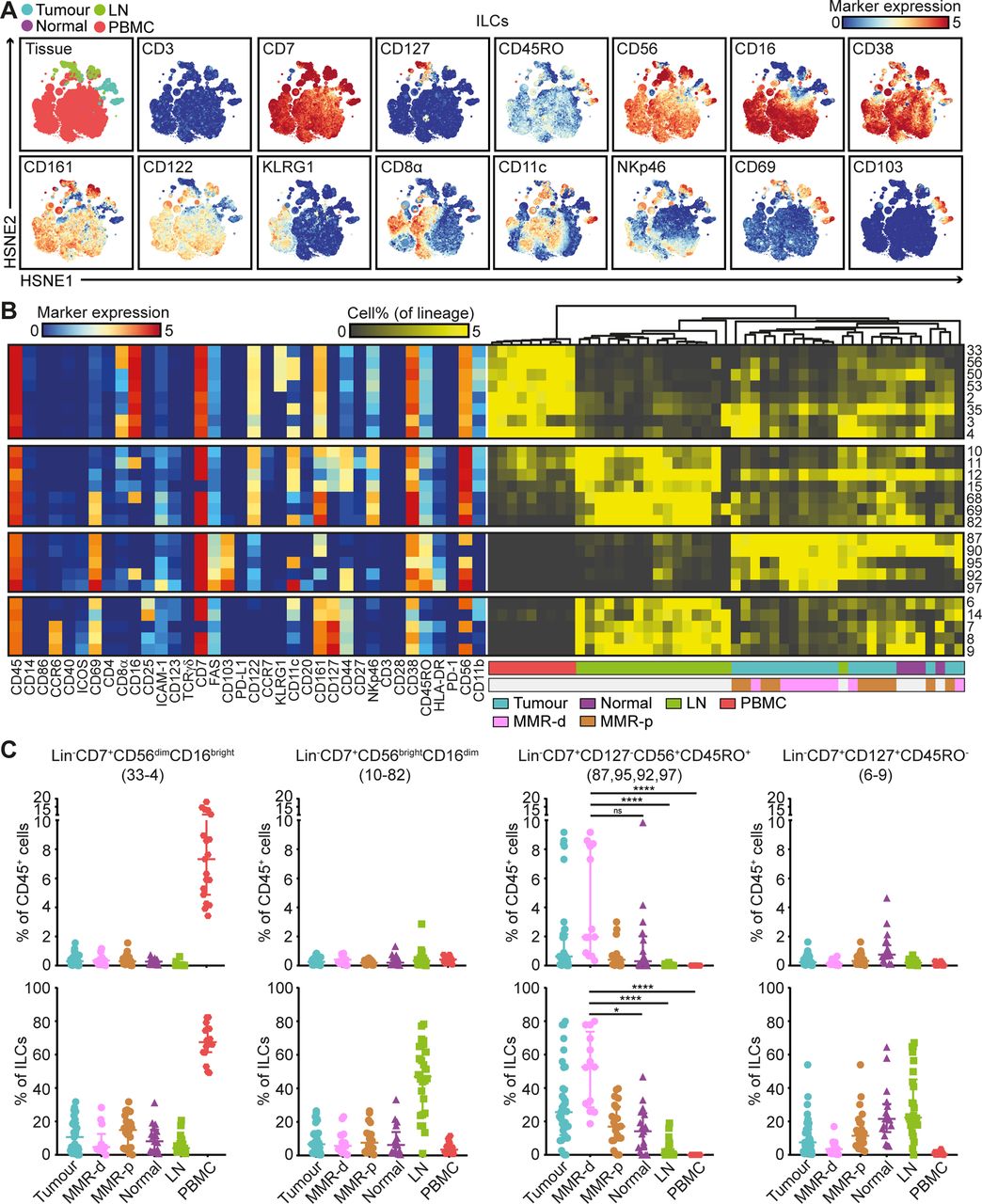
CD127–CD56+CD45RO+ ILCs are the prevalent ILC population in mismatch repair-deficient colorectal tumours. (A) HSNE embedding of 5.5×104 landmarks representing the innate lymphoid compartment (0.4×106 cells) from the discovery cohort of patients with CRC coloured by tissue type (first plot) and relative expression of indicated markers. (B) A heatmap showing median marker expression values (left) and frequencies of selected ILC clusters (right). Hierarchical clustering was performed on cluster frequencies using Spearman’s rank correlation. Colour bars indicate tissue type. (C) Frequencies of selected innate lymphoid clusters among CRC tissues (n=35, MMR-deficient (n=13) and MMR-proficient (n=22)), colorectal healthy mucosa (n=17), tumour-associated lymph nodes (n=26) and peripheral blood (n=19) as percentage of total CD45+ cells (upper panel) and ILCs (lower panel). Cluster IDs correspond to the ones in (B). Bars indicate median±IQR. Each dot represents an individual sample. Data from 22 independent experiments with mass cytometry. NS, not significant, *P<0.05, ****p<0.0001 by Kruskal-Wallis test with Dunn’s test for multiple comparisons. CRC, colorectal cancer; HSNE, hierarchical stochastic neighbour embedding; ILC, innate lymphoid cell; LN, lymph node; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient; PBMC, peripheral blood mononuclear cell.
