Article Text

Abstract
Objective Adeno-associated virus (AAV) is a defective mono-stranded DNA virus, endemic in human population (35%–80%). Recurrent clonal AAV2 insertions are associated with the pathogenesis of rare human hepatocellular carcinoma (HCC) developed on normal liver. This study aimed to characterise the natural history of AAV infection in the liver and its consequence in tumour development.
Design Viral DNA was quantified in tumour and non-tumour liver tissues of 1461 patients. Presence of episomal form and viral mRNA expression were analysed using a DNAse/TaqMan-based assay and quantitative RT-PCR. In silico analyses using viral capture data explored viral variants and new clonal insertions.
Results AAV DNA was detected in 21% of the patients, including 8% of the tumour tissues, equally distributed in two major viral subtypes: one similar to AAV2, the other hybrid between AAV2 and AAV13 sequences. Episomal viral forms were found in 4% of the non-tumour tissues, frequently associated with viral RNA expression and human herpesvirus type 6, the candidate natural AAV helper virus. In 30 HCC, clonal AAV insertions were recurrently identified in CCNA2, CCNE1, TERT, TNFSF10, KMT2B and GLI1/INHBE. AAV insertion triggered oncogenic overexpression through multiple mechanisms that differ according to the localisation of the integration site.
Conclusion We provided an integrated analysis of the wild-type AAV infection in the liver with the identification of viral genotypes, molecular forms, helper virus relationship and viral integrations. Clonal AAV insertions were positive selected during HCC development on non-cirrhotic liver challenging the notion of AAV as a non-pathogenic virus.
- hepatocellular carcinoma
- oncogenes
- carcinogenesis
- liver
- chronic viral hepatitis
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The seroprevalence of adeno-associated virus (AAV) in general population is 40%–80% and AAV2 is the most frequent serotype in human.
AAV has a biphasic life cycle characterised by latent and lytic phases.
The presence of a helper virus is required for the AAV replication.
It is commonly believed that adenovirus is the natural AAV helper virus.
Although AAV is considered a non-pathogenic virus, recurrent clonal AAV2 insertions were associated with hepatocellular carcinoma (HCC) development.
What are the new findings?
Two viral subtypes are present in 21% of the liver tissues: AAV2 and hybrid AAV2/13 sequences.
Episomal AAV forms are found in 4% of non-tumour liver tissues, mainly in young, female patients without liver fibrosis.
Human herpesvirus type 6 is the most frequent AAV helper virus in the liver.
The 2% of patients with HCC displayed clonal AAV integration in cancer driver genes.
AAV clonal insertion in HCC activates oncogenes using various mechanisms.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
These findings are important to understand wild-type AAV biology and its association with hepatocarcinogenesis.
Our data are particularly relevant considering the large usage of AAV vector in liver-targeted gene therapy.
Even if rare, AAV insertional mutagenesis is a new risk factor of HCC development, therefore the notion of AAV as non-pathogenic virus should be reviewed.
Introduction
Adeno-associated virus (AAV) is a small non-enveloped DNA virus with an icosahedral capsid that contains a 4.7 kb linear single-stranded genome.1 2 AAV genome codes for non-structural proteins (Rep78, 68, 52 and 40), capsid proteins (VP1, VP2, VP3) and the assembly activating protein (AAP).3 4 At the extremities, inverse tandem repeats (ITR) are important for the integration in host genome.5 6 AAV is a defective virus that requires a helper virus for an active infection, otherwise it can establish a latent infection through integration into host genome or maintenance as circular episomal form.7–9 AAV seroprevalence showed that the infection is endemic in human populations (30%–80%) starting during childhood.10–12 Twelve distinct serotypes and more than 100 natural variants have been identified, among which AAV2 is the most frequent type in human.13–16
This small virus is attractive for gene therapy because of the lack of identifiable associated disease and the remarkable ability of recombinant AAV (rAAV) vectors to transduce dividing and non-dividing cells with high efficiency, long-term transgene expression, low immunogenicity and specific tissue tropism.17 Although AAV was discovered in 1965, many questions regarding the natural history of AAV infection in human remain unanswered.2 It is well known that the vector predominantly persists in the nucleus as episomal form with sustained RNA expression raising question on putative episomal AAV form in wild-type infection.8 Several helper viruses have been identified but their precise association with wild-type liver AAV infection remains unclear. The frequency of the different AAV genotypes in the human population and AAV persistence in tissues after first infection remains to be determined.18 Moreover, AAV link with tumour development is controversial, with some studies reporting an oncogenic effect of AAV infection in animal model and others suggesting a tumour suppressive role.19–24
Recently, we reported the involvement of AAV2 in the pathogenesis of human hepatocellular carcinoma (HCC) developed on normal liver in the absence of classical HCC risk factors such as infection with HBV and HCV, high alcohol intake, haemochromatosis or aflatoxin B1 exposure.25 Similar to HBV, recurrent AAV2 clonal insertions were described in TERT, CCNE1 and CCNA2 cancer driver genes, leading to their overexpression.25–28 The AAV insertions can activate oncogenes located nearby in the human genome by a liver promoter recently identified within the minimal common AAV inserted sequence adjacent to the 3’ITR of the virus.29
In this work, we investigated the natural history of wild-type AAV infection in the liver and its consequences in tumour development in a large cohort of 1461 patients with benign or malignant liver tumours.
Materials and methods
Patients and tissue samples
A series of 1461 patients was included in the study approved by our local institutional review board (IRB) committees (CCPRB Paris Saint-Louis, 1997 and 2004; Bordeaux 2010-A00498-31, Ile-de-France VII: projects C0-15-003 and PP 16–001). Liver tissues were frozen immediately after surgery in French hospitals. Tumour and non-tumour counterparts were analysed in 1269 patients, only the tumour or non-tumour tissues were investigated for 138 and 54 patients, respectively. The present series included HCC (n=936), hepatocellular adenomas (HCA, n=225), focal nodular hyperplasia (FNH, n=97), hepatoblastoma or transitional tumours (n=87), cholangiocarcinoma (n=46), fibrolamellar carcinoma (n=36) and other tumours (n=34, online supplementary table 1).
Supplementary file 1
Viral DNA screening
Genomic DNA were analysed for the presence of viral DNA by quantitative RT-PCR (qRT-PCR) on Fluidigm 96.96 dynamic arrays using the BioMark Real-Time PCR system with TaqMan probe sets designed with Primer3Plus software (online supplementary figure 1A and table 2). Results were analysed using the Fluidigm Real-Time PCR Analysis software (V.4.1.3) and reported to a reference gene, HMBS. The quantification was expressed in viral copy number/cell. Copy number/cell values were tested for unimodal and bimodal distribution using normalmixEM function of mixtools package in R.30
Isolation of human AAV using viral capture sequencing
Viral capture of genomic DNA was performed for tumour and matched normal sample, sequence as previously described using 120-mer primers recognising all AAV genotypes 1–13 already described with around 305 probes/genotype.25 Viral reads were mapped to all AAV1 to AAV13 reference sequences using Burrows-Wheeler Aligner (V.0.7.15).31 The number of AAV reads correlates with the number of viral copies/cell (online supplementary figure 1B). Read pairs with at least one read aligned on the virus were extracted using samtools (V.1.3),32 and aligned to a custom reference genome including human chromosomes and virus sequences. We calculated the number of reads mapping the AAV/human chimeric and mate regions in each samples by generating a 20k-bin size bed for hg19 genome, which was used for computations with bedtools multicov utility.33 For each bin, we calculated the mean of coverage in the samples displayed in a pan genomic plot. We used chimeric reads to identify insertion breakpoints at base resolution by mapping sequences on both sides of the junctions. Clonal events were considered when >25 reads overlapped the same locus, putative subclonal insertions when 4–24 overlapping reads were identified. All viral insertions were validated by visual inspection on Integrative Genomics Viewer. Sequences have been deposited in the Genbank database MK231253 to MK231264 and KT258720 to KT258730.
The analysis of full-length human-AAV sequences is detailed in online supplementary materials and methods. Sequences have been deposited in the Genbank database MK139243 to MK139299 and MK163929 to MK163942.
RNAseq
Samples enriched in poly(A)+ RNA were sequenced using Illumina TruSeq Stranded mRNA kit on HiSeq2000 sequencer, yielding approximately 45 million 100 base pair (bp) paired-end reads (IntegraGen, Evry).34 Reads were aligned and chimeric sequences reconstructed with TopHat235 and Cufflinks V.2.2.1.36 We used ElemeNT37 to predict transcription start sites (TSS), Alamut Visual software (Interactive Biosoftware) to identify splicing signals on the chimeric DNA sequence, ATGpr38 to identify translation initiation sites and Poly(A) Signal Miner to identify PolyA sites.39 Sequences were deposited in EGA database (EGAS00001002879, EGAS00001001284 and EGAS00001003310).
Detection of viral episomal form
A specific DNAse/TaqMan-based assay was adapted from protocol by Werle-Lapostolle et al 40 to detect AAV episomal form (detailed procedures in online supplementary materials and methods). Junctions of the circular AAV were amplified using two couples of primers surrounding the ITRs (online supplementary table 2) in 2.5% glycerol and 5% dimethyl sulfoxide. PCR products were sequenced by Sanger after ExoSAP-IT (Applied Biosystem) purification.41
Quantitative RT-PCR
AAV mRNA and inserted target genes expressions were analysed using qRT-PCR. Specifically, we used seven AAV custom made and human catalogue TaqMan probes (online supplementary table 2) with AB7900HT PCR System (Applied Biosystem) and BioMark Real-Time PCR system. Expression data were normalised with the 2−ΔCt method relative to ribosomal 18S (Hs03928990_g1). Five normal tissues were used as reference.
Site-directed mutagenesis
The role of the viral polyA signal in AAV-induced gene overexpression was investigated in two plasmids containing AAV insertions in the 3’UTR of TNFSF10. 25 QuikChange Lightning site-directed mutagenesis kit (Agilent) was used to introduce four point mutations in the viral polyA signal (NC_001401: 4424 A>C, 4426 T>G, 4427 A>C, 4429 A>C). All mutations were verified using Sanger sequencing.
Cell culture, transfection and dual luciferase assay
HuH7, HepG2 and HuH6 cells were purchased from ATCC and cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin. Cells were tested for mycoplasma contamination. Identity was verified by exome sequencing. Cells were transfected using Lipofectamine 3000 (Life Technologies) with pmirGLO plasmid (Promega) containing wild-type TNFSF10 3’UTR, the 3’UTR with AAV2 insertions or scrambled AAV2 sequence downstream a luciferase reporter gene. Luminescence from firefly luciferase was normalised on the corresponding renilla luciferase activity. Fold change was calculated relative to the wild-type TNFSF10 3’UTR construct.
Statistical analysis
Statistical analyses were performed using RStudio (V.1.0.136) and GraphPad Prism (V.6.0a). Relationship between AAV and clinical, histological features of the patients was investigated using Χ2 test. P values adjustment was computed for a Monte Carlo test with 2000 permutations. Statistical significance of quantitative variable was determined by Wilcoxon rank-sum test. Association among variables was modelled by a multinomial logistic regression. Luciferase activity of transfected versus control cells was compared using Student’s t-test. All tests were two-tailed and a p value <0.05 was considered as statistically significant.
Results
Identification of two major AAV genotypes in the liver
Screening of frozen liver tissues from 1319 patients with 6 Taqman probes distributed along the genome that collectively recognise all AAV genotypes 1–13 identified AAV DNA in 18% (n=233) of non-tumour liver tissues (online supplementary figure 1). For viral AAV DNA capture of all known genotypes 1–13, we selected 80 non-tumour liver samples including 68 positive samples ranging from 2×10–4 to 0.18 copy number/cell. After sequencing, a full-length AAV sequence was reconstructed in 57 samples and two major AAV subtypes were identified (figure 1A-B). The first subtype (n=25) is highly similar to AAV2 reference sequence (NC_001401) and to VP1 clade B genotype isolated in human14 42 (online supplementary table 2). The second subtype (n=32) showed hybrid sequences including various parts of the AAV13 capsid (similar to clade C14 42 and c-ter in the context of an AAV2 5’ part, it was named AAV2/13 (figure 1B and online supplementary figure 2). We identified along the viral genome 42 silent variants shared by both AAV subtypes, but different from the AAV2 reference NC_001401 (figure 1C). In contrast, several nucleotide variants leading to amino acid substitutions in AAV2/13 sequences were located in the hypervariable regions (HVRs) 5, 6 7 and 10 and originated from AAV13 sequence (figure 1B-C). Screening the overall series of 1319 samples with two probes specific of AAV2/13 subtype and located in the CAP2 region (online supplementary figure 1), identified 47.6% AAV2 and 52.4% AAV2/13 genotypes among 143 samples positive for the variable region.