Tumour-resident ILCs are involved in the antitumour immune response
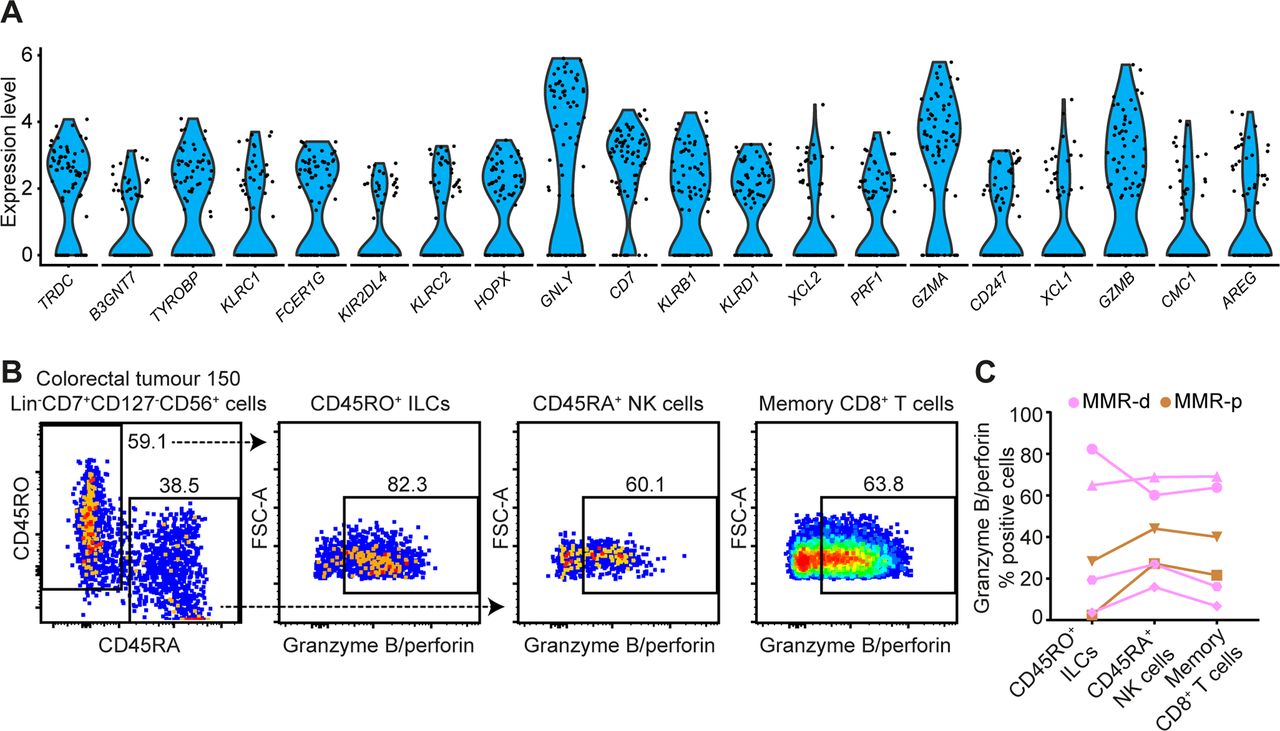
Single-cell RNA-sequencing unveiled high expression levels of cytotoxic molecules (eg, GNLY, PRF1, GZMA, GZMB) in the ILC cluster (figure 5A). In addition, we observed the presence of transcripts for a member of the killer-cell immunoglobulin-like receptor (KIR) family, KIR2DL4 (figure 5A). We performed additional single-cell RNA-sequencing on CD45+ cells from one MMR-deficient tumour with high numbers of Lin–CD7+CD127–CD56+CD45RO+ ILCs (70% of the ILC cluster), as revealed by mass cytometry data. Here, we also observed high expression levels of cytotoxic molecules (eg, GNLY, PRF1, GZMA) as well as the expression of KIR2DL4 and KIR3DL2 in the ILC cluster (online supplementary figure S7). Cell surface expression of KIRs was confirmed by flow cytometry in Lin–CD7+CD127–CD56+CD45RO+ ILCs from this tumour (online supplementary figure S7).

Tumour-resident ILCs are involved in the antitumour immune response. (A) Violin plot showing log-transformed expression levels of the top 20 differentially expressed genes within ILCs (n=74) analysed by single-cell RNA-sequencing on CD45+ cells from CRC tissues (n=7) (figure 1D). Each dot represents a single cell. (B) Representative plots of a MMR-deficient tumour analysed by flow