Adeno-associated virus (AAV) full-length sequences in 57 human liver tissues. (A) Schematic representation of AAV genome (reference NC_001401) with location of the two open reading frames encoding replication proteins (Rep78, Rep68, Rep52 and Rep40), structural proteins (VP1, VP2 and VP3) and assembly activating protein (AAP) protein. Inverted terminal repeats (ITR) are represented on the 5’ and 3’ ends. Promoters (p5, p19 and p40) are indicated with arrows. (B) Nucleotides sequences (4679 bp) from 57 full-length AAV isolated from human liver tissues (ID number indicated with #) multialigned with the ClustalW algorithm compared with reference sequences on the top, AAV2 (NC_001401, in white), AAV3 (NC_001729.1) and AAV13 (EU285562.1). Two distinct viral genotypes, AAV2 and AAV2/13 were identified. Colour bars indicated nucleotide divergence with the AAV2 reference genome similar to AAV3 and/or AAV13 genomes (green) or not (grey), similarities with NC_001401 are in white. Variations due to flip-flop ITR configurations compared with AAV2 reference are labelled in light grey. The liver-specific enhancer-promoter element (LSP) described by Logan et al is indicated.29 (C) Amino acid variations compared with the AAV2 reference are indicated. The triangles indicate genome location of specific AAV2/13 (top) or AAV2 (middle) variants in the series of 57 human liver AAV isolates. Common variants shared by both genotypes are shown (bottom). Grey and black colours refer to silent and missense AAV variants, respectively; numbers correspond to wild-type AAV2 nucleotide sequence coordinates (NC_001401).
AAV infection and episomal form
In the 233 AAV positive liver samples, quantification of the viral DNA showed a bimodal distribution: 97% of the tissues exhibited a low number of copy/cell (ranging from 4.6×10–5 to 0.04) and only 8 patients showed a higher quantity of AAV ranging from 0.07 to 0.18 copy/cell (figure 2A). AAV was significantly enriched in female (p<0.001), young patients (p=0.016) and occurred more frequently in a background of non-fibrotic liver (p<0.001; figure 2B).

Adeno-associated virus (AAV) DNA in non-tumour tissues and viral episomal form. (A) Copy number/cell distribution in 233 samples. The density line defines the low and high positivity groups in blue and red, respectively. (B) Contingency analysis of AAV positive and negative patients according to gender, age and Metavir fibrosis score. Frequency of AAV-positive patients is displayed (χ² test with Monte Carlo simulation and χ² test for trend in proportions for Metavir score). (C) Frequency of RNA expression according to REP and CAP viral transcripts in patients with episomal and not-episomal AAV (χ² test with Monte Carlo simulation). (D) Viral copy number/cell (log10) in AAV-positive samples according to the episomal status and the transcriptional activity of the episome (Wilcoxon rank-sum test). (E) Distribution of the different viral molecular forms according to the age of the patients. *P<0.05, **p<0.01, ***p<0.001.
In 64/233 (27.5%) of the tissues positive for AAV, all the genomic AAV regions were amplified suggesting the presence of the entire viral genome. We designed a DNAse/TaqMan-based assay (online supplementary figure 3A), which allowed to detect episomal AAV in 60 patients, corresponding to 26% of AAV positive samples and 4.6% of all patients. Using in silico analyses of the AAV capture sequencing, among the 57 cases with a complete reconstructed AAV genomic sequence, we identified 14 cases with 3’ITR–5’ITR junctions. Circularised concatemeric structures may escape from our experimental method to identify episomal form,43 however, we did not identified insertion of concatemer in silico. The 3’ITR–5’ITR junctions showed various sequences presenting a double-D ITR structure, in flip or flop configuration, with a 125 bp deletion confirmed by Sanger sequencing (online supplementary figure 3C-D and 4).
AAV transcription is associated with episomal form
Then, we screened for AAV RNA expression in 101 non-tumour liver tissues positive for AAV by qRT-PCR. AAV transcript was identified in 64% of the tested liver tissues. Either AAV REP or CAP expression were enriched in liver tissues with episomal form (p<0.001) and both transcripts were more frequently associated in presence of episomal than not-episomal AAV form (p=0.022), defining a population of patients with an ‘episomal-expressed AAV’ (figure 2C). A higher AAV copies per cell was identified in liver tissues with episomal-expressed AAV, supporting the hypothesis of a viral active infection in these liver samples (figure 2D). Episomal AAV were also more frequent in female patients (p<0.001) and patients without cirrhosis (p<0.001; online supplementary figure 5A). Analysis of AAV positivity in function of age showed a peak of frequency at 25% in the 30–40 years class. AAV episomal form was more frequent in young patients (aged <40 years old) reaching the highest frequency level in the twenties (figure 2E and online supplementary figure 5B). These results suggest that AAV active infection is more frequent in the second and third decade during life, while inactive not-episomal forms subsist after the primary infection.
Co-infection with AAV helper viruses
As AAV is a defective virus, we searched for the presence of potential AAV helper viruses by screening the entire cohort of 1319 liver tissues for human adenoviruses (AdV types A–F), human herpesviruses (HHV type 1, 2, 4, 5, 6, 7 and 8) and human papillomavirus type 16 (HPV16) by qRT-PCR. At least one of these viruses was detected in 43% of the patients (n=570), and only one per patient in 39% (n=520). HHV6 was the most frequent (39%), then HHV4 (Epstein-Barr virus, 6%), while HHV7 and adenovirus were only rarely detected (2% and 0.5%, respectively, figure 3A). No HPV16 and HHV type 1 (HSV1), 2 (HSV2), 5 (CMV) and 8 (KSHV) were found in our cohort of liver tissues. HHV6 was the only helper virus enriched in AAV-positive patients (37.3% vs 44.8%, p=0.039), in particular in patients with episomal or expressed-episomal forms (52.5% and 67.9%, respectively, p<0.001; figure 3B).

Helper viruses according to adeno-associated virus (AAV) status. (A) Frequency of helper viruses’ infections and co-infection in non-tumour tissues (n=1319). (B) Global frequency of human herpesvirus (HHV)6, Epstein-Barr virus (EBV), HHV7 and human adenovirus (AdV) infection according to AAV presence and form (χ² test for trend in proportions). (C) Multivariate analysis for global AAV positivity (left) including the variables closely related to AAV presence in the univariate analysis (logistic regression). The same analysis was performed for the presence of episomal AAV (middle) and episomal and expressed form (right). *P<0.05, ***p<0.001. ns, not significant.
To identify independent features associated with AAV infection in the overall cohort of patients, we performed a multivariate analysis (figure 3C). Female gender (OR=1.83, p<0.001), the age (OR=1.42, p=0.044), non-cirrhotic liver (OR=1.96, p<0.001) and co-infection with HHV6 (OR=1.15, p=0.031) were independently associated with AAV positivity. Three factors were also significantly associated with the presence of episomal and expressed AAV: female gender (OR=4.71, p=0.013), non-fibrotic liver (OR=12.13, p=0.018) and co-infection with HHV6 (OR=1.61, p=0.01).
AAV in tumour tissues
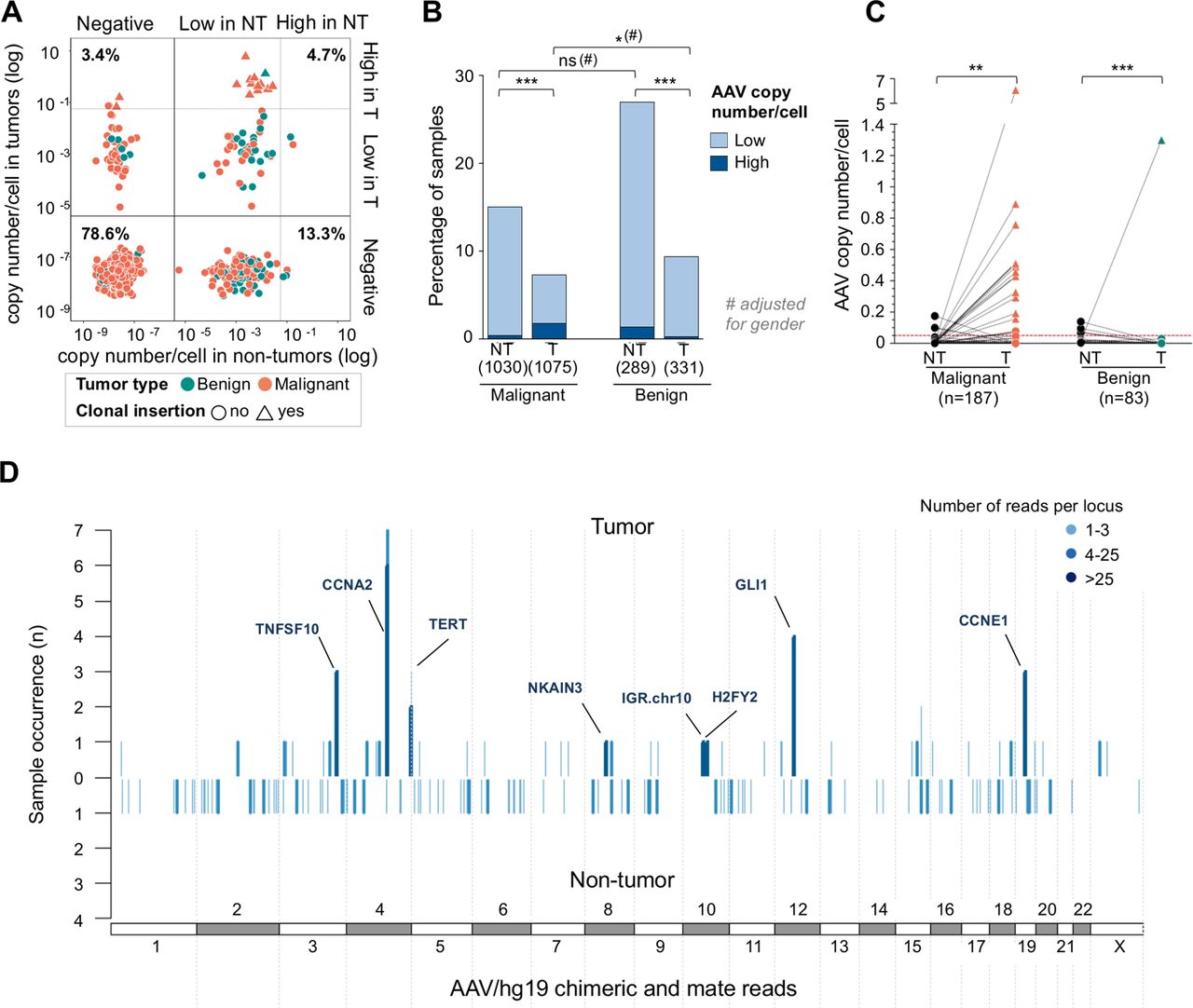
AAV DNA positivity was less frequently identified in the tumour tissues (n=109, 8%) compared with non-tumour liver tissues (n=233, 18%) with only 4.7% of patients presenting AAV in both tumour and non-tumour compartments (figure 4A). Twenty out of the 109 positive tumours showed a high number of AAV copies/cell ranging from 0.07 to 6.08. This value might be underestimated considering both potential contamination by normal cells and ploidy of tumour hepatocytes. The vast majority (n=83, 76%) had only one or two amplified viral regions with an enrichment for the 3’ITR region of the virus (online supplementary figure 6A-B). AAV was detected with a similar frequency in malignant and benign tumours, but with a higher number of copies/cell in malignant tumours corresponding to the clonal AAV insertion events (figure 4B-C; online supplementary table 3). Conversely, in all patients with benign tumours except one with focal nodular hyperplasia (FNH), AAV was more highly positive in the non-tumour counterpart than in the corresponding tumour (figure 4C). Finally, viral episomal forms were rarely identified in tumours (n=8, 0.6%), mostly in benign tumours (four HCA and two FNH) and only two HCC (supplementary figure 3C-D and 4).