cytometry without stimulation showing the distinction between CD45RO+ ILCs and CD45RA+ NK cells within Lin–CD7+CD127–CD56+ cells (first plot) and their expression of cytotoxic molecules. (C) Granzyme B/perforin expression in different immune cell populations of CRC tissues (n=6, of which 4 MMR-deficient and 2 MMR-proficient). Dot shape indicates similar tumour samples. Data from three independent experiments with flow cytometry. CRC, colorectal cancer; ILC, innate lymphoid cell; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient; NK, natural killer.
To further investigate functional properties of tumour-resident lymphocytes, we designed a flow cytometry antibody panel to analyse the cytotoxic potential of Lin–CD7+CD127–CD56+CD45RO+ ILCs, Lin–CD7+CD127–CD56+CD45RA+ NK cells and memory CD8+ T cells in CRC tissues. Strikingly, up to 82.3% of unstimulated CD127–CD56+CD45RO+ ILCs displayed granzyme B/perforin expression in the tumour tissues (figure 5B). Granzyme B/perforin expression by the ILCs was most abundant in MMR-deficient cancers as compared with MMR-proficient cancers (figure 5C). Interestingly, the cytotoxic capacity of CD127–CD56+CD45RO+ ILCs was accompanied by similar profiles in CD127–CD56+CD45RA+ NK cells and memory CD8+ T cells across samples (figure 5C), suggesting a coordinated cytotoxic innate and adaptive immune response in CRC tissues.