Adeno-associated virus (AAV) in tumour tissues and non-tumour liver counterparts. (A) Copy number/cell (log10) of paired tumour (T) and non-tumour (NT) tissues of each patient (n=1269). Solid and dashed line define, respectively, the threshold of positivity and the boundaries between high and low number of viral copies/cell. The frequency of patients with AAV in both tumour and non-tumour counterparts or only in one of them is indicated. (B) Frequency of AAV in tumour and non-tumour tissues of patients with malignant and benign tumours (χ² test with Monte Carlo simulation and Cochran-Mantel-Haenszel for gender adjustment). (C) AAV copy number/cell of paired tumour and non-tumour tissues of 270 AAV-positive patients grouped in malignant and benign tumour patients. Triangles represent the tumours with clonal AAV insertions (Wilcoxon rank-sum test). (D) Pan-genomic views of genomic location of the human/virus matching chimeric and mate reads in tumour (top) and non-tumour (bottom) samples. A line corresponds to a 20k-bin region, colour refers to the average number of reads counted per bin. The height of the lines corresponds to the frequency of presence of reads in the series of samples, considering 94 tumours and 82 non-tumours investigated with viral capture deepseq. *P<0.05, **p<0.01, ***p<0.001. ns, not significant.
AAV insertion in liver tissues
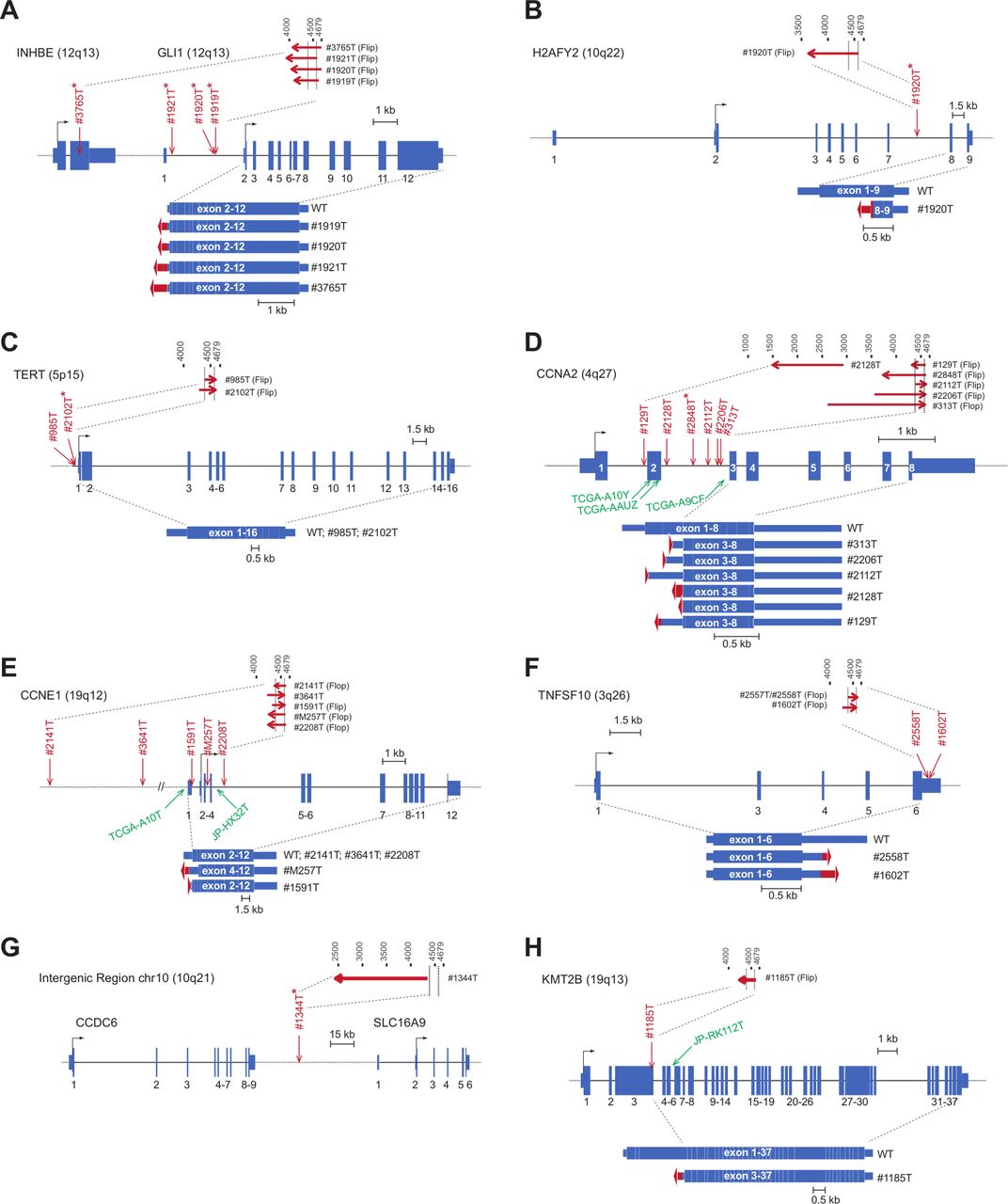
We identified seven novel clonal insertions in six HCCs, in GLI1/INHBE, (figure 5A-B) TERT (figure 5C) and CCNA2 (figure 5D). Only one clonal insertion was identified in a benign focal nodular hyperplasia, it occurred in an intergenic region of chromosome 10 (figure 5G) without consequences on the expression of the nearest genes (figures 4D and 5). Combining with AAV insertions identified in TCGA and ICGC sequenced HCC44 45 and previously described cases in our cohort,25 34 we re-analysed a total of 30 independent AAV insertions in liver tumours (online supplementary table 4). Viral insertions occurred in both directions, AAV2 and AAV2/13 subtypes were equally represented (55% vs 45% of the interpretable cases, respectively) and the minimal AAV region commonly inserted (nucleotide 4390–4570) was identified in 25 out of the 30 insertions.