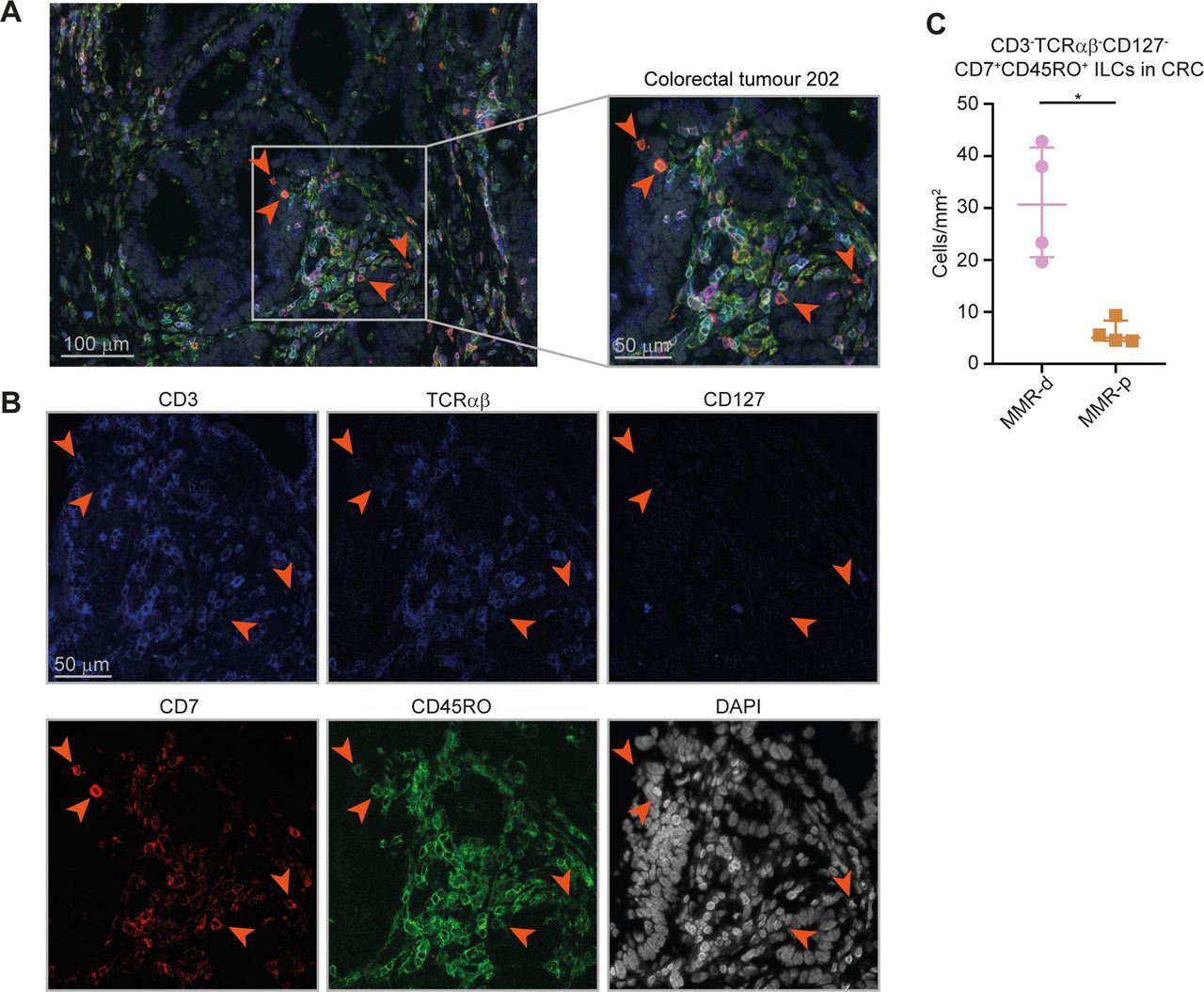
To investigate the spatial localisation of the ILCs in CRCs, we applied 6-colour multispectral immunofluorescence to frozen tissue sections of four MMR-deficient and four MMR-proficient CRCs. We simultaneously detected CD3, TCRαβ, CD127, CD7, CD45RO and DAPI. We identified CD3–TCRαβ–CD127–CD7+CD45RO+ ILCs in the tumours (figure 6A,B) and observed an increased presence of these cells in MMR-deficient as compared with MMR-proficient CRCs (figure 6C). Interestingly, the CD3–TCRαβ–CD127–CD7+CD45RO+ ILCs frequently displayed an intraepithelial localisation in agreement with their CD103+CD69+ tissue-resident phenotype (figure 6A).