Adeno-associated virus (AAV) clonal integration sites and transcripts consequences in tumours. Genes structure are schematised with boxes referring to exons and lines to introns regions. Transcription start sites location is shown on 5’ of the gene. Arrows indicate viral insertion sites in our series, in red, and in TCGA and ICGC tumours, in green. Asterisks refer to new inserted cases. Top lines refer to inserted AAV viral regions and arrows to 5’>3’ sequence orientation. Flip or flop 3’ITR are indicated. Observed transcripts are represented at the bottom of the gene structure with fusion viral sequences in red. WT, wild type.
Six oncogenes were recurrently activated by AAV (online supplementary figure 7). Insertions in GLI1/INHBE (four adenomas transformed into HCC, figure 5A-B), TERT (two HCC, figure 5C), CCNE1 (seven HCC, figure 5E), TNFSF10 (two HCC, figure 5F) and KMT2B (two HCC, figure 5H) led in almost all the cases to an overexpression of full-length coding region of these oncogenes by a promoter and/or a enhancer cis mechanism (figure 5). CCNA2 was inserted in nine HCC; all insertions but one clustered in CCNA2 intron 2, they resulted in an abnormal AAV-CCNA2 transcript leading to a stable oncogenic truncated protein lacking the N-terminal regulatory domain (figure 5D).34 The 3’UTR of TNFSF10 showed AAV insertions in two HCC inducing TNFSF10 overexpression with transcripts that prematurely ended at the viral polyadenylation (figure 5F). Here, using site-directed mutagenesis of both insertions, we demonstrated that the viral polyA signal is required to ensure a strong luciferase overexpression in three different tested cell lines (online supplementary figure 8).
In the non-tumour liver tissues, no clonal AAV insertions were identified; non-clonal insertions were significantly associated with the presence of episomal AAV (p<0.001), in contrast to the tumour samples. In both non-tumour and tumour tissues, non-clonal AAV insertions were randomly distributed along the genome (figure 4D and online supplementary figure 9). No specific enrichment was found in major target of AAV previously described in cell lines.46 47
AAV features and tumour heterogeneity
We explored intertumour heterogeneity by analysing multinodules (n=475) from 186 patients for the presence of viral DNA, clonal insertions and episomal form. Of those, AAV DNA was detected in 25 patients (online supplementary figure 6C), including 4 patients with clonal AAV insertion in at least one nodule. Two patients with HCC displayed clonal AAV integrations in all nodules. Thanks to the next generation sequencing (NGS) data, we were able to predict the evolution of these tumours by looking at the common and private somatic mutations and copy number alterations (CNA) in each nodule. Interestingly, the two tumours from patient #2557 showed the same viral insertion in TNFSF10, similar gene mutations and CNA profiles, demonstrating that AAV insertion is a truck alteration occurring before intrahepatic metastasis (figure 6A). Conversely, the three tumours from patients #1919, resulting from a malignant transformation of adenoma in carcinoma, harboured three different clonal insertions all targeting GLI1, with different gene mutation profile and no CNA suggesting that the three nodules have an independent origin (figure 6B).

Tumour development in patients with multiple nodules and clonal adeno-associated virus (AAV) insertions. The relation between the tumours is determined according to gene mutation profile and copy number alteration (CNA) of each nodule. The number of shared and private alterations is indicated above each branch. The major alterations with amino acid consequences are listed; mutations in driver genes and main CNAs are in bold. The AAV status, diagnosis and sources of genomic information (WGS, WES) are specified for each nodule. The thickness of the branch indicates the number of alterations. The position of the nodules for each patient is represented on the right. (A) The two hepatocellular carcinoma (HCC) nodules of patients #2557 display the same AAV insertion in TNFSF10 and they share 199 somatic mutations and several CNAs. This profile suggests that the nodules originate from the same primary tumour. (B) The three nodules of patient #1919 are heterogeneous for mutation profile and AAV insertions suggesting an independent origin of the tumours. NT, non-tumour.
Discussion
In this study, we provided a comprehensive description at large scale of the different AAV viral forms in the liver and of its oncogenic consequences, contributing to better understand the natural history of AAV infection in human.
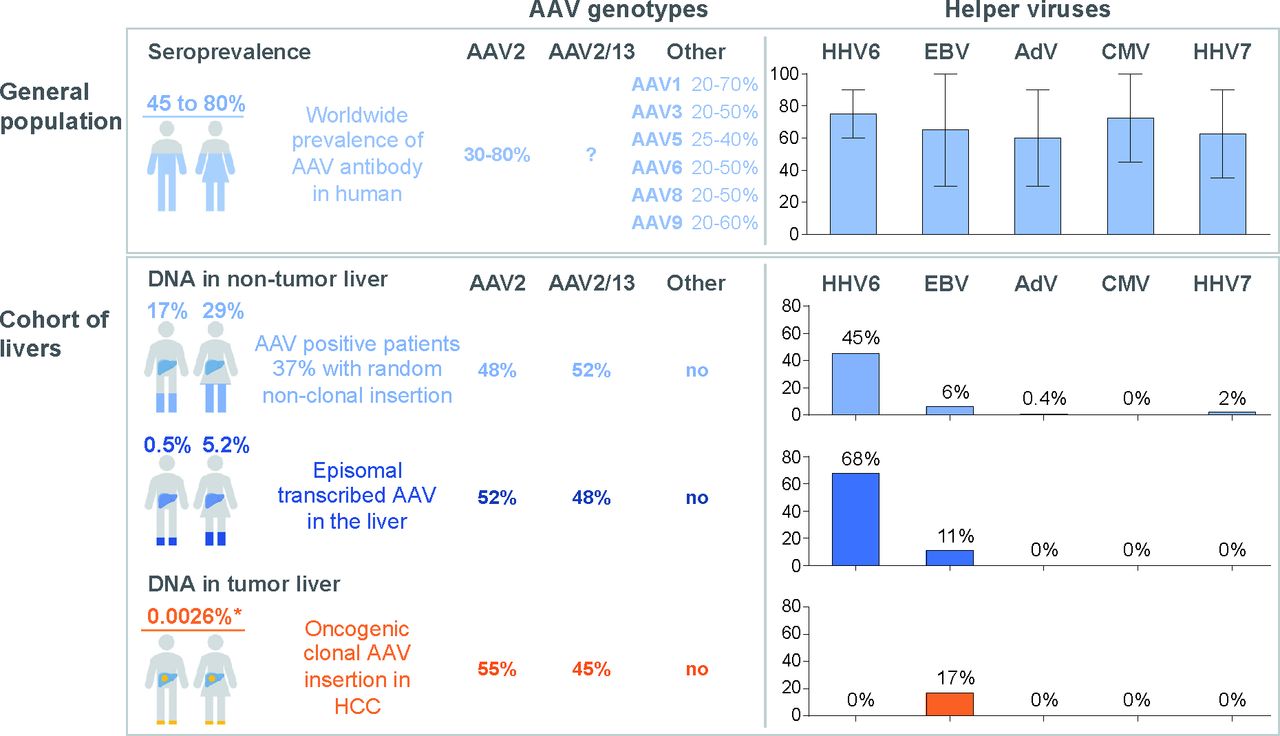
The prevalence of AAV was observed in 21% of patients in non-tumour and/or tumour liver in agreement with the seroprevalence of antibody against AAV identified in 30%–80% of the general population.10–12 48 Our result showed that one out of five patients demonstrates persistent AAV DNA in the liver during life, mainly in the population of young and female patients without liver fibrosis (figure 7). However, since most of our liver tissues were sampled from patients with liver diseases, the exact prevalence of AAV DNA in the liver of healthy individuals remains to be evaluated.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adeno-associated virus (AAV) and helper viruses in the general population and in human liver. Frequency of different AAV genotypes and seroprevalence of AAV10–12 and helper viruses50 51 in the general population are showed in the upper panel. The error bar in the histogram represents the range of helper viruses seroprevalence according to the literature. The bottom panel summarises the results found in this study, with estimated frequencies in the general population. For men and women, the global AAV frequency, the presence of episomal transcribed AAV and the prevalence of oncogenic clonal AAV insertions are indicated. *This prevalence is normalised according to the frequency of clonal AAV insertion in hepatocellular carcinoma (HCC) (2%) and the prevalence of HCC in France (0.013%). AdV, human adenovirus; EBV, Epstein Barr virus; HHV, human herpesvirus.
Only two AAV genotypes, AAV2 and hybrids AAV2/13, were identified in our cohort equally distributed among the patients. AAV2/13 sequences were hybrids between AAV2 in the 5’ part and AAV13 in the 3’ corresponding to the previous clade C of the VP1 classification.14 Since only one full-length AAV sequence from clade C was publicly available,42 our work significantly increased the number of human AAV full-length sequences enlightening the genomic variants associated with an efficient natural AAV infection in the liver. In contrast to previous serological analysis,10 11 49 we did not identified other AAV genotypes in the liver, even if AAV5 and AAV8 were frequent in circulating monocytes.48
AAV episomal form was identified in the non-tumour tissues of 4.6% of the patients, representing 26% of all AAV-positive liver samples, whereas episomal AAV has only been described in human tonsil and adenoid previously.9 It was frequently associated with viral mRNA expression suggesting that the episomal AAV are also transcriptionally active in a significant proportion of the population in the liver (figure 7). Several viruses50 51 are able to support AAV replication in vitro, and it was commonly admitted that adenovirus is the natural AAV helper. Here, we identified HHV6 as the virus most frequently associated with episomal and transcribed AAV in the liver. This co-occurrence was previously described in healthy blood donors48 and HHV6 is able to infect hepatocytes.52–54 The increased frequency of HHV6 in patients with episomal-expressed AAV form could indicate an ongoing active infection in the liver of 2.1% of the patients. In contrast, only very rare patients showed an association with adenovirus or other candidate helper viruses even in livers with episomal and expressed AAV (figure 7). All these results may suggest the role of HHV6 as the natural helper virus of AAV in the liver. However, co-infection with other helper viruses could occur at the initial acute AAV infection, followed by its clearance. Replication-competent infectious AAV has been rescued from human tonsil and adenoid tissue and lymphocytes, it remains to be searched in fresh liver tissues.48 55 Viral clones were isolated and their infectivity was tested in vitro in HeLa cells showing that only AAV clones with a complete double-D ITR structure were able to replicate and gave rise to infectious virus.55 Interestingly, the analysis of the ITRs junctions of the episomal form in our series highlighted the presence of the same double-D structure supporting its role in an active AAV infection. Moreover, here a peculiar link between episomal-expressed AAV in the liver and age suggested that AAV active infection occurs during the first three decades of life and then remains latent.
Analyses of the tumour tissues confirmed the selection of clonal AAV insertion in HCC development in non-cirrhotic liver. Recurrent somatic viral integrations were identified in 2% of our HCC cohort, targeting CCNA2 (33.3%), CCNE1 (27.8%), GLI1/INHBE (11.1%), TERT (11.1%), TNFSF10 (11.1%) and KMT2B (5.6%). AAV insertion induced the overexpression of the target genes through multiple mechanisms that differ according to the target and the localisation of the integration. Clonal insertions upstream the TSS or within the 5’ region of the gene lead to the gain of a positive regulatory mechanism such as the usage of viral enhancers and transcription factor binding sites (TFBS). Interestingly, a recent work by Logan et al has described a liver-specific enhancer-promoter element in wild-type AAV genome within the common inserted region in HCC tumours.29 It consists of 124 nucleotides sequence that contains TFBSs for HNF1-α, HNF6 and GATA6. Noteworthy, this region is absent in many AAV vectors currently in use and should raise a biosafety flag or be deleted in the remaining. In line with our finding, this result strongly supports the mechanism of AAV-induced overexpression of the target gene. In addition, viral insertions in CCNA2 and TNFSF10 genes led the expression of a truncated protein or the premature ending of the transcript within the viral polyA, respectively.