Higher cell density of CD127–CD45RO+ ILCs in mismatch repair-deficient colorectal cancers. (A,B) Representative image of the immunofluorescence microscopic detection of CD3–TCRαβ–CD127–CD7+CD45RO+ ILCs in a MMR-deficient tumour, showing CD3 (coloured in blue), TCRαβ (coloured in blue), CD127 (coloured in blue), CD7 (coloured in red), CD45RO (coloured in green) and DAPI (coloured in grey) as nuclear counterstain. (C) Frequencies of CD3–TCRαβ–CD127–CD7+CD45RO+ ILCs in four MMR-deficient and four MMR-proficient CRCs. *P<0.05 by Mann-Whitney U test. ILC, innate lymphoid cell; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient.
Immune-system-wide analysis reveals correlations between innate and adaptive immune cell subsets in colorectal cancer
Last, we integrated the identified immune cell clusters across all major immune lineages (n=218) in one immune-system-wide analysis to characterise the samples according to tissue type, MMR status and available clinicopathological parameters. The integrated t-SNE analysis confirmed the unique immune composition in the different tissue types and visualised the top 10 ranked immune cell clusters contributing to the distinctive clustering patterns of the samples (online supplementary figure S8). No association was observed with clinical stage while differences related to tumour location and HLA class I expression can be attributed to features that distinguish MMR-deficient and MMR-proficient CRCs (online supplementary figure S8).
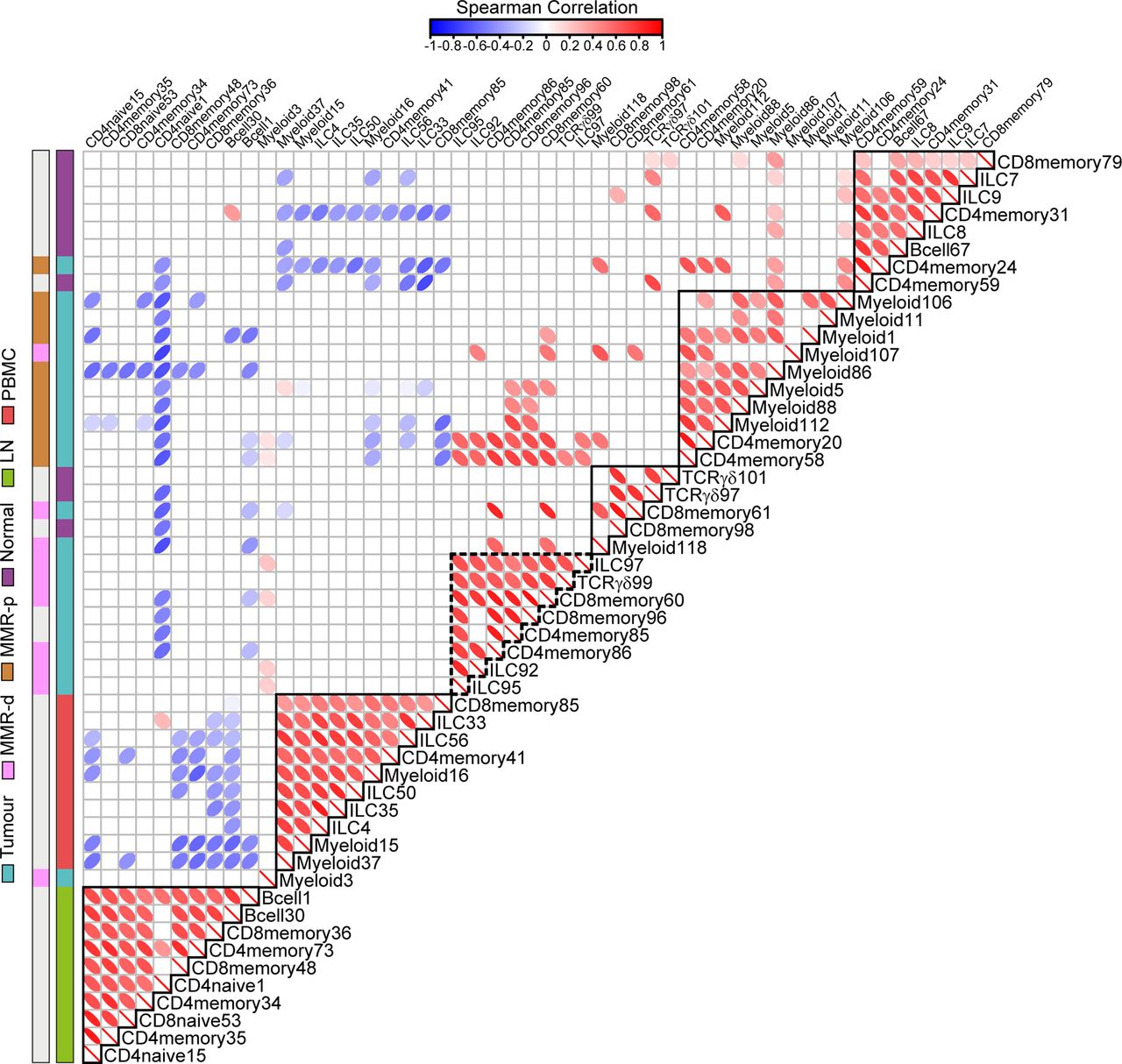
Spearman’s rank correlation analysis performed on the top 10 ranked unique immune cell clusters of each tissue type revealed strong correlations between the presence of CD127–CD56+CD45RO+ ILCs (ILC97,92,95) and the presence of CD103+PD-1+ cytotoxic (CD8memory60,96), helper (CD4memory85,86) and γδ (TCRγδ99) T cell populations in MMR-deficient CRCs (figure 7, online supplementary table S5). In contrast, MMR-proficient tumours were characterised by the presence of several myeloid populations (figure 7, online supplementary table S5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immune-system-wide analysis reveals correlations between innate and adaptive immune cell subsets in colorectal cancer. Matrix showing correlations (Spearman’s ρ, see online supplementary methods) between unique top 10 ranked immune cell clusters for each tissue type (shown in online supplementary figure S8) based on cell percentages (of total CD45+ cells) corresponding to 97 samples from 31 patients with CRC. Colour and shape of the ellipses indicate the strength of the correlation. Only significant correlation coefficients are shown. Colour bars indicate tissue type. Coefficient and p values of correlations for CRC tissues are shown in online supplementary table S5. P values were adjusted for multiple testing using the Benjamini-Hochberg procedure. Data from 22 independent experiments with mass cytometry. CRC, colorectal cancer; ILC, innate lymphoid cell; LN, lymph node; MMR-d, mismatch repair-deficient; MMR-p, mismatch repair-proficient; PBMC, peripheral blood mononuclear cell.
Discussion
We applied mass cytometry to comprehensively analyse the immune landscape of CRCs at single-cell level in tumour and healthy tissues. Our analysis revealed tumour tissue-specific immune signatures across the innate and adaptive immune compartments of CRC. Immunohistochemistry, flow cytometry and recent transcriptomic approaches have provided insight into the complexity of tumour immune landscapes.25–29 However, the number of markers that can be simultaneously assayed in immunohistochemistry or flow cytometry is limited, and bulk transcriptomic studies do not allow for discrimination of phenotypes at the cellular level.30 31 In mass cytometry, over 40 markers can be simultaneously analysed at single-cell level, providing a unique opportunity to obtain a comprehensive overview of tumour-resident lymphocytes.32 33 Here, we combined mass cytometry phenotypes with functional, transcriptional and spatial analyses of tumour-resident immune cell populations in CRC.