AAV oncogenic integrations were identified in our cohort of European patients with HCC. They were also observed in the ICGC-Japan cohort in 3 HCC cases out of 268 HCC (1.1%),45 in 4 out of 334 HCC (1.2%) of the TCGA cohort34 and in 2 out of 289 HCC (0.7%) from Korea.56 Interestingly, the most frequent AAV integrated oncogenes are similar to HBV, that are, CCNA2, CCNE1, TERT and KMT2B. The lower prevalence of AAV could be due to the lack of chronic liver disease associated with active AAV replication in contrast to chronic HBV infection. In the present series, we reinforced the link between AAV oncogenic insertion and the occurrence of HCC in normal liver, including recurrent AAV insertions in the malignant transformation of hepatocellular adenoma in carcinoma targeting GLI1 that defines the activated sonic hedgehog molecular subgroup of adenoma, shHCA.57 In the same line, AAV insertions in cyclin A2 or E1 in HCC are associated with unique chromosomal rearrangement signature and poor prognosis mainly occurring in HCC developed in normal liver.34 These results underline the role of AAV insertion in the development of a specific subgroup of HCC without other aetiologies.
In conclusion, we provided a portrait of AAV infection in the liver with a description of viral genotypes, molecular forms and helper virus paving the way for a renovated interest in wild-type AAV biology. New highlights on the understanding of the oncogenic consequences of AAV integration in HCC tumours emerged from this work. However, further studies are necessary to clarify the impact of AAV infection in additional cohort of patients and the frequency of insertional mutagenesis across different countries.
Acknowledgments
The authors would like to thank surgeons, pathologists and all the clinicians who collected samples and clinical data. Sophie Prevost, Service d’anatomie pathologique, APHP, Hôpital Antoine-Béclère, Clamart, France. Anne de Muret, Service d’anatomopathologie, Centre Hospitalier Régional Universitaire de Tours, Tours, France. Eric Viber, Centre Hépato-Biliaire, INSERM U785, Hôpital Paul Brousse, Villejuif, France. Philippe Merle, Department of Hepatology, Hospices Civils de Lyon, Croix-Rousse University Hospital, Lyon, France. Monique Fabre, Service Anatomie, HU-Necker Enfants Malades APHP, Paris. Nathalie Sturm, Department of Anatomie et Cytologie Pathologiques, CHU de Grenoble, Grenoble, France. Thomas Decaens, Service Hépato-gastroentérologie et Tumeurs du foie, CHU de Grenoble, Grenoble, France. Sophie Michalak, Département de Pathologie cellulaire et tissulaire, CHU ANGERS. Georges-Philippe Pageaux, service d’hépato-gastro-entérologie Hôpital St Eloi CHU Montpellier. Jean-Michel Fabre, service de chirurgie digestive Hôpital St Eloi CHU Montpellier. Emmanuel Boleslawski, service de chirurgie digestive et transplantation. Hôpital Huriez. Chru de lille. 59037 lille cedex. Marie Christine Saint Paul, service d’Anatomie Pathologique, CHU de Nice, Nice. Dominique Wendum, Department of Pathology, Saint-Antoine Hospital, APHP, Paris, France. Olivier Rosmorduc, Department of Gastroenterology and Hepatology, Hôpital de la Pitié-Salpêtrière, APHP, Université Pierre et Marie Curie UPMC, Paris. Jean Christophe Vaillant, service de chirurgie hépato-bilio- pancréatique, CHU Pitié-Salpetriere, Université Pierre et Marie Curie UPMC, Paris. Marianne Ziol, Service d’Anatomopathologie, Hôpital Jean Verdier, Hôpitaux universitaires Paris-Seine- Saint-Denis, APHP, Bondy, France. Nathalie Ganne, Department of Hepatogastroenterology, Hôpital Jean Verdier, APHP, Bondy, France. Luigi Terraciano, Basel University Hospital, Department of Pathology, Basel, Switzerland. Vincenzo Mazzaferro, University of Milan at the Istituto Nazionale Tumori IRCCS (National Cancer Institute). Celine Bazille, Service d’Anatomie Pathologie, CHU de Caen, Caen, France.
References
Footnotes
TLB and SI contributed equally.
Contributors Study design: J-CN, JZ-R, TLB, SI. Generation of experimental data: TLB, SI, CP, IM, SD. Analysis and interpretation of data: TLB, SI, CP, IM, SD, QB, SC, TZH, EL, J-CN, JZ-R. Collection of samples and related histological and clinical data: J-CN, JZ-R, JC, GM, CG, VP, GA, AL, LP, LC, PB-S, J-FB and investigators. Drafting of the manuscript: JZ-R, TLB, SI. Revision of the manuscript and approval of the final version of the manuscript: JZ-R, J-CN, TLB, SI, IM, SD, QB, SC, TZH, EL, JC, GM, CG, VP, GA, AL, LP, LC, PB-S, J-FB.
Funding This work was supported by INSERM, by INCa within the ICGC project, France Génomique, Cancéropole Ile de France (ExhauTrans project), ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé, National Alliance for Life Sciences & Health) within the framework of the Cancer Plan (“HTE program-HetColi network” and “Cancer et environnement program”), the Réseau national CRB Foie, Ligue Nationale contre le cancer: project équipe Labellisée, Fondation Schueller Bettencourt “coup d’élan”, Prix Ligue contre le Cancer comité de Paris René et Andrée Duquesne 2018, the SIRIC CARPEM and Fondation Mérieux, Labex OncoImmunology (investissement d’avenir) ANRS and the French Liver Biobanks network—INCa, BB-0033-00085, Hepatobio bank. QB is supported by a fellowship from the HOB doctoral school and the ministry of Education and Research, TLB is supported by an “Attractivité IDEX" fellowship from IUH and CP is supported by a doctoral fellowship funded by ANRS.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The sequencing data reported in this paper have been deposited to Genbank (accessions: KT258720-KT258730, MK139243-MK139299, MK163929-MK163942 and MK231253-MK231264) and EGA (European Genome-phenome Archive) database (RNA-seq accessions: EGAS00001002879, EGAS00001001284 and EGAS00001003310). All supplemental data and sequences are available at http://zucmanlab.com/wp-content/uploads/2019/05/Zucman-CaptureVirusProbes.xlsx.