Within the innate compartment, we observed that a previously unappreciated innate lymphoid population, Lin–CD7+CD127–CD56+CD45RO+ ILCs, is enriched in MMR-deficient tumours and displayed cytotoxic activity. In-situ detection of the ILCs confirmed a higher cell density in MMR-deficient CRCs and showed a frequent intraepithelial localisation. This is in line with their tissue-resident phenotype (CD103+CD69+) and supports an active role for these cells in the antitumour immune response. The ILCs resemble previous descriptions of TCR–CD103+ cells in mice that were found to express granzyme B.34 Additionally, a unique subset of NK cells has been found in several human tissues and was described as NKp44+CD103+ intraepithelial ILC1-like.35 36 In contrast to NKp44+CD103+ ILC1, the CD127–CD56+CD45RO+ ILCs identified here lacked CD122 and NKp46 expression (figure 4B) and showed low levels of NKp44 (data not shown). These variable marker expression patterns most likely represent additional levels of plasticity and heterogeneity within ILC subsets. Single-cell RNA-sequencing revealed the presence of transcripts for KIR2DL4 and KIR3DL2 in the ILC cluster, which hints towards potential activation mechanisms.37 Common ligands of KIRs include HLA class I molecules,38 39 and loss of HLA class I expression has been described to occur in the majority of MMR-deficient CRCs.40–42 It is tempting to speculate that CD127–CD56+CD45RO+ ILC-mediated cytotoxicity towards such HLA-loss variants may contribute to the antitumour response in MMR-deficient CRCs, a link that requires further investigation.
The presence of CD127–CD56+CD45RO+ ILCs strongly correlated with tissue-resident CD103+CD69+ γδ T cells coexpressing activation markers HLA-DR, CD38 and PD-1 in MMR-deficient CRCs. It has been shown that human peripheral blood γδ T cells can express PD-1 and exhibit natural killer-like activity.43 The expression of PD-1, in conjunction with their cytotoxic potential, suggest an active role of tumour-resident γδ T cells in the antitumour immune response and potentially as targets for PD-1 checkpoint blockade. This will be subject of further studies.
Within the adaptive compartment, we found dominant, tumour tissue-specific CD8+ and CD4+ T cell populations that displayed a highly similar activated tissue-resident phenotype. Such CD8+ T cell populations have been described in ovarian cancer,44 45 lung cancer46 and recently in melanoma,47 cervical carcinoma48 and CRC,22 23 and their presence was associated with an improved clinical prognosis. Single-cell RNA-sequencing revealed that CD8+ T cells in colorectal tumours showed a cytotoxic profile, indicative of potential antitumour reactivity. In addition, we found a dominant tumour tissue-specific population of ICOS+ CD4+ T cells. ICOS belongs to the CD28/CTLA-4 family and serves as a costimulatory molecule for T cell activation.49 Activation of ICOS by agonists has been proposed for anticancer treatment.50 Here, we identified a CD161+PD-1+ as well as a CD25+ population of tumour-resident ICOS+ CD4+ T cells. The latter corresponds to a regulatory T cell subset displaying high levels of FOXP3 expression, that, interestingly, expressed higher levels of ICOS as compared with the CD161+PD-1+ counterpart. The use of ICOS agonists may, therefore, also result in activation of ICOS+ T cells with suppressive and regulatory properties in the tumour microenvironment. In contrast to the tumour-resident CD8+ T cells, ICOS+ CD4+ T cells were present in both MMR-deficient and MMR-proficient tumours to a similar extent.
We observed CD161+ and CD161– counterparts of tumour-resident cytotoxic and helper T cells and CD127–CD56+CD45RO+ ILCs. CD161 has been shown to mark a subset of tissue-resident memory CD8+ T cells with enhanced effector function and cytokine production.51 52 In our study, the CD161– counterpart of the tumour-resident T cell and ILC populations was particularly enriched in MMR-deficient CRCs as compared with MMR-proficient CRCs. The functional relevance of this observation will be subject of future studies. Nevertheless, we observed increased CD161 expression in PD-1 high cells as compared with PD-1 intermediate/negative cells for tumour-resident CD8+ and CD4+ T cell populations (online supplementary figure S9). As PD-1 high cells in human cancer have been associated with a state of T cell dysfunction,53–55 CD161 expression could be an additional marker for this functional state.
Interestingly, we identified what could be the non-activated counterparts of the CD103+PD-1+ cytotoxic and γδ T cells in both tumour and healthy colorectal tissues. Mobilisation and activation of these cells from the colorectal healthy mucosa to the tumour tissue may be beneficial for immunotherapy in CRC. Strikingly, while lymph nodes are traditionally viewed as key players of antitumour immune responses, we did not detect non-activated precursors of tumour-resident immune cell populations in the lymph node samples, with the exception of tumour-positive lymph nodes. Furthermore, we observed that lymph nodes harboured a large population of CD4+CD25+CD127– Tregs, suggesting they might be a primary source of Tregs in the cancer microenvironment. The tumour-resident immune cell populations were also not mirrored in peripheral blood, although the in-depth investigation of their presence in these tissues with complementary approaches should be conducted.
It should be noted that the mass cytometry antibody panel was primarily developed to characterise T cell, γδ T cell and ILC compartments, and in future studies additional efforts are required to further explore the myeloid and B cell compartment. Furthermore, the number and pattern of infiltrating lymphocytes can be influenced by various tumour characteristics. In this study, we have shown profound differences in lymphocytic infiltration that distinguish MMR-deficient from MMR-proficient CRCs. Other factors not investigated in this study that can influence the infiltration of lymphocytes in tumours include, for instance, occurrence of somatic mutations (neoantigens) and the co-occurrence of inflammatory bowel disease. Although the results are of preliminary nature, they point to the involvement of additional subsets than T cells in immune responses to CRC, particularly ILCs and γδ T cells. This is especially relevant in the context of responses to checkpoint blockade therapy in absence of HLA class I expression.56 Future approaches might opt for an in-depth investigation of these specific lineages for a detailed characterisation of phenotypes that complement the markers used in this study. The next step will be to investigate the involvement of these subsets in the clinical setting of patients treated by checkpoint blockade.
In conclusion, we identified a previously unappreciated innate immune cell population that was specifically enriched in CRC tissues, displayed cytotoxic activity and strongly contributed to a data-driven distinction between immunogenic (MMR-deficient) and non-immunogenic (MMR-proficient) tumours. Furthermore, we revealed strong correlations between the presence of these innate cells and tumour-resident CD8+, CD4+ and γδ T cells with an activated phenotype in MMR-deficient tumours that together may play a critical role in tumour control.
Acknowledgments
We thank M G Kallenberg-Lantrua, A M E G Voet-van den Brink and F A Holman for their help in collecting and providing samples from CRC patients; J van den Bulk for the isolation of PBMCs from CRC patients; W E Corver for flow cytometric cell sorting, R J McLaughlin for providing help with mass cytometry experiments, the Leiden Genome Technology Center for their help with single-cell RNA-sequencing and J Oosting for help with statistical analysis.
References
Footnotes
VU, MEI and TA contributed equally.
FK and NFCCM contributed equally.
Contributors NLdV conceived the study, performed experiments and wrote the manuscript. NLdV, VvU, MEI, TA and AM analysed the data. AFS and KCMJP provided samples from patients. MEI, RvdB and NFCCdM processed the samples and performed experiments. TH, VvU and BPFL developed Cytosplore and HSNE applications. FK and NFCCdM initiated and led the project and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Funding The authors acknowledge funding from the Fight Colorectal Cancer-Michael’s Mission-AACR Fellowship in Young Onset, Late-Stage Colorectal Cancer Research 2015 (15-40-1645-DEMI), the KWF Bas Mulder Award UL (2015-7664), the ZonMw Veni grant (016.176.l44), the NWO-AES grant (12720: VANPIRE) and the European Commission under a MSCA-ITN award (675743: ISPIC).
Competing interests None declared.
Ethics approval Medical Ethical Committee of the Leiden University Medical Centre (protocol P15.282).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.